
Modeling Alzheimer’s Disease in Caenorhabditis elegans

 ,
,  ,
,  ,
,  and
and
Abstract
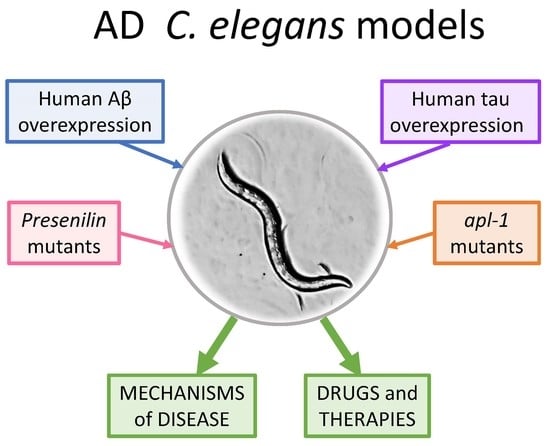
1. Introduction
2. Models to Study Alzheimer’s Disease
3. Genes Implicated in Alzheimer’s Disease: Homologs in C. elegans
4. The C. elegans Model: Advantages and Limitations
5. C. elegans Models to Study the Mechanism of Toxicity of Aβ
5.1. Mechanism of Toxicity of Aβ Oligomers
5.2. Aβ Toxicity and Insulin Signaling
5.3. Aβ Toxicity and Proteostasis
5.4. Toxicity of Aβ and Gene Expression
5.5. Aβ Toxicity and Oxidative Stress
5.6. Other Models of AD with Aβ Overexpression in C. elegans
6. C. elegans Models to Study the Function of APP
7. C. elegans Models to Study the Function of Presenilins
8. C. elegans Models to Study the Function and Mechanism of Toxicity of Tau
8.1. Models of Tau Expression in the Nervous System of C. elegans
8.2. Effects of Tau Phosphorylation in C. elegans
8.3. Models with Tau Expression in Muscle Cells
8.4. Models Using the Endogenous ptl-1 Gene
9. Use of C. elegans Models of Alzheimer’s Disease to Find New Drugs and Therapies
9.1. Antidiabetics
9.2. Antioxidants
9.3. Compounds with Anti-Amyloidogenic Mechanism by Direct Interaction with Oligomers
9.4. Modulators of Enzymes and Receptors Involved in AD
9.5. Other Compounds and Mechanisms Studied in C. elegans Models of AD

{kind=link}
| Drug | Action | C. elegans AD Model | References |
|---|---|---|---|
| Metformin | Antidiabetic | Muscle Aβ (CL2006, CL4176) Pan-neuronal Aβ (CL2355, GRU102) | [45,46] |
| NT219 | Antidiabetic | Muscle Aβ (CL2006) | [128] |
| Metformin + Lithium | Antidiabetic | Pan-neuronal Aβ (GRU102) | [129] |
| Clioquinol (8-hydroxyquinoline) | Antioxidant | Aβ in glutamatergic neurons (UA166) | [130] |
| MitoQ | Antioxidant | Muscle Aβ (CL2006) | [132] |
| diphenyldiselenide (PhSe)2 | Antioxidant | Muscle Aβ (CL2006, CL4176) Pan-neuronal Aβ (CL2355) | [133] |
| Antibodies anti-Aβ | Anti-amyloidogenic | Muscle Aβ (GMC101) | [134] |
| Humanin peptide | Anti-amyloidogenic | Muscle Aβ (CL4176) | [135] |
| Lactoferrin-derived peptides | Anti-amyloidogenic | Muscle Aβ (CL4176) | [136] |
| Bicyclic peptides | Anti-amyloidogenic | Muscle Aβ (GMC101) | [137] |
| Peptides from C. ternatea | Anti-amyloidogenic | Muscle Aβ (CL4176). Pan-neuronal Aβ (CL2355) | [138] |
| CNI-1493 and C1213 | Anti-amyloidogenic | Muscle Aβ (CL2006, CL4176) | [141] |
| Aminosterol trodusquemin | Anti-amyloidogenic | Muscle Aβ (GMC101) | [142] |
| Thioflavin | Anti-amyloidogenic | Tau-V337M (aex-3/T337) | [143] |
| PNR502 (tubulin binding compound) | Anti-amyloidogenic | Muscle Aβ (CL4176). Pan-neuronal Aβ (CL2355) | [144] |
| BIBA (antiaggregating + anti-inflammatory) | Anti-amyloidogenic | Muscle Aβ (CL4176) | [145] |
| CHF11 | Anti-amyloidogenic | Muscle Aβ (CL4176) | [146] |
| Frondoside A | Anti-amyloidogenic | Muscle Aβ (CL2006, CL4176). Pan-neuronal Aβ (CL2355, GRU102) | [147] |
| Photo-oxygenation | Anti-amyloidogenic | Muscle Aβ (CL2006) | [148] |
| Carnosine and kynuric acid | Anti-amyloidogenic | Muscle Aβ (GMC101) | [149] |
| Alkaloids of Lycoris radiata | Acetylcholinesterase gene expression inhibition | Muscle Aβ (CL4176) | [154] |
| SAS-0132 and JVW-1009 | Sig2R antagonists | Pan-neuronal APP overexpression | [155] |
| Buckwheat trypsin inhibitor | Autophagy activation | Muscle Aβ (CL4176) | [156] |
| MICA | Dihydrolipoamide dehydrogenase inhibitor | Muscle Aβ (CL2006, CL4176) Pan-neuronal Aβ (CL2355) | [59] |
| NP103 | GSK-3 inhibitor | Tau-V337M (aex-3/T337) | [143] |
| STX64 | Steroid sulfatase inhibitor | Muscle Aβ (CL2006, GMC101) | [157] |
| Cannabidiol | Cannabinoid receptor 1 activation | Pan-neuronal Aβ (CL2355) | [158] |
| Vitamine B12 | Methionine synthase activation | Muscle Aβ (CL4176, GMC101) | [159,160] |
| Betaine | Cystathionine-β-synthase activation | Muscle Aβ (CL2006) | [161] |
| Resveratrol | Mitochondrial and ER UPR activation | Muscle Aβ (CL2006) | [162] |
| Dauricin | ER UPR activation | Muscle Aβ (CL2120, GMC101) | [163,164] |
| Spermidine | Mitophagy activation | Pan-neuronal Aβ and tau overexpression (UM0001) | [165] |
| Caffeic acid | Antioxidant, antiaggregating | Muscle Aβ (CL4176) | [166] |
| Nicotine | SKN-1 pathway activation | Muscle Aβ (CL4176, CL2120) | [167] |
| Swimming exercise | Antioxidant, others | Muscle Aβ (CL2120). Pan-neuronal Aβ (CL2355) | [168,169] |
| Bacillus Subtilis | Gut microbiota | Muscle Aβ (CL2120, GMC101) | [170] |
10. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Asik, R.; Suganthy, N.; Aarifa, M.; Kumar, A.; Szigeti, K.; Mathe, D.; Gulyás, B.; Archunan, G.; Padmanabhan, P. Alzheimer’s Disease: A Molecular View of β-Amyloid Induced Morbific Events. Biomedicines 2021, 9, 1126. [Google Scholar] [CrossRef] [PubMed]
- Selkoe, D.J.; Hardy, J. The amyloid hypothesis of Alzheimer’s disease at 25 years. EMBO Mol. Med. 2016, 8, 595–608. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Li, C. Understanding the molecular basis of Alzheimer’s disease using a Caenorhabditis elegans model system. Brain Struct. Funct. 2010, 214, 263–283. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Holtzman, D.M. Alzheimer Disease: An Update on Pathobiology and Treatment Strategies. Cell 2019, 179, 312–339. [Google Scholar] [CrossRef]
- Cavanaugh, S.E.; Pippin, J.J.; Barnard, N.D. Animal models of Alzheimer disease: Historical pitfalls and a path forward. ALTEX Altern. Anim. Exp. 2014, 31, 279–302. [Google Scholar]
- Sharma, N.; Khurana, N.; Muthuraman, A. Lower vertebrate and invertebrate models of Alzheimer’s disease—A review. Eur. J. Pharmacol. 2017, 815, 312–323. [Google Scholar] [CrossRef]
- Van Dam, D.; De Deyn, P.P. Animal models in the drug discovery pipeline for Alzheimer’s disease. Br. J. Pharmacol. 2011, 164, 1285–1300. [Google Scholar] [CrossRef]
- Wentzell, J.; Kretzschmar, D. Alzheimer’s Disease and tauopathy studies in flies and worms. Neurobiol. Dis. 2010, 40, 21–28. [Google Scholar] [CrossRef]
- Mhatre, S.D.; Paddock, B.E.; Saunders, A.J.; Marenda, D.R. Invertebrate models of Alzheimer’s disease. J. Alzheimer’s Dis. 2013, 33, 3–16. [Google Scholar] [CrossRef]
- Lim, C.H.; Mathuru, A.S. Modeling Alzheimer’s and other age related human diseases in embryonic systems. J. Dev. Biol. 2018, 6, 1. [Google Scholar] [CrossRef]
- Lai, C.H.; Chou, C.Y.; Ch’ang, L.Y.; Liu, C.S.; Lin, W.C. Identification of novel human genes evolutionarily conserved in Caenorhabditis elegans by comparative proteomics. Genome Res. 2000, 10, 703–713. [Google Scholar] [CrossRef] [PubMed]
- Kim, W.; Underwood, R.S.; Greenwald, I.; Shaye, D.D. OrthoList 2: A New Comparative Genomic Analysis of Human and Caenorhabditis elegans Genes. Genetics 2018, 210, 445–461. [Google Scholar] [CrossRef]
- Sims, R.; Hill, M.; Williams, J. The multiplex model of the genetics of Alzheimer’s disease. Nat. Neurosci. 2020, 23, 311–322. [Google Scholar] [CrossRef] [PubMed]
- Godini, R.; Pocock, R.; Fallahi, H. Caenorhabditis elegans hub genes that respond to amyloid beta are homologs of genes involved in human Alzheimer’s disease. PLoS ONE 2019, 14, e0219486. [Google Scholar] [CrossRef] [PubMed]
- Hornsten, A.; Lieberthal, J.; Fadia, S.; Malins, R.; Ha, L.; Xu, X.; Daigle, I.; Markowitz, M.; Plasterk, R.; Li, C.; et al. APL-1, a Caenorhabditis elegans protein related to the human-amyloid precursor protein, is essential for viability. Proc. Natl. Acad. Sci. USA 2007, 104, 1971–1976. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Li, C. Caenorhabditis elegans as a model organism to study APP function. Exp. Brain Res. 2012, 217, 397–411. [Google Scholar] [CrossRef]
- Ochiishi, T.; Doi, M.; Yamasaki, K.; Hirose, K.; Kitamura, A.; Urabe, T.; Hattori, N.; Kinjo, M.; Ebihara, T.; Shimura, H. Development of new fusion proteins for visualizing amyloid-β oligomers in vivo. Sci. Rep. 2016, 6, 22712. [Google Scholar] [CrossRef]
- Sulston, J.E.; Schierenberg, E.; White, J.G.; Thomson, J.N. The embryonic cell lineage of the nematode Caenorhabditis elegans. Dev. Biol. 1983, 100, 64–119. [Google Scholar] [CrossRef]
- White, J.; Southgate, E.; Thomson, J.; Brenner, S. The structure of the nervous system of the nematode Caenorhabditis elegans. Philos. Trans. R. Soc. Lond. B Biol. Sci. 1986, 314, 1–340. [Google Scholar]
- Kamath, R.S.; Fraser, A.G.; Dong, Y.; Poulin, G.; Durbin, R.; Gotta, M.; Kanapin, A.; Le Bot, N.; Moreno, S.; Sohrmann, M.; et al. Systematic functional analysis of the Caenorhabditis elegans genome using RNAi. Nature 2003, 421, 231–237. [Google Scholar] [CrossRef]
- Caldwell, K.A.; Willicott, C.W.; Caldwell, G.A. Modeling neurodegeneration in Caenorhabditis elegans. DMM Dis. Model. Mech. 2020, 13, dmm046110. [Google Scholar] [CrossRef] [PubMed]
- Volovik, Y.; Marques, F.C.; Cohen, E. The nematode Caenorhabditis elegans: A versatile model for the study of proteotoxicity and aging. Methods 2014, 68, 458–464. [Google Scholar] [CrossRef] [PubMed]
- Machino, K.; Link, C.D.; Wang, S.; Murakami, H.; Murakami, S. A semi-automated motion-tracking analysis of locomotion speed in the C. elegans transgenics overexpressing beta-amyloid in neurons. Front. Genet. 2014, 5, 202. [Google Scholar] [CrossRef] [PubMed]
- Apostolakou, A.E.; Sula, X.K.; Nastou, K.C.; Nasi, G.I.; Iconomidou, V.A. Exploring the conservation of Alzheimer-related pathways between H. sapiens and C. elegans: A network alignment approach. Sci. Rep. 2021, 11, 4572. [Google Scholar] [CrossRef]
- Wang, H.; Robinson, J.L.; Kocabas, P.; Gustafsson, J.; Anton, M.; Cholley, P.-E.; Huang, S.; Gobom, J.; Svensson, T.; Uhlen, M.; et al. Genome-scale metabolic network reconstruction of model animals as a platform for translational research. Proc. Natl. Acad. Sci. USA 2021, 118, e2102344118. [Google Scholar] [CrossRef] [PubMed]
- Ermolaeva, M.A.; Schumacher, B. Insights from the worm: The C. elegans model for innate immunity. Semin. Immunol. 2014, 26, 303–309. [Google Scholar] [CrossRef]
- Oikonomou, G.; Shaham, S. The glia of Caenorhabditis elegans. Glia 2011, 59, 1253–1263. [Google Scholar] [CrossRef]
- Link, C.D. Expression of human beta-amyloid peptide in transgenic Caenorhabditis elegans. Proc. Natl. Acad. Sci. USA 1995, 92, 9368–9372. [Google Scholar] [CrossRef]
- McColl, G.; Roberts, B.R.; Gunn, A.P.; Perez, K.A.; Tew, D.J.; Masters, C.L.; Barnham, K.J.; Cherny, R.A.; Bush, A.I. The Caenorhabditis elegans Aβ1-42 model of Alzheimer disease predominantly Expresses Aβ3-42. J. Biol. Chem. 2009, 284, 22697–22702. [Google Scholar] [CrossRef]
- Mccoll, G.; Roberts, B.R.; Pukala, T.L.; Kenche, V.B.; Roberts, C.M.; Link, C.D.; Ryan, T.M.; Masters, C.L.; Barnham, K.J.; Bush, A.I.; et al. Utility of an improved model of amyloid-beta (Aβ1-42) toxicity in Caenorhabditis elegans for drug screening for Alzheimer’s disease. Mol. Neurodegener. 2012, 7, 57. [Google Scholar] [CrossRef]
- Link, C.D. C. elegans models of age-associated neurodegenerative diseases: Lessons from transgenic worm models of Alzheimer’s disease. Exp. Gerontol. 2006, 41, 1007–1013. [Google Scholar] [CrossRef] [PubMed]
- Griffin, E.F.; Caldwell, K.A.; Caldwell, G.A. Genetic and Pharmacological Discovery for Alzheimer’s Disease Using Caenorhabditis elegans. ACS Chem. Neurosci. 2017, 8, 2596–2606. [Google Scholar] [CrossRef] [PubMed]
- Dosanjh, L.E.; Brown, M.K.; Rao, G.; Link, C.D.; Luo, Y. Behavioral phenotyping of a transgenic Caenorhabditis elegans expressing neuronal amyloid-β. J. Alzheimer’s Dis. 2010, 19, 681–690. [Google Scholar] [CrossRef]
- Rebolledo, D.L.; Aldunate, R.; Kohn, R.; Neira, I.; Minniti, A.N.; Inestrosa, N.C. Copper reduces Aβ oligomeric species and ameliorates neuromuscular synaptic defects in a C. elegans model of inclusion body myositis. J. Neurosci. 2011, 31, 10149–10158. [Google Scholar] [CrossRef]
- Cohen, E.; Bieschke, J.; Perciavalle, R.M.; Kelly, J.W.; Dillin, A. Opposing activities protect against age-onset proteotoxicity. Science 2006, 313, 1604–1610. [Google Scholar] [CrossRef] [PubMed]
- Shea, D.; Hsu, C.C.; Bi, T.M.; Paranjapye, N.; Childers, M.C.; Cochran, J.; Tomberlin, C.P.; Wang, L.; Paris, D.; Zonderman, J.; et al. α-Sheet secondary structure in amyloid β-peptide drives aggregation and toxicity in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2019, 116, 8895–8900. [Google Scholar] [CrossRef]
- Meinen, B.A.; Gadkari, V.V.; Stull, F.; Ruotolo, B.T.; Bardwell, J.C.A. SERF engages in a fuzzy complex that accelerates primary nucleation of amyloid proteins. Proc. Natl. Acad. Sci. USA 2019, 116, 23040–23049. [Google Scholar] [CrossRef]
- Pras, A.; Houben, B.; Aprile, F.A.; Seinstra, R.; Gallardo, R.; Janssen, L.; Hogewerf, W.; Gallrein, C.; De Vleeschouwer, M.; Mata-Cabana, A.; et al. The cellular modifier MOAG-4/SERF drives amyloid formation through charge complementation. EMBO J. 2021, 40, e107568. [Google Scholar] [CrossRef]
- Lim, C.H.; Kaur, P.; Teo, E.; Lam, V.Y.M.; Zhu, F.; Kibat, C.; Gruber, J.; Mathuru, A.; Tolwinski, N.S. Application of optogenetic amyloid-β distinguishes between metabolic and physical damage in neurodegeneration. Elife 2020, 9, e52589. [Google Scholar] [CrossRef]
- Aprile, F.; Sormanni, P.; Podpolny, M.; Chhangur, S.; Needham, L.; Ruggeri, F.; Perni, M.; Limbocker, R.; Heller, G.; Sneideris, T.; et al. Rational design of a conformation-specific antibody for the quantification of Aβ oligomers. Proc. Natl. Acad. Sci. USA 2020, 117, 13509–13518. [Google Scholar] [CrossRef]
- Gallrein, C.; Iburg, M.; Michelberger, T.; Koçak, A.; Puchkov, D.; Liu, F.; Ayala Mariscal, S.M.; Nayak, T.; Kaminski Schierle, G.S.; Kirstein, J. Novel amyloid-beta pathology C. elegans model reveals distinct neurons as seeds of pathogenicity. Prog. Neurobiol. 2021, 198, 101907. [Google Scholar] [CrossRef] [PubMed]
- Quartey, M.O.; Nyarko, J.N.K.; Maley, J.M.; Barnes, J.R.; Bolanos, M.A.C.; Heistad, R.M.; Knudsen, K.J.; Pennington, P.R.; Buttigieg, J.; De Carvalho, C.E.; et al. The Aβ(1–38) peptide is a negative regulator of the Aβ(1–42) peptide implicated in Alzheimer disease progression. Sci. Rep. 2021, 11, 431. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Paulsson, J.F.; Blinder, P.; Burstyn-Cohen, T.; Du, D.; Estepa, G.; Adame, A.; Pham, H.M.; Holzenberger, M.; Kelly, J.W.; et al. Reduced IGF-1 Signaling Delays Age-Associated Proteotoxicity in Mice. Cell 2009, 139, 1157–1169. [Google Scholar] [CrossRef] [PubMed]
- Cohen, E.; Du, D.; Joyce, D.; Kapernick, E.A.; Volovik, Y.; Kelly, J.W.; Dillin, A. Temporal requirements of insulin/IGF-1 signaling for proteotoxicity protection. Aging Cell 2010, 9, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Ebert, P.R. Metformin Attenuates Aβ Pathology Mediated Through Levamisole Sensitive Nicotinic Acetylcholine Receptors in a C. elegans Model of Alzheimer’s Disease. Mol. Neurobiol. 2017, 54, 5427–5439. [Google Scholar] [CrossRef]
- Teo, E.; Ravi, S.; Barardo, D.; Kim, H.-S.; Fong, S.; Cazenave-Gassiot, A.; Yin Tan, T.; Ching, J.; Kovalik, J.-P.; Wenk, M.R.; et al. Metabolic stress is a primary pathogenic event in transgenic Caenorhabditis elegans expressing pan-neuronal human amyloid beta. Elife 2019, 8, e50069. [Google Scholar] [CrossRef]
- Ahmad, W. Overlapped metabolic and therapeutic links between Alzheimer and diabetes. Mol. Neurobiol. 2013, 47, 399–424. [Google Scholar] [CrossRef]
- Voisine, C.; Pedersen, J.S.; Morimoto, R.I. Chaperone networks: Tipping the balance in protein folding diseases. Neurobiol. Dis. 2010, 40, 12–20. [Google Scholar] [CrossRef]
- Kikis, E.A. The struggle by Caenorhabditis elegans to maintain proteostasis during aging and disease. Biol. Direct 2016, 11, 58. [Google Scholar] [CrossRef]
- Jensen, L.T.; Møller, T.H.; Larsen, S.A.; Jakobsen, H.; Olsen, A. A new role for laminins as modulators of protein toxicity in Caenorhabditis elegans. Aging Cell 2012, 11, 82–92. [Google Scholar] [CrossRef]
- Papaevgeniou, N.; Sakellari, M.; Jha, S.; Tavernarakis, N.; Holmberg, C.I.; Gonos, E.S.; Chondrogianni, N. 18α-Glycyrrhetinic acid proteasome activator decelerates aging and Alzheimer’s disease progression in Caenorhabditis elegans and neuronal cultures. Antioxid. Redox Signal. 2016, 25, 855–869. [Google Scholar] [CrossRef]
- Lehrbach, N.J.; Ruvkun, G. Endoplasmic reticulum-associated SKN-1A/Nrf1 mediates a cytoplasmic unfolded protein response and promotes longevity. Elife 2019, 8, e44425. [Google Scholar] [CrossRef] [PubMed]
- Groh, N.; Bühler, A.; Huang, C.; Li, K.W.; van Nierop, P.; Smit, A.B.; Fändrich, M.; Baumann, F.; David, D.C. Age-dependent protein aggregation initiates amyloid-β aggregation. Front. Aging Neurosci. 2017, 9, 138. [Google Scholar] [CrossRef] [PubMed]
- Sorrentino, V.; Romani, M.; Mouchiroud, L.; Beck, J.S.; Zhang, H.; D’Amico, D.; Moullan, N.; Potenza, F.; Schmid, A.W.; Rietsch, S.; et al. Enhancing mitochondrial proteostasis reduces amyloid-β proteotoxicity. Nature 2017, 552, 187–193. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.M.; Merin, D.A.; Fonte, V.; Link, C.D. AIP-1 ameliorates β-amyloid peptide toxicity in a Caenorhabditis elegans Alzheimer’s disease model. Hum. Mol. Genet. 2009, 18, 2739–2747. [Google Scholar] [CrossRef] [PubMed]
- Vaz Bravo, F.; Da Silva, J.; Chan, R.B.; Di Paolo, G.; Teixeira-Castro, A.; Oliveira, T.G. Phospholipase D functional ablation has a protective effect in an Alzheimer’s disease Caenorhabditis elegans model. Sci. Rep. 2018, 8, 3540. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Bai, H.; Huang, H.; Zhu, M.; Zhang, D.; Huang, X. Forward genetic screening of a novel gene hmgs-1 involved in Alzheimer Disease Pathogenesis in a transgenic Caenorhabditis elegans model. Biochem. Biophys. Res. Commun. 2020, 525, 141–147. [Google Scholar] [CrossRef]
- Leiteritz, A.; Baumanns, S.; Wenzel, U. Amyloid-beta (Aβ1–42)-induced paralysis in Caenorhabditis elegans is reduced through NHR-49/PPARalpha. Neurosci. Lett. 2020, 730, 135042. [Google Scholar] [CrossRef]
- Ahmad, W.; Ebert, P.R. 5-Methoxyindole-2-carboxylic acid (MICA) suppresses Aβ-mediated pathology in C. elegans. Exp. Gerontol. 2018, 108, 215–225. [Google Scholar] [CrossRef]
- Ahmad, W.; Ebert, P.R. Suppression of a core metabolic enzyme dihydrolipoamide dehydrogenase (dld) protects against amyloid beta toxicity in C. elegans model of Alzheimer’s disease. Genes Dis. 2021, 8, 849–866. [Google Scholar] [CrossRef]
- Mukherjee, S.; Russell, J.C.; Carr, D.T.; Burgess, J.D.; Allen, M.; Serie, D.J.; Boehme, K.L.; Kauwe, J.S.K.; Naj, A.C.; Fardo, D.W.; et al. Systems biology approach to late-onset Alzheimer’s disease genome-wide association study identifies novel candidate genes validated using brain expression data and Caenorhabditis elegans experiments. Alzheimer’s Dement. 2017, 13, 1133–1142. [Google Scholar] [CrossRef] [PubMed]
- Sutphin, G.L.; Backer, G.; Sheehan, S.; Bean, S.; Corban, C.; Liu, T.; Peters, M.J.; van Meurs, J.B.J.; Murabito, J.M.; Johnson, A.D.; et al. Caenorhabditis elegans orthologs of human genes differentially expressed with age are enriched for determinants of longevity. Aging Cell 2017, 16, 672–682. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Saar, V.; Leung, K.L.; Chen, L.; Wong, G. Human amyloid β peptide and tau co-expression impairs behavior and causes specific gene expression changes in Caenorhabditis elegans. Neurobiol. Dis. 2018, 109, 88–101. [Google Scholar] [CrossRef] [PubMed]
- Benbow, S.J.; Strovas, T.J.; Darvas, M.; Saxton, A.; Kraemer, B.C. Synergistic toxicity between tau and amyloid drives neuronal dysfunction and neurodegeneration in transgenic C. elegans. Hum. Mol. Genet. 2020, 29, 495–505. [Google Scholar] [CrossRef]
- Beeg, M.; Stravalaci, M.; Romeo, M.; Carrá, A.D.; Cagnotto, A.; Rossi, A.; Diomede, L.; Salmona, M.; Gobbi, X.M. Clusterin binds to Aβ1-42 Oligomers with high affinity and interferes with peptide aggregation by inhibiting primary and secondary nucleation. J. Biol. Chem. 2016, 291, 6958–6966. [Google Scholar] [CrossRef] [PubMed]
- Beeg, M.; Battocchio, E.; de Luigi, A.; Colombo, L.; Natale, C.; Cagnotto, A.; Corbelli, A.; Fiordaliso, F.; Diomede, L.; Salmona, M.; et al. Nonphosphorylated tau slows down Aβ1-42 aggregation, binds to Aβ1-42 oligomers, and reduces Aβ1-42 toxicity. J. Biol. Chem. 2021, 296, 100664. [Google Scholar] [CrossRef] [PubMed]
- Hassan, W.M.; Dostal, V.; Huemann, B.N.; Yerg, J.E.; Link, C.D. Identifying Aβ-specific pathogenic mechanisms using a nematode model of Alzheimer’s disease. Neurobiol. Aging 2015, 36, 857–866. [Google Scholar] [CrossRef]
- Ayyadevara, S.; Balasubramaniam, M.; Suri, P.; Mackintosh, S.; Tackett, A.; Sullivan, D.; Shmookler Reis, R.; Dennis, R. Proteins that accumulate with age in human skeletal-muscle aggregates contribute to declines in muscle mass and function in Caenorhabditis elegans. Aging 2016, 8, 3486–3497. [Google Scholar] [CrossRef]
- Yuan, J.; Chang, S.Y.; Yin, S.G.; Liu, Z.Y.; Cheng, X.; Liu, X.J.; Jiang, Q.; Gao, G.; Lin, D.Y.; Kang, X.L.; et al. Two conserved epigenetic regulators prevent healthy ageing. Nature 2020, 579, 118–122. [Google Scholar] [CrossRef]
- Sesti, F.; Liu, S.; Cai, S.Q. Oxidation of potassium channels by ROS: A general mechanism of aging and neurodegeneration? Trends Cell Biol. 2010, 20, 45–51. [Google Scholar] [CrossRef]
- Wan, L.; Nie, G.; Zhang, J.; Luo, Y.; Zhang, P.; Zhang, Z.; Zhao, B. β-Amyloid peptide increases levels of iron content and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic. Biol. Med. 2011, 50, 122–129. [Google Scholar] [CrossRef] [PubMed]
- Huang, J.; Chen, S.; Hu, L.; Niu, H.; Sun, Q.; Li, W.; Tan, G.; Li, J.; Jin, L.J.; Lyu, J.; et al. Mitoferrin-1 is Involved in the Progression of Alzheimer’s Disease Through Targeting Mitochondrial Iron Metabolism in a Caenorhabditis elegans Model of Alzheimer’s Disease. Neuroscience 2018, 385, 90–101. [Google Scholar] [CrossRef] [PubMed]
- Cacho-Valadez, B.; Muñoz-Lobato, F.; Pedrajas, J.R.; Cabello, J.; Fierro-González, J.C.; Navas, P.; Swoboda, P.; Link, C.D.; Miranda-Vizuete, A. The characterization of the Caenorhabditis elegans mitochondrial thioredoxin system uncovers an unexpected protective role of thioredoxin reductase 2 in β-Amyloid peptide toxicity. Antioxid. Redox Signal. 2012, 16, 1384–1400. [Google Scholar] [CrossRef] [PubMed]
- Muñoz-Lobato, F.; Rodríguez-Palero, M.J.; Naranjo-Galindo, F.J.; Shephard, F.; Gaffney, C.J.; Szewczyk, N.J.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Link, C.D.; et al. Protective Role of DNJ-27/ERdj5 in Caenorhabditis elegans models of human neurodegenerative diseases. Antioxid. Redox Signal. 2014, 20, 217–235. [Google Scholar] [CrossRef] [PubMed]
- Lu, T.; Aron, L.; Zullo, J.; Pan, Y.; Kim, H.; Chen, Y.; Yang, T.H.; Kim, H.M.; Drake, D.; Liu, X.S.; et al. REST and stress resistance in ageing and Alzheimer’s disease. Nature 2014, 507, 448–454. [Google Scholar] [CrossRef]
- Zullo, J.M.; Drake, D.; Aron, L.; O’Hern, P.; Dhamne, S.C.; Davidsohn, N.; Mao, C.A.; Klein, W.H.; Rotenberg, A.; Bennett, D.A.; et al. Regulation of lifespan by neural excitation and REST. Nature 2019, 574, 359–364. [Google Scholar] [CrossRef] [PubMed]
- Luo, Y.; Yue, W.; Quan, X.; Wang, Y.; Zhao, B.; Lu, Z. Asymmetric dimethylarginine exacerbates Aβ-induced toxicity and oxidative stress in human cell and Caenorhabditis elegans models of Alzheimer disease. Free Radic. Biol. Med. 2015, 79, 117–126. [Google Scholar] [CrossRef]
- Di Fede, G.; Catania, M.; Morbin, M.; Rossi, G.; Suardi, S.; Mazzoleni, G.; Merlin, M.; Giovagnoli, A.R.; Prioni, S.; Erbetta, A.; et al. A Recessive Mutation in the APP Gene with Dominant-Negative Effect on Amyloidogenesis. Science 2009, 323, 1473–1477. [Google Scholar] [CrossRef]
- Diomede, L.; Di Fede, G.; Romeo, M.; Bagnati, R.; Ghidoni, R.; Fiordaliso, F.; Salio, M.; Rossi, A.; Catania, M.; Paterlini, A.; et al. Expression of A2V-mutated Aβ in Caenorhabditis elegans results in oligomer formation and toxicity. Neurobiol. Dis. 2014, 62, 521–532. [Google Scholar] [CrossRef][Green Version]
- Fong, S.; Teo, E.; Ng, L.F.; Chen, C.B.; Lakshmanan, L.N.; Tsoi, S.Y.; Moore, P.K.; Inoue, T.; Halliwell, B.; Gruber, J. Energy crisis precedes global metabolic failure in a novel Caenorhabditis elegans Alzheimer Disease model. Sci. Rep. 2016, 6, 33781. [Google Scholar] [CrossRef]
- Sinnige, T.; Ciryam, P.; Casford, S.; Dobson, C.M.; De Bono, M.; Vendruscolo, M. Expression of the amyloid-β peptide in a single pair of C. elegans sensory neurons modulates the associated behavioural response. PLoS ONE 2019, 14, e0217746. [Google Scholar] [CrossRef] [PubMed]
- Griffin, E.F.; Scopel, S.E.; Stephen, C.A.; Holzhauer, A.C.; Vaji, M.A.; Tuckey, R.A.; Berkowitz, L.A.; Caldwell, K.A.; Caldwell, G.A. ApoE-associated modulation of neuroprotection from Aβ-mediated neurodegeneration in transgenic Caenorhabditis elegans. DMM Dis. Model. Mech. 2019, 12, dmm037218. [Google Scholar] [CrossRef] [PubMed]
- Wiese, M.; Antebi, A.; Zheng, H. Intracellular trafficking and synaptic function of APL-1 in Caenorhabditis elegans. PLoS ONE 2010, 5, e12790. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Raps, D.A.; Li, C. APL-1, the Alzheimer’s Amyloid precursor protein in Caenorhabditis elegans, modulates multiple metabolic pathways throughout development. Genetics 2012, 191, 493–507. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Cheng, R.; Tolen, L.; Shah, V.; Gillani, A.; Nasrin, A.; Li, C. Pan-neuronal expression of APL-1, an APP-related protein, disrupts olfactory, gustatory, and touch plasticity in Caenorhabditis elegans. J. Neurosci. 2012, 32, 10156–10169. [Google Scholar] [CrossRef] [PubMed]
- Ewald, C.Y.; Li, C. The secreted Alzheimer-related amyloid precursor protein fragment has an essential role in C. elegans. Prion 2012, 6, 433–436. [Google Scholar] [CrossRef]
- Ewald, C.Y.; Marfil, V.; Li, C. Alzheimer-related protein APL-1 modulates lifespan through heterochronic gene regulation in Caenorhabditis elegans. Aging Cell 2016, 15, 1051–1062. [Google Scholar] [CrossRef]
- Balklava, Z.; Niehage, C.; Currinn, H.; Mellor, L.; Guscott, B.; Poulin, G.; Hoflack, B.; Wassmer, T. The amyloid precursor protein controls PIKfyve function. PLoS ONE 2015, 10, e0130485. [Google Scholar] [CrossRef]
- Wang, B.; Li, H.; Mutlu, S.A.; Bowser, D.A.; Moore, M.J.; Wang, M.C.; Zheng, H. The amyloid precursor protein is a conserved receptor for slit to mediate axon guidance. eNeuro 2017, 4, e0185-17. [Google Scholar] [CrossRef]
- Sae-Lee, W.; Scott, L.L.; Brose, L.; Encarnacion, A.J.; Shi, T.; Kore, P.; Oyibo, L.O.; Ye, C.; Rozmiarek, S.K.; Pierce, J.T. APP-induced patterned neurodegeneration is exacerbated by APOE4 in Caenorhabditis elegans. G3 Genes Genomes Genet. 2020, 10, 2851–2861. [Google Scholar] [CrossRef]
- Smialowska, A.; Baumeister, R. Presenilin function in Caenorhabditis elegans. Neurodegener. Dis. 2006, 3, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Sarasija, S.; Norman, K.R. A γ-secretase independent role for presenilin in calcium homeostasis impacts mitochondrial function and morphology in Caenorhabditis elegans. Genetics 2015, 201, 1453–1466. [Google Scholar] [CrossRef] [PubMed]
- Sarasija, S.; Laboy, J.T.; Ashkavand, Z.; Bonner, J.; Tang, Y.; Norman, K.R. Presenilin mutations deregulate mitochondrial Ca2+ homeostasis and metabolic activity causing neurodegeneration in Caenorhabditis elegans. Elife 2018, 7, e33052. [Google Scholar] [CrossRef] [PubMed]
- Sarasija, S.; Norman, K.R. Role of presenilin in mitochondrial oxidative stress and neurodegeneration in Caenorhabditis elegans. Antioxidants 2018, 7, 111. [Google Scholar] [CrossRef]
- Ashkavand, Z.; Sarasija, S.; Ryan, K.C.; Laboy, J.T.; Norman, K.R. Corrupted ER-mitochondrial calcium homeostasis promotes the collapse of proteostasis. Aging Cell 2020, 19, e13065. [Google Scholar] [CrossRef]
- Ryan, K.C.; Ashkavand, Z.; Sarasija, S.; Laboy, J.T.; Samarakoon, R.; Norman, K.R. Increased mitochondrial calcium uptake and concomitant mitochondrial activity by presenilin loss promotes mTORC1 signaling to drive neurodegeneration. Aging Cell 2021, 35, 2871–2884. [Google Scholar] [CrossRef]
- Mun, M.J.; Kim, J.H.; Choi, J.Y.; Jang, W.C. Calcium homeostasis modulator 1 gene P86L polymorphism and the risk for alzheimer’s disease: A meta-analysis. Neurosci. Lett. 2016, 619, 8–14. [Google Scholar] [CrossRef]
- Dreses-Werringloer, U.; Lambert, J.C.; Vingtdeux, V.; Zhao, H.; Vais, H.; Siebert, A.; Jain, A.; Koppel, J.; Rovelet-Lecrux, A.; Hannequin, D.; et al. A polymorphism in CALHM1 influences Ca2+ homeostasis, Abeta levels, and Alzheimer’s disease risk. Cell 2008, 133, 1149–1161. [Google Scholar] [CrossRef]
- Tanis, J.E.; Ma, Z.; Krajacic, P.; He, L.; Foskett, K.J.; Lamitina, T. CLHM-1 is a functionally conserved and conditionally toxic Ca2+-permeable ion channel in Caenorhabditis elegans. J. Neurosci. 2013, 33, 12275–12286. [Google Scholar] [CrossRef]
- Strang, K.H.; Golde, T.E.; Giasson, B.I. MAPT mutations, tauopathy, and mechanisms of neurodegeneration. Lab. Investig. 2019, 99, 912–928. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Mandelkow, E. Caenorhabditis elegans models of tauopathy. FASEB J. 2017, 31, 5137–5148. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Zhang, B.; Leverenz, J.B.; Thomas, J.H.; Trojanowski, J.Q.; Schellenberg, G.D. Neurodegeneration and defective neurotransmission in a Caenorhabditis elegans model of tauopathy. Proc. Natl. Acad. Sci. USA 2003, 100, 9980–9985. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Burgess, J.K.; Chen, J.H.; Thomas, J.H.; Schellenberg, G.D. Molecular pathways that influence human tau-induced pathology in Caenorhabditis elegans. Hum. Mol. Genet. 2006, 15, 1483–1496. [Google Scholar] [CrossRef] [PubMed]
- Kraemer, B.C.; Schellenberg, G.D. SUT-1 enables tau-induced neurotoxicity in C. elegans. Hum. Mol. Genet. 2007, 16, 1959–1971. [Google Scholar] [CrossRef]
- Guthrie, C.R.; Schellenberg, G.D.; Kraemer, B.C. SUT-2 potentiates tau-induced neurotoxicity in Caenorhabditis elegans. Hum. Mol. Genet. 2009, 18, 1825–1838. [Google Scholar] [CrossRef]
- Guthrie, C.R.; Greenup, L.; Leverenz, J.B.; Kraemer, B.C. MSUT2 is a determinant of susceptibility to tau neurotoxicity. Hum. Mol. Genet. 2011, 20, 1989–1999. [Google Scholar] [CrossRef]
- Kow, R.L.; Strovas, T.J.; McMillan, P.J.; Jacobi, A.M.; Behlke, M.A.; Saxton, A.D.; Latimer, C.S.; Keene, C.D.; Kraemer, B.C. Distinct Poly(A) nucleases have differential impact on sut-2 dependent tauopathy phenotypes. Neurobiol. Dis. 2021, 147, 105148. [Google Scholar] [CrossRef]
- Miyasaka, T.; Ding, Z.; Gengyo-Ando, K.; Oue, M.; Yamaguchi, H.; Mitani, S.; Ihara, Y. Progressive neurodegeneration in C. elegans model of tauopathy. Neurobiol. Dis. 2005, 20, 372–383. [Google Scholar] [CrossRef]
- Fatouros, C.; Pir, G.J.; Biernat, J.; Koushika, S.P.; Mandelkow, E.; Mandelkow, E.M.; Schmidt, E.; Baumeister, R. Inhibition of tau aggregation in a novel Caenorhabditis elegans model of tauopathy mitigates proteotoxicity. Hum. Mol. Genet. 2012, 21, 3587–3603. [Google Scholar] [CrossRef]
- Pir, G.J.; Choudhary, B.; Kaniyappan, S.; Chandupatla, R.R.; Mandelkow, E.; Mandelkow, E.M.; Wang, Y. Suppressing Tau Aggregation and Toxicity by an Anti-Aggregant Tau Fragment. Mol. Neurobiol. 2019, 56, 3751–3767. [Google Scholar] [CrossRef]
- Xie, C.; Miyasaka, T.; Yoshimura, S.; Hatsuta, H.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Murayama, S.; Ihara, Y. The homologous carboxyl-terminal domains of microtubule-associated protein 2 and Tau induce neuronal dysfunction and have differential fates in the evolution of neurofibrillary tangles. PLoS ONE 2014, 9, e89796. [Google Scholar] [CrossRef] [PubMed]
- Pir, G.J.; Choudhary, B.; Mandelkow, E.; Mandelkow, E.M. Tau mutant A152T, a risk factor for FTD/PSP, induces neuronal dysfunction and reduced lifespan independently of aggregation in a C. elegans Tauopathy model. Mol. Neurodegener. 2016, 11, 33. [Google Scholar] [CrossRef] [PubMed]
- Choudhary, B.; Mandelkow, E.; Mandelkow, E.M.; Pir, G.J. Glutamatergic nervous system degeneration in a C. elegans TauA152T tauopathy model involves pathways of excitotoxicity and Ca2+ dysregulation. Neurobiol. Dis. 2018, 117, 189–202. [Google Scholar] [CrossRef] [PubMed]
- Miyasaka, T.; Shinzaki, Y.; Yoshimura, S.; Yoshina, S.; Kage-Nakadai, E.; Mitani, S.; Ihara, Y. Imbalanced expression of tau and tubulin induces neuronal dysfunction in C. elegans models of tauopathy. Front. Neurosci. 2018, 12, 415. [Google Scholar] [CrossRef]
- Cummins, N.; Tweedie, A.; Zuryn, S.; Bertran-Gonzalez, J.; Götz, J. Disease-associated tau impairs mitophagy by inhibiting Parkin translocation to mitochondria. EMBO J. 2019, 38, e99360. [Google Scholar] [CrossRef]
- Fang, E.F.; Hou, Y.; Palikaras, K.; Adriaanse, B.A.; Kerr, J.S.; Yang, B.; Lautrup, S.; Hasan-Olive, M.M.; Caponio, D.; Dan, X.; et al. Mitophagy inhibits amyloid-β and tau pathology and reverses cognitive deficits in models of Alzheimer’s disease. Nat. Neurosci. 2019, 22, 401–412. [Google Scholar] [CrossRef]
- Guha, S.; Fischer, S.; Johnson, G.V.W.; Nehrke, K. Tauopathy-associated tau modifications selectively impact neurodegeneration and mitophagy in a novel C. elegans single-copy transgenic model. Mol. Neurodegener. 2020, 15, 65. [Google Scholar] [CrossRef]
- Brandt, R.; Gergou, A.; Wacker, I.; Fath, T.; Hutter, H. A Caenorhabditis elegans model of tau hyperphosphorylation: Induction of developmental defects by transgenic overexpression of Alzheimer’s disease-like modified tau. Neurobiol. Aging 2009, 30, 22–33. [Google Scholar] [CrossRef]
- Ahmad, W. Dihydrolipoamide dehydrogenase suppression induces human tau phosphorylation by increasing whole body glucose levels in a C. elegans model of Alzheimer’s Disease. Exp. Brain Res. 2018, 236, 2857–2866. [Google Scholar] [CrossRef]
- Taylor, L.M.; McMillan, P.J.; Liachko, N.F.; Strovas, T.J.; Ghetti, B.; Bird, T.D.; Dirk Keene, C.; Kraemer, B.C. Pathological phosphorylation of tau and TDP-43 by TTBK1 and TTBK2 drives neurodegeneration. Mol. Neurodegener. 2018, 13, 7. [Google Scholar] [CrossRef]
- Russell, J.C.; Lei, H.; Chaliparambil, R.K.; Fish, S.; Markiewicz, S.M.; Lee, T.I.; Noori, A.; Kaeberlein, M. Generation and characterization of a tractable C. elegans model of tauopathy. GeroScience 2021, 43, 2621–2631. [Google Scholar] [CrossRef] [PubMed]
- Tien, N.W.; Wu, G.H.; Hsu, C.C.; Chang, C.Y.; Wagner, O.I. Tau/PTL-1 associates with kinesin-3 KIF1A/UNC-104 and affects the motor’s motility characteristics in C. elegans neurons. Neurobiol. Dis. 2011, 43, 495–506. [Google Scholar] [CrossRef] [PubMed]
- Chew, Y.L.; Fan, X.; Götz, J.; Nicholas, H.R. PTL-1 regulates neuronal integrity and lifespan in C. elegans. J. Cell Sci. 2013, 126, 2079–2091. [Google Scholar] [PubMed]
- Chew, Y.L.; Fan, X.; Götz, J.; Nicholas, H.R. Regulation of age-related structural integrity in neurons by protein with tau-like repeats (PTL-1) is cell autonomous. Sci. Rep. 2014, 4, 5185. [Google Scholar] [CrossRef] [PubMed]
- Chen, X.; Barclay, J.W.; Burgoyne, R.D.; Morgan, A. Using C. elegans to discover therapeutic compounds for ageing-associated neurodegenerative diseases. Chem. Cent. J. 2015, 9, 65. [Google Scholar] [CrossRef] [PubMed]
- Ma, L.; Zhao, Y.; Chen, Y.; Cheng, B.; Peng, A.; Huang, K. Caenorhabditis elegans as a model system for target identification and drug screening against neurodegenerative diseases. Eur. J. Pharmacol. 2018, 819, 169–180. [Google Scholar] [CrossRef]
- Perni, M.; Challa, P.K.; Kirkegaard, J.B.; Limbocker, R.; Koopman, M.; Hardenberg, M.C.; Sormanni, P.; Müller, T.; Saar, K.L.; Roode, L.W.Y.; et al. Massively parallel C. elegans tracking provides multi-dimensional fingerprints for phenotypic discovery. J. Neurosci. Methods 2018, 306, 57–67. [Google Scholar] [CrossRef]
- El-Ami, T.; Moll, L.; Carvalhal Marques, F.; Volovik, Y.; Reuveni, H.; Cohen, E. A novel inhibitor of the insulin/IGF signaling pathway protects from age-onset, neurodegeneration-linked proteotoxicity. Aging Cell 2014, 13, 165–174. [Google Scholar] [CrossRef]
- Teo, E.; Fong, S.; Tolwinski, N.; Gruber, J. Drug synergy as a strategy for compression of morbidity in a Caenorhabditis elegans model of Alzheimer’s disease. GeroScience 2020, 42, 849–856. [Google Scholar] [CrossRef]
- Matlack, K.E.S.; Tardiff, D.F.; Narayan, P.; Hamamichi, S.; Caldwell, K.A.; Caldwell, G.A.; Lindquist, S. Clioquinol promotes the degradation of metal-dependent amyloid-ß (Aß) oligomers to restore endocytosis and ameliorate Aß toxicity. Proc. Natl. Acad. Sci. USA 2014, 111, 4013–4018. [Google Scholar] [CrossRef]
- Ryan, T.M.; Roberts, B.R.; McColl, G.; Hare, D.J.; Doble, P.A.; Li, Q.X.; Lind, M.; Roberts, A.M.; Mertens, H.D.T.; Kirb, N.; et al. Stabilization of nontoxic Ajβ-oligomers: Insights into the mechanism of action of hydroxyquinolines in Alzheimer’s disease. J. Neurosci. 2015, 35, 2871–2884. [Google Scholar] [CrossRef] [PubMed]
- Ng, L.F.; Gruber, J.; Cheah, I.K.; Goo, C.K.; Cheong, W.F.; Shui, G.; Sit, K.P.; Wenk, M.R.; Halliwell, B. The mitochondria-targeted antioxidant MitoQ extends lifespan and improves healthspan of a transgenic Caenorhabditis elegans model of Alzheimer disease. Free Radic. Biol. Med. 2014, 71, 390–401. [Google Scholar] [CrossRef] [PubMed]
- Zamberlan, D.C.; Arantes, L.P.; Machado, M.L.; Golombieski, R.; Soares, F.A.A. Diphenyl-diselenide suppresses amyloid-β peptide in Caenorhabditis elegans model of Alzheimer’s disease. Neuroscience 2014, 278, 40–50. [Google Scholar] [CrossRef] [PubMed]
- Aprile, F.A.; Sormanni, P.; Perni, M.; Arosio, P.; Linse, S.; Knowles, T.P.J.; Dobson, C.M.; Vendruscolo, M. Selective targeting of primary and secondary nucleation pathways in Ab42 aggregation using a rational antibody scanning method. Sci. Adv. 2017, 3, e1700488. [Google Scholar] [CrossRef]
- Romeo, M.; Stravalaci, M.; Beeg, M.; Rossi, A.; Fiordaliso, F.; Corbelli, A.; Salmona, M.; Gobbi, M.; Cagnotto, A.; D’Iomede, L. Humanin Specifically Interacts with Amyloid-β Oligomers and Counteracts Their in vivo Toxicity. J. Alzheimer’s Dis. 2017, 57, 857–871. [Google Scholar] [CrossRef]
- Manzanares, P.; Martínez, R.; Garrigues, S.; Genovés, S.; Ramón, D.; Marcos, J.F.; Martorell, P. Tryptophan-Containing Dual Neuroprotective Peptides: Prolyl Endopeptidase Inhibition and Caenorhabditis elegans Protection from β-Amyloid Peptide Toxicity. Int. J. Mol. Sci. 2018, 19, 1491. [Google Scholar] [CrossRef]
- Ikenoue, T.; Aprile, F.A.; Sormanni, P.; Ruggeri, F.S.; Perni, M.; Heller, G.T.; Haas, C.P.; Middel, C.; Limbocker, R.; Mannini, B.; et al. A rationally designed bicyclic peptide remodels Aβ42 aggregation in vitro and reduces its toxicity in a worm model of Alzheimer’s disease. Sci. Rep. 2020, 10, 15280. [Google Scholar] [CrossRef]
- Kalmankar, N.V.; Hari, H.; Sowdhamini, R.; Venkatesan, R. Disulfide-Rich Cyclic Peptides from Clitoria ternatea Protect against β-Amyloid Toxicity and Oxidative Stress in Transgenic Caenorhabditis elegans. J. Med. Chem. 2021, 64, 7422–7433. [Google Scholar] [CrossRef]
- Habchi, J.; Chia, S.; Limbocker, R.; Mannini, B.; Ahn, M.; Perni, M.; Hansson, O.; Arosio, P.; Kumita, J.R.; Challa, P.K.; et al. Systematic development of small molecules to inhibit specific microscopic steps of Aβ42 aggregation in Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2017, 114, E200–E208. [Google Scholar] [CrossRef]
- Michaels, T.C.T.; Weber, C.A.; Mahadevan, L. Optimal control strategies for inhibition of protein aggregation. Proc. Natl. Acad. Sci. USA 2019, 116, 14593–14598. [Google Scholar] [CrossRef]
- Sankowski, R.; Herring, A.; Keyvani, K.; Frenzel, K.; Wu, J.; Röskam, S.; Noelker, C.; Bacher, M.; Al-Abed, Y. The multi-target effects of cni-1493: Convergence of antiamylodogenic and antiinflammatory properties in animal models of alzheimer’s disease. Mol. Med. 2016, 22, 776–788. [Google Scholar] [CrossRef] [PubMed]
- Limbocker, R.; Chia, S.; Ruggeri, F.S.; Perni, M.; Cascella, R.; Heller, G.T.; Meisl, G.; Mannini, B.; Habchi, J.; Michaels, T.C.T.; et al. Trodusquemine enhances Aβ 42 aggregation but suppresses its toxicity by displacing oligomers from cell membranes. Nat. Commun. 2019, 10, 225. [Google Scholar] [CrossRef] [PubMed]
- Gamir-Morralla, A.; Sacristán, S.; Medina, M.; Iglesias, T. Effects of Thioflavin T and GSK-3 Inhibition on Lifespan and Motility in a Caenorhabditis elegans Model of Tauopathy. J. Alzheimer’s Dis. Rep. 2019, 3, 47–57. [Google Scholar] [CrossRef] [PubMed]
- Kakraba, S.; Ayyadevara, S.; Penthala, N.R.; Balasubramaniam, M.; Ganne, A.; Liu, L.; Alla, R.; Bommagani, S.B.; Barger, S.W.; Griffin, W.S.T.; et al. A Novel Microtubule-Binding Drug Attenuates and Reverses Protein Aggregation in Animal Models of Alzheimer’s Disease. Front. Mol. Neurosci. 2019, 12, 310. [Google Scholar] [CrossRef] [PubMed]
- Yang, T.; Zhu, Z.; Yin, E.; Wang, Y.; Zhang, C.; Yuan, H.; Zhang, H.; Jin, S.; Guo, Z.; Wang, X. Alleviation of symptoms of alzheimer’s disease by diminishing aβ neurotoxicity and neuroinflammation. Chem. Sci. 2019, 10, 10149–10158. [Google Scholar] [CrossRef] [PubMed]
- Wang, H.; Yue, Y.; Zhao, H.; Wu, H.; Jiang, K.; Li, S.; Zhao, M.; Lin, F. Neuroprotective Effects of 2-Substituted 1,3-Selenazole Amide Derivatives on Amyloid-Beta-Induced Toxicity in a Transgenic Caenorhabditis elegans Model of Alzheimer’s Disease. Neurotox. Res. 2021, 39, 841–850. [Google Scholar] [CrossRef] [PubMed]
- Tangrodchanapong, T.; Sobhon, P.; Meemon, K. Frondoside A Attenuates Amyloid-β Proteotoxicity in Transgenic Caenorhabditis elegans by Suppressing Its Formation. Front. Pharmacol. 2020, 11, 553579. [Google Scholar] [CrossRef] [PubMed]
- Liu, Z.; Ma, M.; Yu, D.; Ren, J.; Qu, X. Target-driven supramolecular self-assembly for selective amyloid-β photooxygenation against Alzheimer’s disease. Chem. Sci. 2020, 11, 11003–11008. [Google Scholar] [CrossRef]
- Joshi, P.; Perni, M.; Limbocker, R.; Mannini, B.; Casford, S.; Chia, S.; Habchi, J.; Labbadia, J.; Dobson, C.M.; Vendruscolo, M. Two human metabolites rescue a C. elegans model of Alzheimer’s disease via a cytosolic unfolded protein response. Commun. Biol. 2021, 4, 843. [Google Scholar] [CrossRef]
- Van Assche, R.; Temmerman, L.; Dias, D.A.; Boughton, B.; Boonen, K.; Braeckman, B.P.; Schoofs, L.; Roessner, U. Metabolic profiling of a transgenic Caenorhabditis elegans Alzheimer model. Metabolomics 2015, 11, 477–486. [Google Scholar] [CrossRef]
- Akan, I.; Olivier-Van Stichelen, S.; Bond, M.R.; Hanover, J.A. Nutrient-driven O-GlcNAc in proteostasis and neurodegeneration. J. Neurochem. 2018, 144, 7–34. [Google Scholar] [CrossRef] [PubMed]
- Ahmad, W.; Shabbiri, K.; Ahmad, I. Prediction of human tau 3D structure, and interplay between O-β-GlcNAc and phosphorylation modifications in Alzheimer’s disease: C. elegans as a suitable model to study these interactions in vivo. Biochem. Biophys. Res. Commun. 2020, 528, 466–472. [Google Scholar] [CrossRef] [PubMed]
- Hanover, J.A.; Wang, P. O-GlcNAc cycling shows neuroprotective potential in C. elegans models of neurodegenerative disease. Worm 2013, 2, e27043. [Google Scholar] [CrossRef] [PubMed]
- Xin, L.; Yamujala, R.; Wang, Y.; Wang, H.; Wu, W.H.; Lawton, M.A.; Long, C.; Di, R. Acetylcholineestarase-Inhibiting Alkaloids from Lycoris radiata Delay Paralysis of Amyloid Beta-Expressing Transgenic C. elegans CL4176. PLoS ONE 2013, 8, e63874. [Google Scholar] [CrossRef]
- Yi, B.; Sahn, J.J.; Ardestani, P.M.; Evans, A.K.; Scott, L.L.; Chan, J.Z.; Iyer, S.; Crisp, A.; Zuniga, G.; Pierce, J.T.; et al. Small molecule modulator of sigma 2 receptor is neuroprotective and reduces cognitive deficits and neuroinflammation in experimental models of Alzheimer’s disease. J. Neurochem. 2017, 140, 561–575. [Google Scholar] [CrossRef]
- Li, J.; Cui, X.; Ma, X.; Wang, Z. rBTI reduced β-amyloid-induced toxicity by promoting autophagy-lysosomal degradation via DAF-16 in Caenorhabditis elegans. Exp. Gerontol. 2017, 89, 78–86. [Google Scholar] [CrossRef]
- Pérez-Jiménez, M.M.; Monje-Moreno, J.M.; Brokate-Llanos, A.M.; Venegas-Calerón, M.; Sánchez-García, A.; Sansigre, P.; Valladares, A.; Esteban-García, S.; Suárez-Pereira, I.; Vitorica, J.; et al. Steroid hormones sulfatase inactivation extends lifespan and ameliorates age-related diseases. Nat. Commun. 2021, 12, 49. [Google Scholar] [CrossRef]
- Wang, Z.; Zheng, P.; Xie, Y.; Chen, X.; Solowij, N.; Green, K.; Chew, Y.L.; Huang, X.F. Cannabidiol regulates CB1-pSTAT3 signaling for neurite outgrowth, prolongs lifespan, and improves health span in Caenorhabditis elegans of Aβ pathology models. FASEB J. 2021, 35, e21537. [Google Scholar] [CrossRef]
- Lam, A.B.; Kervin, K.; Tanis, J.E. Vitamin B12 impacts amyloid beta-induced proteotoxicity by regulating the methionine/S-adenosylmethionine cycle. Cell Rep. 2021, 36, 109753. [Google Scholar] [CrossRef]
- Andra, A.; Tanigawa, S.; Bito, T.; Ishihara, A.; Watanabe, F.; Yabuta, Y. Effects of vitamin B12 deficiency on amyloid-β toxicity in Caenorhabditis elegans. Antioxidants 2021, 10, 962. [Google Scholar] [CrossRef]
- Leiteritz, A.; Dilberger, B.; Wenzel, U.; Fitzenberger, E. Betaine reduces β-amyloid-induced paralysis through activation of cystathionine-β-synthase in an Alzheimer model of Caenorhabditis elegans. Genes Nutr. 2018, 13, 21. [Google Scholar] [CrossRef] [PubMed]
- Regitz, C.; Fitzenberger, E.; Mahn, F.L.; Dußling, L.M.; Wenzel, U. Resveratrol reduces amyloid-beta (Aβ1–42)-induced paralysis through targeting proteostasis in an Alzheimer model of Caenorhabditis elegans. Eur. J. Nutr. 2016, 55, 741–747. [Google Scholar] [CrossRef] [PubMed]
- Pu, Z.; Ma, S.; Wang, L.; Li, M.; Shang, L.; Luo, Y.; Chen, W. Amyloid-beta Degradation and Neuroprotection of Dauricine Mediated by Unfolded Protein Response in a Caenorhabditis elegans Model of Alzheimer’s disease. Neuroscience 2018, 392, 25–37. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Pu, Z.; Li, M.; Wang, K.; Deng, L.; Chen, W. Antioxidative and antiapoptosis: Neuroprotective effects of dauricine in Alzheimer’s disease models. Life Sci. 2020, 243, 117237. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, M.; Dai, Y.; Sun, Y.; Aman, Y.; Xu, Y.; Yu, P.; Zheng, Y.; Yang, J.; Zhu, X. Spermidine inhibits neurodegeneration and delays aging via the PINK1-PDR1-dependent mitophagy pathway in C. elegans. Aging 2020, 12, 16852–16866. [Google Scholar] [CrossRef]
- Li, H.; Yu, X.; Li, C.; Ma, L.; Zhao, Z.; Guan, S.; Wang, L. Caffeic acid protects against Aβ toxicity and prolongs lifespan in: Caenorhabditis elegans models. Food Funct. 2021, 12, 1219–1231. [Google Scholar] [CrossRef]
- Lu, X.; Zhang, Y.; Li, H.; Jin, Y.; Zhao, L.; Wang, X. Nicotine prevents in vivo Aβ toxicity in Caenorhabditis elegans via SKN-1. Neurosci. Lett. 2021, 761, 136114. [Google Scholar] [CrossRef]
- Chuang, H.S.; Kuo, W.J.; Lee, C.L.; Chu, I.H.; Chen, C.S. Exercise in an electrotactic flow chamber ameliorates age-related degeneration in Caenorhabditis elegans. Sci. Rep. 2016, 6, 28064. [Google Scholar] [CrossRef]
- Laranjeiro, R.; Harinath, G.; Hewitt, J.E.; Hartman, J.H.; Royal, M.A.; Meyer, J.N.; Vanapalli, S.A.; Driscoll, M. Swim exercise in Caenorhabditis elegans extends neuromuscular and gut healthspan, enhances learning ability, and protects against neurodegeneration. Proc. Natl. Acad. Sci. USA 2019, 116, 23829–23839. [Google Scholar] [CrossRef]
- Cogliati, S.; Clementi, V.; Francisco, M.; Crespo, C.; Argañaraz, F.; Grau, R. Bacillus Subtilis Delays Neurodegeneration and Behavioral Impairment in the Alzheimer’s Disease Model Caenorhabditis elegans. J. Alzheimer’s Dis. 2020, 73, 1035–1052. [Google Scholar] [CrossRef]
| Human Genes Associated with AD [13] | C. elegans Orthologue [12] | FAD/LOAD |
|---|---|---|
| Amyloid Beta Precursor Protein (APP) | apl-1 | FAD |
| Amyloid Beta Precursor-Like Protein 1 (APLP1) | apl-1 | - |
| Amyloid Beta Precursor-Like Protein 2 (APLP2) | apl-1 | - |
| α-secretase (ADAM10 and ADAM17) | sup-17 and adm-4 | LOAD |
| β-secretase (BACE1) | none | - |
| γ-secretase complex, Presenilin 1 (PSEN1) | sel-12, hop-1 and spe-4 | FAD |
| γ-secretase complex, Presenilin 2 (PSEN2) | sel-12, hop-1 and spe-4 | FAD |
| γ-secretase complex, Nicastrin (NCSTN) | aph-2 | - |
| γ-secretase complex, Anterior pharynx-defective-1 (APH1A) | aph-1 | - |
| γ-secretase complex, Presenilin enhancer 2-subunit (PSENEN) | pen-2 | - |
| Microtubule associated protein MAP2/MAP4/MAPT/Tau | ptl-1 | LOAD |
| Apolipoprotein E, APOE | none | LOAD |
| Glycogen synthase kinase 3 beta, GSK3β | gsk-3 | LOAD |
| Phosphoinositide-binding clathrin adaptor, domain 2 (SNAP91, PICALM) | unc-11 | LOAD |
| Bridging integrator 1, Amphiphysin family (BIN1, BIN2, AMPH) | amph-1 | LOAD |
| Clusterin-associated protein-1 (CLUAP1) | dyf-3 | LOAD |
| Ephrin Type-A Receptor 1 (EPHA1) | vab-1 | LOAD |
| Clusterin (CLU/TRPM2) | ced-11 | LOAD |
| Inositol Polyphosphate-5-Phosphatase D (INPP5D) | inpp-1 | LOAD |
| Complement component receptor 1 (CR1) | lev-9 | LOAD |
| ABI Family Member 3 (ABI3) | abi-1 | LOAD |
| Phospholipase Cγ2 (PLCG2) | plc-3 | LOAD |
| Myocyte-Specific Enhancer Factor 2C (MEF2C) | mef-2 | LOAD |
| CD2-Associated Protein (CD2AP) | Y44E3A.4 | LOAD |
| Nuclear Polyadenylated RNA-Binding Protein (CELF1) | etr-1 | LOAD |
| PH Domain-Containing Family C1 (FERMT2) | unc-112 | LOAD |
| Thioredoxin Domain-Containing Protein (NME8) | ndk-1 | LOAD |
| Sortilin Related Receptor 1 (SORL1) | F14B4.1 | LOAD |
| Phospholipid-Transporting ATPase (ABCA7) | abt-2 | LOAD |
| Sodium/Potassium/Calcium Exchanger (SLC24A2/A4) | ncx-4, ncx-5 | LOAD |
| Ras Additionally, Rab Interactor 3 (RIN3) | rin-1 | LOAD |
| Protein Tyrosine Kinase 2 Beta (PTK2B) | kin-32 | LOAD |
| Enoyl-CoA Hydratase Domain Containing 3 (ECHDC3) | ech-2 | LOAD |
| Angiotensin I Converting Enzyme (ACE) | acn-1 | LOAD |
| A Disintegrin Additionally, Metalloproteinase With Thrombospondin Motifs 1 (ADAMTS1) | gon-1 | LOAD |
| Thyroid Hormone Receptor Interactor 4 (TRIP4) | asc-1 | LOAD |
| Retinoic Acid Receptor-Related Orphan Receptor A (RORA) | nhr-23 | LOAD |
| Zinc Finger Protein 423 (ZNF423) | lin-13 | LOAD |
| Zinc Finger Protein 655 (ZNF655) | ztf-2 | LOAD |
| Benzodiazepine Receptor-Associated Protein 1 (TSPOAP1, BZRAP1) | rimb-1 | LOAD |
| Trophoblast Glycoprotein (TPBG) | lron-3 | LOAD |
| Heparan Sulfate-Glucosamine 3-Sulfotransferase 1 (HS3ST1) | hst-3.1 | LOAD |
| Protein Kinase D3 (PRKD3) | dkf-2 | LOAD |
| NADH:Ubiquinone Oxidoreductase Complex Assembly Factor 7 (NDUFAF7) | ZK1128.1 | LOAD |
| Nicotinic acetylcholine receptor Epsilon Subunit (CHRNE) | acr-2, acr-3, acr-6, acr-8, acr-12, lev-1, lev-8, unc-29, unc-38, unc-63 | LOAD |
| Repressor element 1-silencing transcription factor (REST) | spr-3 and spr-4 | LOAD |
| Pros |
|---|
| Many human genes possess orthologues in C. elegans, among them most (but not all) of the genes involved in Alzheimer’s disease |
| Aβ expression affects similar pathways in worm, mouse and human |
| Short generation and life cycle, around 3 weeks, and low maintenance and propagation costs |
| Small nervous system, only 302 neurons, with an invariant neuronal network |
| Transparent body, allows visualization of fluorescent proteins at all stages of its life |
| Complete characterization of cell fate lineage and neuronal connectivity |
| Complete genome sequence and very powerful genetic manipulation tools |
| Wide availability of mutant strains of most of the genes |
| Availability of extensive RNAi libraries able to silence most of the genes |
| Conserved protein interaction networks involved in AD |
| Numerous methods available for the functional characterization of neurodegeneration, motility disturbances or protein aggregation |
| Ability to make high throughput chemical screens for drug assay |
| Cons |
| Lacks β-secretase and β-amyloid peptide sequence in APP. Unable to generate endogenous Aβ |
| Lacks APOE gene |
| Lack of many specific mammalian features: circulatory system, myelinated neurons, defined brain structures such as hippocampus or cortex, complex connections of the human brain, adaptative immune system, among others |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alvarez, J.; Alvarez-Illera, P.; Santo-Domingo, J.; Fonteriz, R.I.; Montero, M. Modeling Alzheimer’s Disease in Caenorhabditis elegans. Biomedicines 2022, 10, 288. https://doi.org/10.3390/biomedicines10020288
Alvarez J, Alvarez-Illera P, Santo-Domingo J, Fonteriz RI, Montero M. Modeling Alzheimer’s Disease in Caenorhabditis elegans. Biomedicines. 2022; 10(2):288. https://doi.org/10.3390/biomedicines10020288
Chicago/Turabian StyleAlvarez, Javier, Pilar Alvarez-Illera, Jaime Santo-Domingo, Rosalba I. Fonteriz, and Mayte Montero. 2022. "Modeling Alzheimer’s Disease in Caenorhabditis elegans" Biomedicines 10, no. 2: 288. https://doi.org/10.3390/biomedicines10020288
APA StyleAlvarez, J., Alvarez-Illera, P., Santo-Domingo, J., Fonteriz, R. I., & Montero, M. (2022). Modeling Alzheimer’s Disease in Caenorhabditis elegans. Biomedicines, 10(2), 288. https://doi.org/10.3390/biomedicines10020288