
AAV-Vectored Expression of the Vascular Normalizing Agents 3TSR and Fc3TSR, and the Anti-Angiogenic Bevacizumab Extends Survival in a Murine Model of End-Stage Epithelial Ovarian Carcinoma

 ,
,  , ,
, ,  , and
, and 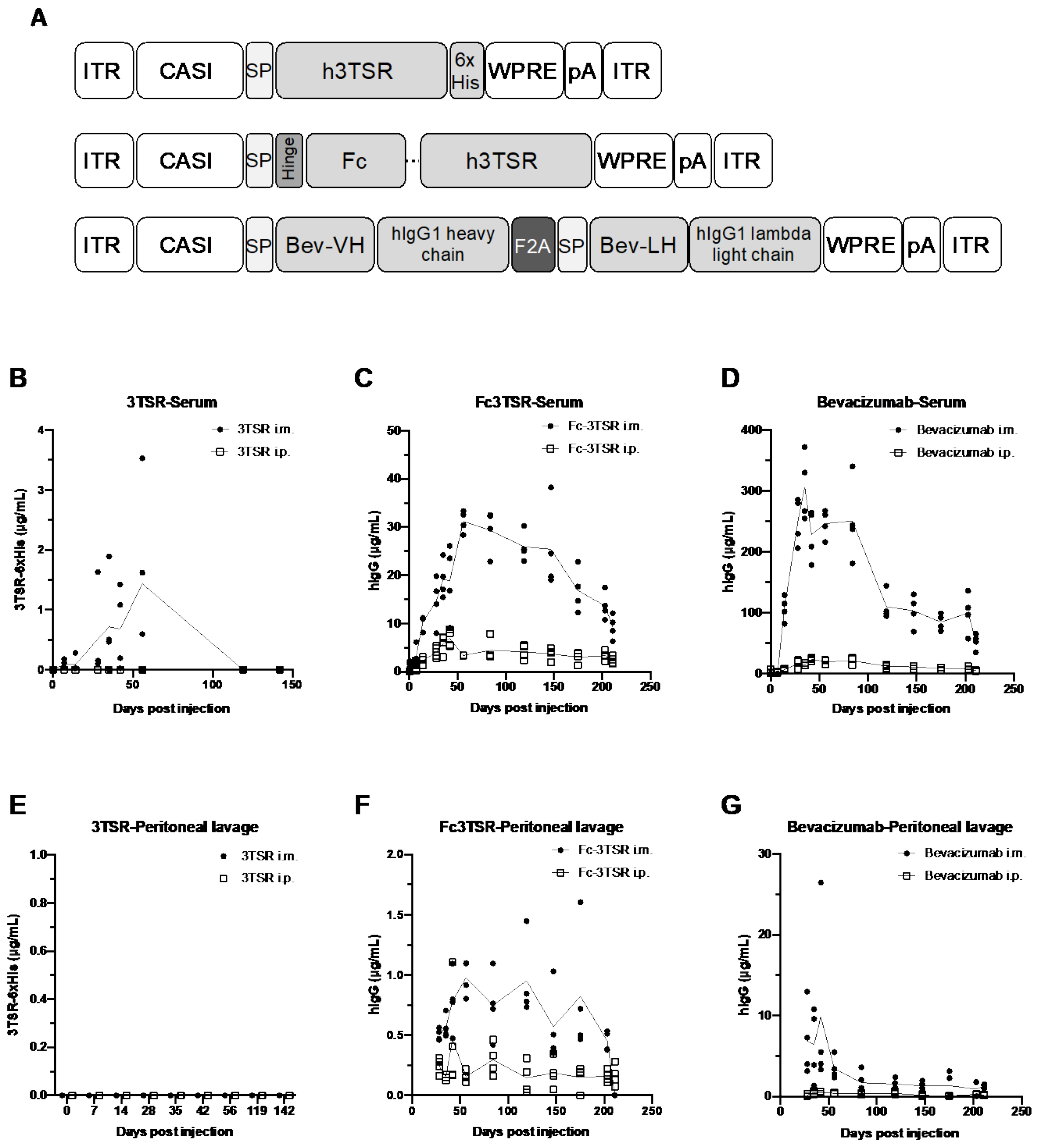{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Ethics
2.2. AAV Vector Construction and Virus Production
2.3. AOaV-1 Production and Purification
2.4. Transgene Expression Monitoring
2.5. ID8 Ovarian Epithelial Cancer Model
2.6. Flow Cytometry
2.7. Tumor-Directed Antibody Responses
2.8. ELISA
2.9. Statistical Analyses
3. Results
3.1. AAV-Mediated Expression of 3TSR, Fc3TSR and Bevacizumab Results in Sustained Transgene Expression in the Blood and Peritoneal Cavity, with Intramuscular Administration Resulting in Higher Expression Levels
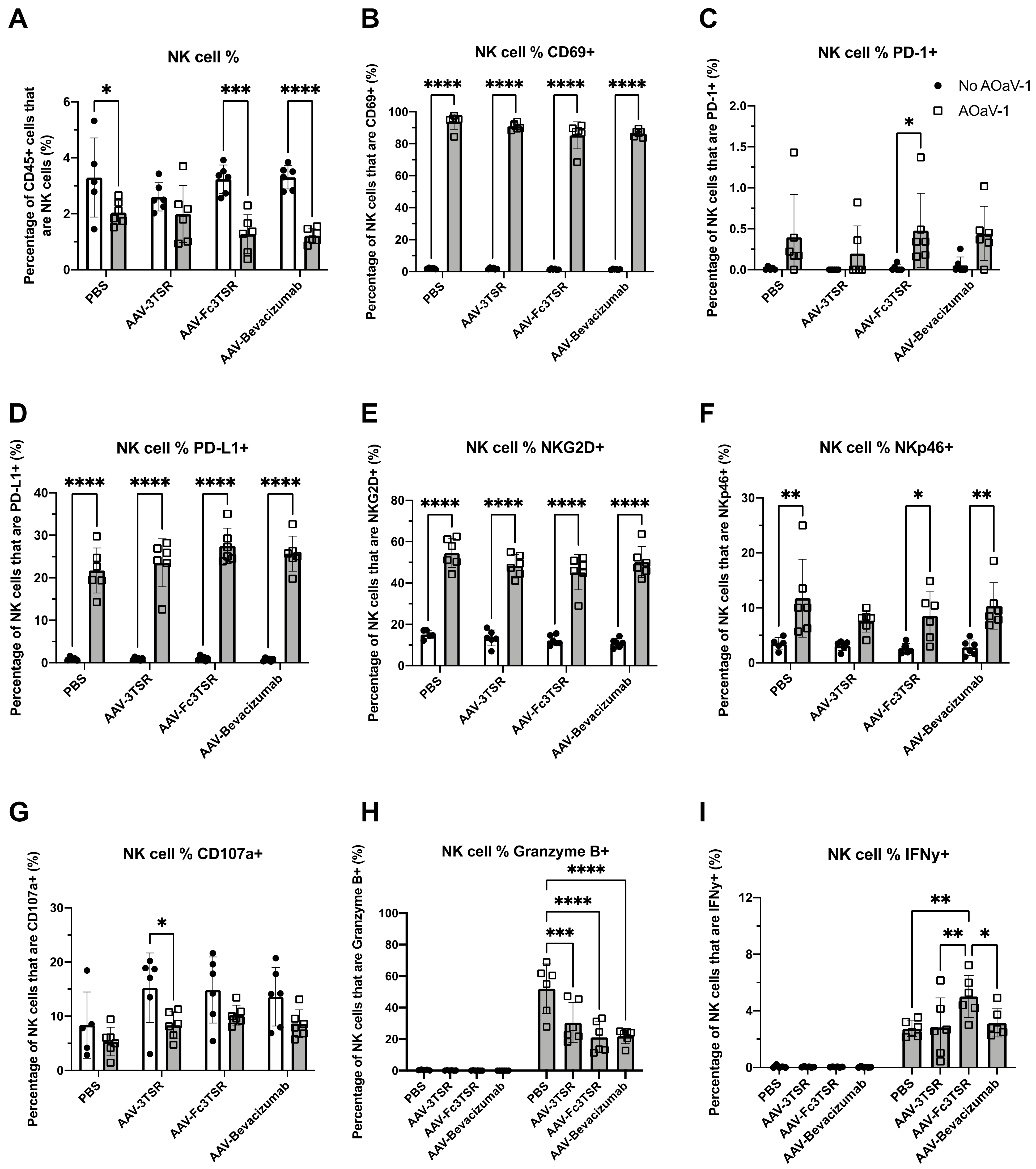
3.2. ID8 Tumor-Bearing Mice Expressing Vectorized Fc3TSR and Treated with Oncolytic AOaV-1 Had Significantly Increased Numbers of Activated NK Cells 36 h Post Treatment
3.3. Tumor Specific TNF-α+IFN-γ+CD8+ T Cells Were Significantly Increased in the AAV-Bevacizumab+AOaV-1 Treatment Group
3.4. AAV-3TSR Monotherapy Resulted in Significantly Higher Tumor-Specific Antibodies
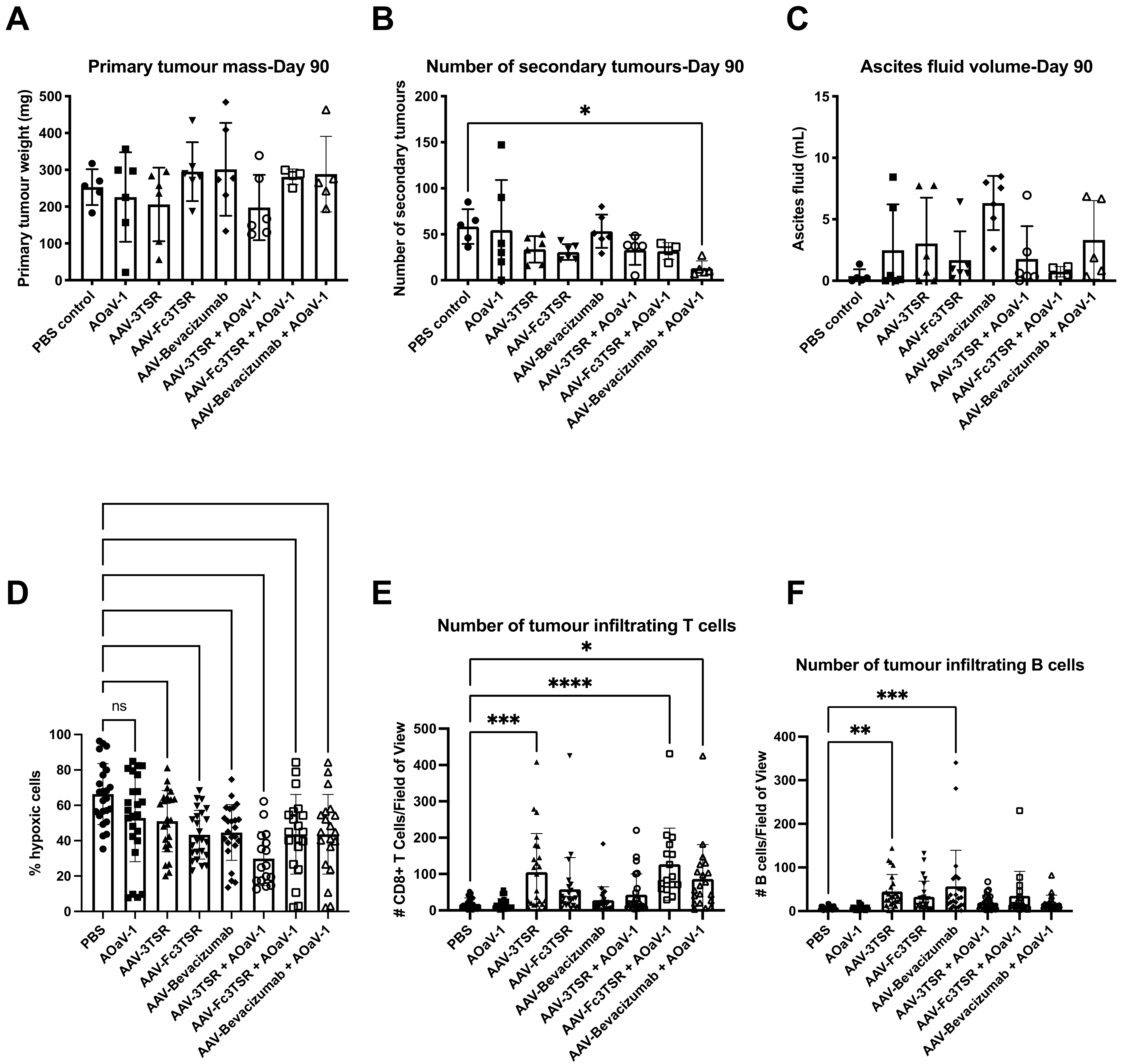
3.5. Analysis of Disease Progression at 90 Days Post Tumor Cell Implantation
3.6. Serum Expression Levels of 3TSR, Fc3TSR and Bevacizumab Diminished More Rapidly in Tumor Bearing Mice Than in Naïve Mice
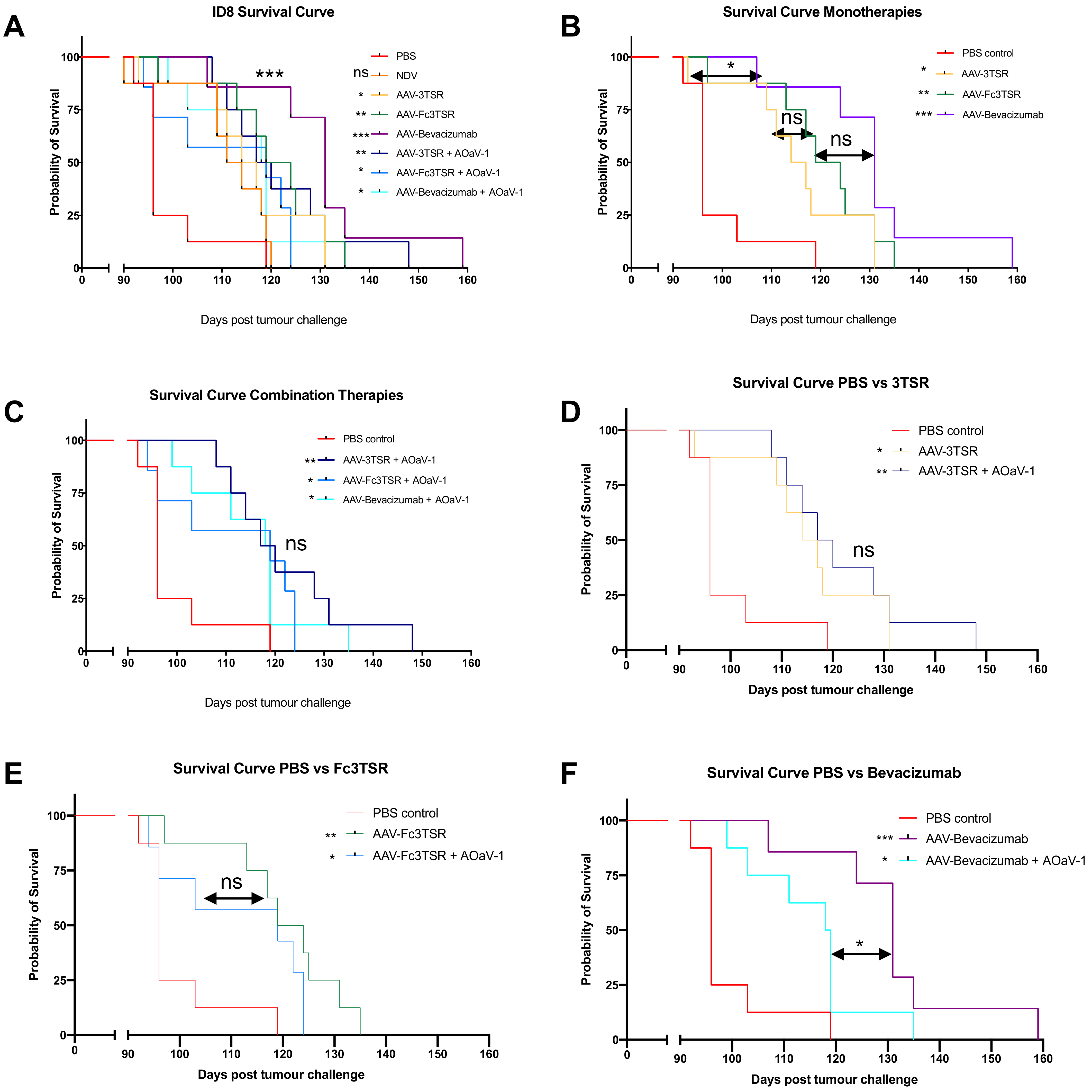
3.7. All Three AAV Treatments Significantly Extended Survival in the ID8 Ovarian Cancer Model with AAV-Bevacizumab Treatment Resulting in the Greatest Extension in Survival
4. Discussion
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Acknowledgments
Conflicts of Interest
References
- Momenimovahed, Z.; Tiznobaik, A.; Taheri, S.; Salehiniya, H. Ovarian cancer in the world: Epidemiology and risk factors. Int. J. Womens Health 2019, 11, 287–299. [Google Scholar] [CrossRef] [PubMed]
- Coburn, S.B.; Bray, F.; Sherman, M.E.; Trabert, B. International patterns and trends in ovarian cancer incidence, overall and by histologic subtype. Int. J. Cancer 2017, 140, 2451–2460. [Google Scholar] [CrossRef] [PubMed]
- Shabir, S.; Gill, P.K. Global scenario on ovarian cancer—Its dynamics, relative survival, treatment, and epidemiology. Adesh Univ. J. Med. Sci. Res. 2020, 2, 17–25. [Google Scholar] [CrossRef]
- Lengyel, E. Ovarian cancer development and metastasis. Am. J. Pathol. 2010, 177, 1053–1064. [Google Scholar] [CrossRef] [PubMed]
- Kyriakides, M.; Rama, N.; Sidhu, J.; Gabra, H.; Keun, H.C.; El-Bahrawy, M. Metabonomic analysis of ovarian tumour cyst fluid by proton nuclear magnetic resonance spectroscopy. Oncotarget 2016, 7, 7216–7226. [Google Scholar] [CrossRef]
- Chung, A.S.; Lee, J.; Ferrara, N. Targeting the tumour vasculature: Insights from physiological angiogenesis. Nat. Rev. Cancer 2010, 10, 505–514. [Google Scholar] [CrossRef]
- Niu, G.; Chen, X. Vascular endothelial growth factor as an anti-angiogenic target for cancer therapy. Curr. Drug Targets 2010, 11, 1000–1017. [Google Scholar] [CrossRef] [PubMed]
- McCormack, P.L.; Keam, S.J. Bevacizumab—A review of its use in metastatic colorectal cancer. Drugs 2008, 68, 487–506. [Google Scholar] [CrossRef] [PubMed]
- Kazazi-Hyseni, F.; Beijnen, J.H.; Schellens, J.H. Bevacizumab. Oncologist 2010, 15, 819–825. [Google Scholar] [CrossRef]
- Shih, T.; Lindley, C. Bevacizumab—An angiogenesis inhibitor for the treatment of solid malignancies. Clin. Ther. 2006, 28, 1779–1802. [Google Scholar] [CrossRef]
- Coleman, R.L.; Brady, M.F.; Herzog, T.J.; Sabbatini, P.; Armstrong, D.K.; Walker, J.L.; Kim, B.-G.; Fujiwara, K.; Tewari, K.S.; O’Malley, D.M.; et al. Bevacizumab and paclitaxel–carboplatin chemotherapy and secondary cytoreduction in recurrent, platinum-sensitive ovarian cancer (NRG oncology/gynecologic oncology group study GOG-0213): A multicentre, open-label, randomised, phase 3 trial. Lancet Oncol. 2017, 18, 779–791. [Google Scholar] [CrossRef]
- Perren, T.J.; Swart, A.M.; Pfisterer, J.; Ledermann, J.A.; Pujade-Lauraine, E.; Kristensen, G.; Carey, M.S.; Beale, P.; Cervantes, A.; Kurzeder, C.; et al. A phase 3 trial of bevacizumab in ovarian cancer. N. Engl. J. Med. 2011, 365, 2484–2496. [Google Scholar] [CrossRef]
- Berton, D.; Floquet, A.; Lescaut, W.; Baron, G.; Kaminsky, M.C.; Toussaint, P.; Largillier, R.; Savoye, A.M.; Alexandre, J.; Delbaldo, C.; et al. Real-World Experience of Bevacizumab as First-Line Treatment for Ovarian Cancer: The GINECO ENCOURAGE Cohort of 468 French Patients. Front. Pharmacol. 2021, 12, 711813. [Google Scholar] [CrossRef] [PubMed]
- Awwad, S.; Angkawinitwong, U. Overview of Antibody Drug Delivery. Pharmaceutics 2018, 10, 83. [Google Scholar] [CrossRef] [PubMed]
- Lambrechts, D.; Lenz, H.J.; de Haas, S.; Carmeliet, P.; Scherer, S.J. Markers of response for the antiangiogenic agent bevacizumab. J. Clin. Oncol. 2013, 31, 1219–1230. [Google Scholar] [CrossRef] [PubMed]
- Burger, R.A.; Brady, M.F.; Bookman, M.A.; Fleming, G.F.; Monk, B.J.; Huang, H.; Mannel, R.S.; Homesley, H.D.; Fowler, J.; Greer, B.E.; et al. Incorporation of bevacizumab in the primary treatment of ovarian cancer. N. Engl. J. Med. 2011, 365, 2473–2483. [Google Scholar] [CrossRef]
- Monk, B.J.; Pujade-Lauraine, E.; Burger, R.A. Integrating bevacizumab into the management of epithelial ovarian cancer: The controversy of front-line versus recurrent disease. Ann. Oncol. 2013, 24 (Suppl. S10), x53–x58. [Google Scholar] [CrossRef] [PubMed]
- Xie, Y.; Hicks, M.J.; Kaminsky, S.M.; Moore, M.A.; Crystal, R.G.; Rafii, A. AAV-mediated persistent bevacizumab therapy suppresses tumor growth of ovarian cancer. Gynecol. Oncol. 2014, 135, 325–332. [Google Scholar] [CrossRef]
- Azzopardi, N.; Lecomte, T.; Ternant, D.; Boisdron-Celle, M.; Piller, F.; Morel, A.; Gouilleux-Gruart, V.; Vignault-Desvignes, C.; Watier, H.; Gamelin, E.; et al. Cetuximab pharmacokinetics influences progression-free survival of metastatic colorectal cancer patients. Clin. Cancer Res. 2011, 17, 6329–6337. [Google Scholar] [CrossRef]
- Huang, Y.; Goel, S.; Duda, D.G.; Fukumura, D.; Jain, R.K. Vascular normalization as an emerging strategy to enhance cancer immunotherapy. Cancer Res. 2013, 73, 2943–2948. [Google Scholar] [CrossRef]
- Matuszewska, K.; Pereira, M.; Petrik, D.; Lawler, J.; Petrik, J. Normalizing Tumor Vasculature to Reduce Hypoxia, Enhance Perfusion, and Optimize Therapy Uptake. Cancers 2021, 13, 4444. [Google Scholar] [CrossRef] [PubMed]
- Tan, K.; Duquette, M.; Liu, J.H.; Dong, Y.; Zhang, R.; Joachimiak, A.; Lawler, J.; Wang, J.H. Crystal structure of the TSP-1 type 1 repeats: A novel layered fold and its biological implication. J. Cell Biol. 2002, 159, 373–382. [Google Scholar] [CrossRef] [PubMed]
- Lawler, J. The functions of thrombospondin-1 and-2. Curr. Opin. Cell Biol. 2000, 12, 634–640. [Google Scholar] [CrossRef]
- Sims, J.N.; Lawler, J. Thrombospondin-1-based antiangiogenic therapy. J. Ocul. Pharmacol. Ther. 2015, 31, 366–370. [Google Scholar] [CrossRef]
- Dawson, D.W.; Pearce, F.A.; Zhong, R.; Silverstein, R.L.; Frazier, W.A.; Bouck, N.P. CD36 mediates the in vitro inhibitory effects of thrombospondin-1 on endothelial cells. J. Cell Biol. 1997, 138, 707–717. [Google Scholar] [CrossRef]
- Miao, W.M.; Seng, W.L.; Duquette, M.; Lawler, P.; Laus, C.; Lawler, J. Thrombospondin-1 type 1 repeat recombinant proteins inhibit tumor growth through transforming growth factor-β-dependent and -independent mechanisms. Cancer Res. 2001, 61, 7830–7839. [Google Scholar]
- Russell, S.; Duquette, M.; Liu, J.; Drapkin, R.; Lawler, J.; Petrik, J. Combined therapy with thrombospondin-1 type I repeats (3TSR) and chemotherapy induces regression and significantly improves survival in a preclinical model of advanced stage epithelial ovarian cancer. FASEB J. 2015, 29, 576–588. [Google Scholar] [CrossRef]
- Yu, D.L.; Stegelmeier, A.A.; Chow, N.; Rghei, A.D.; Matuszewska, K.; Lawler, J.; Bridle, B.W.; Petrik, J.J.; Wootton, S.K. AAV-mediated expression of 3TSR inhibits tumor and metastatic lesion development and extends survival in a murine model of epithelial ovarian carcinoma. Cancer Gene Ther. 2019, 27, 356–367. [Google Scholar] [CrossRef]
- Goel, S.; Duda, D.G.; Xu, L.; Munn, L.L.; Boucher, Y.; Fukumura, D.; Jain, R.K. Normalization of the vasculature for treatment of cancer and other diseases. Physiol. Rev. 2011, 91, 1071–1121. [Google Scholar] [CrossRef]
- Matuszewska, K.; Ten Kortenaar, S.; Pereira, M.; Santry, L.A.; Petrik, D.; Lo, K.M.; Bridle, B.W.; Wootton, S.K.; Lawler, J.; Petrik, J. Addition of an Fc-IgG induces receptor clustering and increases the in vitro efficacy and in vivo anti-tumor properties of the thrombospondin-1 type I repeats (3TSR) in a mouse model of advanced stage ovarian cancer. Gynecol. Oncol. 2021, 164, 154–169. [Google Scholar] [CrossRef]
- Breuer, C.B.; Hanlon, K.S.; Natasan, J.S.; Volak, A.; Meliani, A.; Mingozzi, F.; Kleinstiver, B.P.; Moon, J.J.; Maguire, C.A. In vivo engineering of lymphocytes after systemic exosome-associated AAV delivery. Sci. Rep. 2020, 10, 4544. [Google Scholar] [CrossRef] [PubMed]
- Halbert, C.L.; Allen, J.M.; Miller, A.D. Adeno-associated virus type 6 (AAV6) vectors mediate efficient transduction of airway epithelial cells in mouse lungs compared to that of AAV2 vectors. J. Virol. 2001, 75, 6615–6624. [Google Scholar] [CrossRef] [PubMed]
- Zincarelli, C.; Soltys, S.; Rengo, G.; Rabinowitz, J.E. Analysis of AAV serotypes 1-9 mediated gene expression and tropism in mice after systemic injection. Mol. Ther. 2008, 16, 1073–1080. [Google Scholar] [CrossRef] [PubMed]
- Amer, M.H. Gene therapy for cancer- present status and future perspective. Mol. Cell Ther. 2014, 2, 27. [Google Scholar] [CrossRef]
- Pinto, C.; Silva, G.; Ribeiro, A.S.; Oliveira, M.; Garrido, M.; Bandeira, V.S.; Nascimento, A.; Coroadinha, A.S.; Peixoto, C.; Barbas, A.; et al. Evaluation of AAV-mediated delivery of shRNA to target basal-like breast cancer genetic vulnerabilities. J. Biotechnol. 2019, 300, 70–77. [Google Scholar] [CrossRef]
- Mahendra, G.; Kumar, S.; Isayeva, T.; Mahasreshti, P.J.; Curiel, D.T.; Stockardt, C.R.; Grizzle, W.E.; Alapati, V.; Singh, R.; Siegal, G.P.; et al. Antiangiogenic cancer gene therapy by adeno-associated virus 2-mediated stable expression of the soluble FMS-like tyrosine kinase-1 receptor. Cancer Gene Ther. 2005, 12, 26–34. [Google Scholar] [CrossRef]
- Takei, Y.; Mizukami, H.; Saga, Y.; Yoshimura, I.; Hasumi, Y.; Takayama, T.; Kohno, T.; Matsushita, T.; Okada, T.; Kume, A.; et al. Suppression of ovarian cancer by muscle-mediated expression of soluble VEGFR-1/Flt-1 using adeno-associated virus serotype 1-derived vector. Int. J. Cancer 2007, 120, 278–284. [Google Scholar] [CrossRef]
- Harding, T.; Donahue, B.; Wang, J.; Koprivnikar, K.; Yendluri, S.; Tu, G.-H.; Cordier, L.; Colbern, G.; VanRoey, M.; Gonzalez, M.; et al. AAV vectors for brain cancer gene therapy. Mol. Ther. 2003, 7, S89. [Google Scholar] [CrossRef]
- Santiago-Ortiz, J.L.; Schaffer, D.V. Adeno-associated virus (AAV) vectors in cancer gene therapy. J. Control. Release 2016, 240, 287–301. [Google Scholar] [CrossRef]
- Li, T.; Kang, G.; Wang, T.; Huang, H. Tumor angiogenesis and anti-angiogenic gene therapy for cancer. Oncol. Lett. 2018, 16, 687–702. [Google Scholar] [CrossRef]
- Noro, T.; Miyake, K.; Suzuki-Miyake, N.; Igarashi, T.; Uchida, E.; Misawa, T.; Yamazaki, Y.; Shimada, T. Adeno-associated viral vector-mediated expression of endostatin inhibits tumor growth and metastasis in an orthotropic pancreatic cancer model in hamsters. Cancer Res. 2004, 64, 7486–7490. [Google Scholar] [CrossRef] [PubMed]
- Pan, J.G.; Luo, R.Q.; Zhou, X.; Han, R.F.; Zeng, G.W. Potent antitumor activity of the combination of HSV-TK and endostatin by adeno-associated virus vector for bladder cancer in vivo. Clin. Lab. 2013, 59, 1147–1158. [Google Scholar] [CrossRef] [PubMed]
- Ponnazhagan, S.; Mahendra, G.; Kumar, S.; Shaw, D.R.; Stockard, C.R.; Grizzle, W.E.; Meleth, S. Adeno-associated virus 2-mediated antiangiogenic cancer gene therapy: Long-term efficacy of a vector encoding angiostatin and endostatin over vectors encoding a single factor. Cancer Res. 2004, 64, 1781–1787. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Ma, H.I.; Lin, S.Z.; Chiang, Y.H.; Li, J.; Chen, S.L.; Tsao, Y.P.; Xiao, X. Intratumoral gene therapy of malignant brain tumor in a rat model with angiostatin delivered by adeno-associated viral (AAV) vector. Gene Ther. 2002, 9, 2–11. [Google Scholar] [CrossRef]
- Wu, Q.J.; Gong, C.Y.; Luo, S.T.; Zhang, D.M.; Zhang, S.; Shi, H.S.; Lu, L.; Yan, H.X.; He, S.S.; Li, D.D.; et al. AAV-mediated human PEDF inhibits tumor growth and metastasis in murine colorectal peritoneal carcinomatosis model. BMC Cancer 2012, 12, 129. [Google Scholar] [CrossRef]
- Zhao, S.; Zhang, Y.; Wang, L.; Yang, L.; Zou, L.; Gao, F. Adeno-associated virus 2 mediated gene transfer of vascular endothelial growth factor Trap: A new treatment option for glioma. Cancer Biol. Ther. 2019, 20, 65–72. [Google Scholar] [CrossRef]
- Li, J.; Zhu, P.; Wang, L.; Yang, L.; Zou, L.; Gao, F. Study of diffusion-weighted magnetic resonance imaging in the evaluation of the response to AAV2-VEGF-Trap neoadjuvant treatment in a triple-negative breast cancer animal model. Cancer Med. 2019, 8, 1594–1603. [Google Scholar] [CrossRef]
- Hacker, U.T.; Bentler, M.; Kaniowska, D.; Morgan, M.; Büning, H. Towards Clinical Implementation of Adeno-Associated Virus (AAV) Vectors for Cancer Gene Therapy: Current Status and Future Perspectives. Cancers 2020, 12, 1889. [Google Scholar] [CrossRef]
- Subramanian, I.V.; Devineni, S.; Ghebre, R.; Ghosh, G.; Joshi, H.P.; Jing, Y.; Truskinovsky, A.M.; Ramakrishnan, S. AAV-P125A-endostatin and paclitaxel treatment increases endoreduplication in endothelial cells and inhibits metastasis of breast cancer. Gene Ther. 2011, 18, 145–154. [Google Scholar] [CrossRef]
- Limberis, M.P.; Vandenberghe, L.H.; Zhang, L.; Pickles, R.J.; Wilson, J.M. Transduction efficiencies of novel AAV vectors in mouse airway epithelium in vivo and human ciliated airway epithelium in vitro. Mol. Ther. 2009, 17, 294–301. [Google Scholar] [CrossRef]
- Qiao, C.; Zhang, W.; Yuan, Z.; Shin, J.H.; Li, J.; Jayandharan, G.R.; Zhong, L.; Srivastava, A.; Xiao, X.; Duan, D. Adeno-associated virus serotype 6 capsid tyrosine-to-phenylalanine mutations improve gene transfer to skeletal muscle. Hum. Gene Ther. 2010, 21, 1343–1348. [Google Scholar] [CrossRef] [PubMed]
- Van Lieshout, L.P.; Domm, J.M.; Rindler, T.N.; Frost, K.L.; Sorensen, D.L.; Medina, S.J.; Booth, S.A.; Bridges, J.P.; Wootton, S.K. A novel triple-mutant AAV6 capsid induces rapid and potent transgene expression in the muscle and respiratory tract of mice. Mol. Ther. Methods Clin. Dev. 2018, 9, 323–329. [Google Scholar] [CrossRef] [PubMed]
- Rghei, A.D.; van Lieshout, L.P.; McLeod, B.M.; Pei, Y.; Lopes, J.A.; Zielinska, N.; Baracuhy, E.M.; Stevens, B.A.Y.; Thomas, S.P.; Yates, J.G.E.; et al. Safety and Tolerability of the Adeno-Associated Virus Vector, AAV6.2FF, Expressing a Monoclonal Antibody in Murine and Ovine Animal Models. Biomedicines 2021, 9, 1186. [Google Scholar] [CrossRef] [PubMed]
- Van Lieshout, L.P.; Soule, G.; Sorensen, D.; Frost, K.L.; He, S.; Tierney, K.; Safronetz, D.; Booth, S.A.; Kobinger, G.P.; Qiu, X.; et al. Intramuscular Adeno-Associated Virus-Mediated Expression of Monoclonal Antibodies Provides 100% Protection Against Ebola Virus Infection in Mice. J. Infect. Dis. 2018, 217, 916–925. [Google Scholar] [CrossRef] [PubMed]
- Zamarin, D.; Palese, P. Oncolytic Newcastle disease virus for cancer therapy: Old challenges and new directions. Future Microbiol. 2012, 7, 347–367. [Google Scholar] [CrossRef]
- Santry, L.A.; McAusland, T.M.; Susta, L.; Wood, G.A.; Major, P.P.; Petrik, J.J.; Bridle, B.W.; Wootton, S.K. Production and purification of high-titer Newcastle disease virus for use in preclinical mouse models of cancer. Mol. Ther. Methods Clin. Dev. 2018, 9, 181–191. [Google Scholar] [CrossRef]
- Balazs, A.B.; Chen, J.; Hong, C.M.; Rao, D.S.; Yang, L.; Baltimore, D. Antibody-based protection against HIV infection by vectored immunoprophylaxis. Nature 2012, 481, 81–84. [Google Scholar] [CrossRef]
- Zufferey, R.; Donello, J.E.; Trono, D.; Hope, T.J. Woodchuck hepatitis virus posttranscriptional regulatory element enhances expression of transgenes delivered by retroviral vectors. J. Virol. 1999, 73, 2886–2892. [Google Scholar] [CrossRef]
- Halbert, C.L.; Allen, J.M.; Miller, A.D. Efficient mouse airway transduction following recombination between AAV vectors carrying parts of a larger gene. Nat. Biotechnol. 2002, 20, 697–701. [Google Scholar] [CrossRef]
- Lieshout, L.P.; Domm, L.M.; Wootton, S.K. AAV-mediated gene delivery to the lung. In Adeno-Associated Virus Vectors; Humana Press: New York, NY, USA, 2019; pp. 361–372. [Google Scholar] [CrossRef]
- Rghei, A.D.; Stevens, B.A.Y.; Thomas, S.P.; Yates, J.G.E.; McLeod, B.M.; Karimi, K.; Susta, L.; Bridle, B.W.; Wootton, S.K. Production of Adeno-Associated Virus Vectors in Cell Stacks for Preclinical Studies in Large Animal Models. J. Vis. Exp. 2021, 172. [Google Scholar] [CrossRef]
- Park, M.S.; Steel, J.; Garcia-Sastre, A.; Swayne, D.; Palese, P. Engineered viral vaccine constructs with dual specificity: Avian influenza and Newcastle disease. Proc. Natl. Acad. Sci. USA 2006, 103, 8203–8208. [Google Scholar] [CrossRef] [PubMed]
- Sergel, T.A.; McGinnes, L.W.; Morrison, T.G. A single amino acid change in the Newcastle disease virus fusion protein alters the requirement for HN protein in fusion. J. Virol. 2000, 74, 5101–5107. [Google Scholar] [CrossRef] [PubMed]
- Greenaway, J.; Henkin, J.; Lawler, J.; Moorehead, R.; Petrik, J. ABT-510 induces tumor cell apoptosis and inhibits ovarian tumor growth in an orthotopic, syngeneic model of epithelial ovarian cancer. Mol. Cancer Ther. 2009, 8, 64–74. [Google Scholar] [CrossRef]
- Matuszewska, K.; Santry, L.A.; van Vloten, J.P.; AuYeung, A.W.K.; Major, P.P.; Lawler, J.; Wootton, S.K.; Bridle, B.W.; Petrik, J. Combining Vascular Normalization with an Oncolytic Virus Enhances Immunotherapy in a Preclinical Model of Advanced-Stage Ovarian Cancer. Clin. Cancer Res. 2019, 25, 1624–1638. [Google Scholar] [CrossRef]
- Van Vloten, J.P.; Santry, L.A.; McAusland, T.M.; Karimi, K.; McFadden, G.; Petrik, J.J.; Wootton, S.K.; Bridle, B.W. Quantifying antigen-specific T cell responses when using antigen-agnostic immunotherapies. Mol. Ther. Methods Dev. 2019, 13, 154–166. [Google Scholar] [CrossRef] [PubMed]
- McAusland, T.M.; van Vloten, J.P.; Santry, L.A.; Guilleman, M.M.; Rghei, A.D.; Ferreira, E.M.; Ingrao, J.C.; Arulanandam, R.; Major, P.P.; Susta, L.; et al. Combining vanadyl sulfate with Newcastle disease virus potentiates rapid innate immune-mediated regression with curative potential in murine cancer models. Mol. Ther. Oncolytics 2021, 20, 306–324. [Google Scholar] [CrossRef]
- Alter, G.; Malenfant, J.M.; Altfeld, M. CD107a as a functional marker for the identification of natural killer cell activity. J. Immunol. Methods 2004, 294, 15–22. [Google Scholar] [CrossRef]
- Greenaway, J.; Moorehead, R.; Shaw, P.; Petrik, J. Epithelial-stromal interaction increases cell proliferation, survival and tumorigenicity in a mouse model of human epithelial ovarian cancer. Gynecol. Oncol. 2008, 108, 385–394. [Google Scholar] [CrossRef]
- Rath, T.; Baker, K.; Dumont, J.A.; Peters, R.T.; Jiang, H.; Qiao, S.W.; Lencer, W.I.; Pierce, G.F.; Blumberg, R.S. Fc-fusion proteins and FcRn: Structural insights for longer-lasting and more effective therapeutics. Crit. Rev. Biotechnol. 2015, 35, 235–254. [Google Scholar] [CrossRef]
- Dang, C.V. Links between metabolism and cancer. Genes Dev. 2012, 26, 877–890. [Google Scholar] [CrossRef]
- Romero-Garcia, S.; Lopez-Gonzalez, J.S.; Baez-Viveros, J.L.; Aguilar-Cazares, D.; Prado-Garcia, H. Tumor cell metabolism: An integral view. Cancer Biol. Ther. 2011, 12, 939–948. [Google Scholar] [CrossRef] [PubMed]
- Beauregard, J.M.; Hofman, M.S.; Kong, G.; Hicks, R.J. The tumour sink effect on the biodistribution of 68Ga-DOTA-octreotate: Implications for peptide receptor radionuclide therapy. Eur. J. Nucl. Med. Mol. Imaging 2012, 39, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Bumbaca, D.; Xiang, H.; Boswell, C.A.; Port, R.E.; Stainton, S.L.; Mundo, E.E.; Ulufatu, S.; Bagri, A.; Theil, F.P.; Fielder, P.J.; et al. Maximizing tumour exposure to anti-neuropilin-1 antibody requires saturation of non-tumour tissue antigenic sinks in mice. Br. J. Pharmacol. 2012, 166, 368–377. [Google Scholar] [CrossRef] [PubMed]
- Fliss, C.; Heinzel, A.; Miller, B.; Vogg, A.T.J.; Langen, K.-J.; Mottaghy, F.M. Relevant tumor sink effect in prostate cancer patients receiving 177Lu-PSMA-617 radioligand therapy. Nuklearmedizin 2018, 57, 19–25. [Google Scholar] [CrossRef]
- Tong, J.G.; Evans, A.C.; Ho, M.L.; Guenther, C.M.; Brun, M.J.; Judd, J.; Wu, E.; Suh, J. Reducing off target viral delivery in ovarian cancer gene therapy using a protease-activated AAV2 vector platform. J. Control. Release 2019, 307, 292–301. [Google Scholar] [CrossRef]
- Wensveen, F.M.; Jelencic, V.; Polic, B. NKG2D: A master regulator of immune cell responsiveness. Front. Immunol. 2018, 9, 441. [Google Scholar] [CrossRef]
- Barrow, A.D.; Martin, C.J.; Colonna, M. The natural cytotoxicity receptors in health and disease. Front. Immunol. 2019, 10, 909. [Google Scholar] [CrossRef]
- Chen, H.; Herndon, M.E.; Lawler, J. The cell biology of thrombospondin-1. Matrix Biol. 2000, 19, 597–614. [Google Scholar] [CrossRef]
- Reynolds, A.R. Potential relevance of bell-shaped and u-shaped dose-responses for the therapeutic targeting of angiogenesis in cancer. Dose-Response 2010, 8, 253–284. [Google Scholar] [CrossRef]
- Tjin Tham Sjin, R.M.; Naspinski, J.; Birsner, A.E.; Li, C.; Chan, R.; Lo, K.M.; Gillies, S.; Zurakowski, D.; Folkman, J.; Samulski, J.; et al. Endostatin therapy reveals a U-shaped curve for antitumor activity. Cancer Gene Ther. 2006, 13, 619–627. [Google Scholar] [CrossRef][Green Version]






Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Stegelmeier, A.A.; Santry, L.A.; Guilleman, M.M.; Matuszewska, K.; Minott, J.A.; Yates, J.G.E.; Stevens, B.A.Y.; Thomas, S.P.; Vanderkamp, S.; Hanada, K.; et al. AAV-Vectored Expression of the Vascular Normalizing Agents 3TSR and Fc3TSR, and the Anti-Angiogenic Bevacizumab Extends Survival in a Murine Model of End-Stage Epithelial Ovarian Carcinoma. Biomedicines 2022, 10, 362. https://doi.org/10.3390/biomedicines10020362
Stegelmeier AA, Santry LA, Guilleman MM, Matuszewska K, Minott JA, Yates JGE, Stevens BAY, Thomas SP, Vanderkamp S, Hanada K, et al. AAV-Vectored Expression of the Vascular Normalizing Agents 3TSR and Fc3TSR, and the Anti-Angiogenic Bevacizumab Extends Survival in a Murine Model of End-Stage Epithelial Ovarian Carcinoma. Biomedicines. 2022; 10(2):362. https://doi.org/10.3390/biomedicines10020362
Chicago/Turabian StyleStegelmeier, Ashley A., Lisa A. Santry, Matthew M. Guilleman, Kathy Matuszewska, Jessica A. Minott, Jacob G. E. Yates, Brenna A. Y. Stevens, Sylvia P. Thomas, Sierra Vanderkamp, Kiersten Hanada, and et al. 2022. "AAV-Vectored Expression of the Vascular Normalizing Agents 3TSR and Fc3TSR, and the Anti-Angiogenic Bevacizumab Extends Survival in a Murine Model of End-Stage Epithelial Ovarian Carcinoma" Biomedicines 10, no. 2: 362. https://doi.org/10.3390/biomedicines10020362
APA StyleStegelmeier, A. A., Santry, L. A., Guilleman, M. M., Matuszewska, K., Minott, J. A., Yates, J. G. E., Stevens, B. A. Y., Thomas, S. P., Vanderkamp, S., Hanada, K., Pei, Y., Rghei, A. D., van Vloten, J. P., Pereira, M., Thompson, B., Major, P. P., Petrik, J. J., Bridle, B. W., & Wootton, S. K. (2022). AAV-Vectored Expression of the Vascular Normalizing Agents 3TSR and Fc3TSR, and the Anti-Angiogenic Bevacizumab Extends Survival in a Murine Model of End-Stage Epithelial Ovarian Carcinoma. Biomedicines, 10(2), 362. https://doi.org/10.3390/biomedicines10020362