Abstract
Treatment of pancreatic ductal adenocarcinoma (PDAC), a dismal disease with poor survival rates, is hampered by the high prevalence of chemotherapy resistance. Resistance is accompanied by macrophage infiltration into the tumor microenvironment, and infiltrated macrophages are key players in chemotherapy resistance. In the current manuscript, we identify CCAAT/enhancer-binding protein delta (C/EBPδ) as an important transcription factor driving macrophage-dependent gemcitabine resistance. We show that conditioned medium obtained from wild type macrophages largely diminishes gemcitabine-induced cytotoxicity of PDAC cells, whereas conditioned medium obtained from C/EBPδ-deficient macrophages only minimally affects gemcitabine-induced PDAC cell death. Subsequent analysis of RNA-Seq data identified the pyrimidine metabolism pathway amongst the most significant pathways down-regulated in C/EBPδ-deficient macrophages and size filtration experiments indeed showed that the low molecular weight and free metabolite fraction most effectively induced gemcitabine resistance. In line with a role for pyrimidines, we next show that supplementing macrophage conditioned medium with deoxycytidine overruled the effect of macrophage conditioned media on gemcitabine resistance. Consistently, macrophage C/EBPδ-dependent resistance is specific for gemcitabine and does not affect paclitaxel or 5-FU-induced cytotoxicity. Overall, we thus show that C/EBPδ-dependent deoxycytidine biosynthesis in macrophages induces gemcitabine resistance of pancreatic cancer cells.
1. Introduction
Pancreatic adenocarcinoma (PDAC) is a devastating disease with one of the worst survival rates of all human cancers [1]. Despite improvements in the treatment of cancer in general, hardly any progress has been made in PDAC treatment and the average survival rate remains around 6-8 months. Five years after diagnosis, more than 90% of PDAC patients have died, while 10-year mortality rates approach 99% [2,3]. The dismal prognosis of PDAC patients is largely due to the fact that most patients present with locally advanced or metastatic disease and are consequently ineligible for curative resection [4]. Chemotherapy-based therapies therefore remain standard of care [5], but chemotherapeutic treatments only minimally affect PDAC progression [6,7], therefore improving the efficacy of chemotherapy in PDAC is of utmost importance.
Macrophages are specialized phagocytic cells of the innate immune system critically involved in host defense and tissue homeostasis [8]. In cancer biology, macrophages are traditionally considered anti-tumorigenic, as seminal papers showed that macrophages may kill tumor cells by secreting cytotoxic molecules, such as TNF-α, IL-12, nitric oxide (NO) and reactive oxygen species (ROS) [9,10]. Once recruited to the tumor, however, macrophages are reprogrammed to prevent killing of cancer cells. Such reprogrammed M2 or tumor associated macrophages (TAMs) not only show limited cytotoxicity towards cancer cells but actually become pro-tumorigenic and potentiate tumor growth [11,12,13]. Indeed, TAMs induce epithelial-to-mesenchymal transition (EMT) [14,15,16] and limiting macrophage infiltration decreases the number of metastatic lesions [17]. In addition, TAM numbers are associated with therapy resistance in PDAC [18,19], and pharmacological depletion of TAMs enhanced the therapeutic response to gemcitabine in tumor-bearing KPC mice [20].
CCAAT/enhancer-binding protein delta (C/EBPδ), also known as nuclear factor interleukin (IL)-6β, is a member of the C/EBP family of transcription factors [21]. It modulates many biological processes involved in cancer biology and C/EBPδ acts as tumor suppressor in multiple tumor types [22,23,24,25]. In PDAC, C/EBPδ is down-regulated in cancer cells, and reduced C/EBPδ expression correlates with poor prognosis [26]. As opposed to low C/EBPδ expression in PDAC cells, most stromal cells show strong C/EBPδ expression [27]. In particular, the expression of C/EBPδ in macrophages seems interesting, as C/EBPδ is well-known to regulate macrophage-dependent chemokine and cytokine expression [28], whereas C/EBPδ also may affect macrophage polarization [29]. Considering the importance of cytokine production and macrophage polarization in drug resistance [30], we hypothesized that macrophage C/EBPδ would have an impact on drug resistance of PDAC cells. By using C/EBPδ-deficient macrophages, we show that macrophage C/EBPδ drives gemcitabine, but not 5-FU or paclitaxel, resistance in a deoxycytidine-dependent manner.
2. Materials and Methods
2.1. Cell Culture
PANC-1, MIA PaCa-2 and BxPc3 PDAC cells (ATCC, Manassas, VA, USA) were cultured in DMEM (Lonza, Basel, Switzerland), whereas wild type and C/EBPδ knock out RAW264.7 macrophages, generated and characterized as described previously [31], were cultured in IMDM medium (Gibco, Thermo Fischer Scientific, Waltham, MA, USA). All media were supplemented with 10% fetal calf serum (FCS), 2 mM L-glutamine, 100 units/mL penicillin, and 500 μg/mL streptomycin (all Lonza, Basel, Switzerland) according to routine cell culture procedures. Cells were maintained in a humidified incubator at 37 °C and 5% CO2. All PDAC cell lines were authenticated by STR profiling (Promega PowerPlex, Leiden, Netherlands), and tested for mycoplasma by PCR monthly.
2.2. Conditioned Medium Collection and PDAC Cytotoxicity Assays
Wild type and C/EBPδ knock out RAW264.7 macrophages were grown to 70% confluence in a T75 flask, after which the growth medium was refreshed. After 24 h, medium was collected, sterilized through a 0.2 µm filter and stored at −20 °C. PDAC cells, seeded in 100 μL complete medium in 96-well plates were, upon adherence of the cells, incubated in conditioned medium (diluted 1:1 in complete DMEM) supplemented with gemcitabine, 5-FU or paclitaxel. Based on IC50 analysis (Figures S1 and S3), PANC-1 cells were treated with 20 nM gemcitabine, 5 nM paclitaxel or 4 μM 5-FU, MIA PaCa-2 cells were treated with 15 nM gemcitabine, 20 nM paclitaxel or 2 μM 5-FU, whereas BxPc3 cells were treated with 10 nM gemcitabine, 4 nM paclitaxel or 4 μM 5-FU. After 96 h, drug-induced cytotoxicity was assessed essentially as described before [32]. In detail, PDAC cells were washed and incubated with crystal violet (3% formaldehyde, 0.5% crystal violet in H2O) at room temperature. After 20 min, the crystal violet solution was removed, cells were washed 3 times with tap water, and 75 µL/well DMSO was added to solubilize the formed crystals. After 20 min of incubation on a plate shaker at room temperature, the absorbance was measured at 600 nm on a Synergy HT plate reader (BioTek Instruments, Winooski, VT, USA).
2.3. Size Separation of Proteins in Conditioned Media
RAW264.7 conditioned media, collected as described above, were separated using 10 kD Molecular Weight Cut Off ultra-centrifugal filters (Amicon-Ultra, Millipore, Ireland). The concentrated >10 kD fraction was resuspended in IMDM to the original volume before storage at 4 °C, whereas the flow-through fraction (<10 kD) was stored at 4 °C until used in experiments.
2.4. RT-qPCR and RNA-Seq
RNA was extracted form TriReagent lysed cells according to routine procedures. Eluted RNA was analyzed spectrophotometrically using a NanoDrop 2000 (Thermo Fisher Scientific, Waltham, MA, USA). All samples were treated with RQ1 RNAse-Free DNAse (Promega Benelux BV) and reverse-transcribed into cDNA using M-MLV Reverse Transcriptase (Promega Benelux BV, Leiden, The Netherlands), random hexamers (Fisher scientific) and 10 mM dNTPs (Fermentas, Fisher scientific, Landsmeer, The Netherlands). The Sensi-FAST™ SYBR® No-ROX Kit (GC biotech, Waddinxveen, The Netherlands) was used to perform real-time quantitative RT-PCR on a LightCycler® 480 Instrument II (Roche Molecular Systems, Inc., Almere, The Netherlands). Expression levels were normalized to the expression of the reference genes TBP, B2M and UBC using the primers listed in Supplementary Table S1. RNA-Seq results and the corresponding experimental design were described previously [31] and represent transcriptional differences between wild type and C/EBPδ-deficient RAW264.7 cells. The complete sequence libraries are publicly available through the National Center for Biotechnology Information gene expression omnibus under the following accession number: GSE173552. Differential gene expression was analyzed using the R2 microarray analysis and visualization platform (R2: Genomics Analysis and Visualization Platform. Available online: http://r2.amc.nl; last accessed 12 January 2022) with a false discovery adjusted p-value less than 0.01. To analyze the function of the differentially expressed genes, Kyoto Encyclopedia of Genes and Genomes (KEGG) pathway enrichment analyses were conducted with a p-value cutoff off less than 0.005.
2.5. Statistical Analysis
All data are expressed as means ± SEM. Differences between multiple groups were analyzed by one-way ANOVA with Bonferroni correction for multiple testing, whereas t-tests were used for comparisons between 2 groups. Analyses were performed using GraphPad Prism version 8 (GraphPad Software, Inc., La Jolla, CA, USA). Statistically significant differences were considered with a p-value less than 0.05.
3. Results
3.1. Macrophage C/EBPδ Drives Gemcitabine Resistance of Pancreatic Cancer Cells
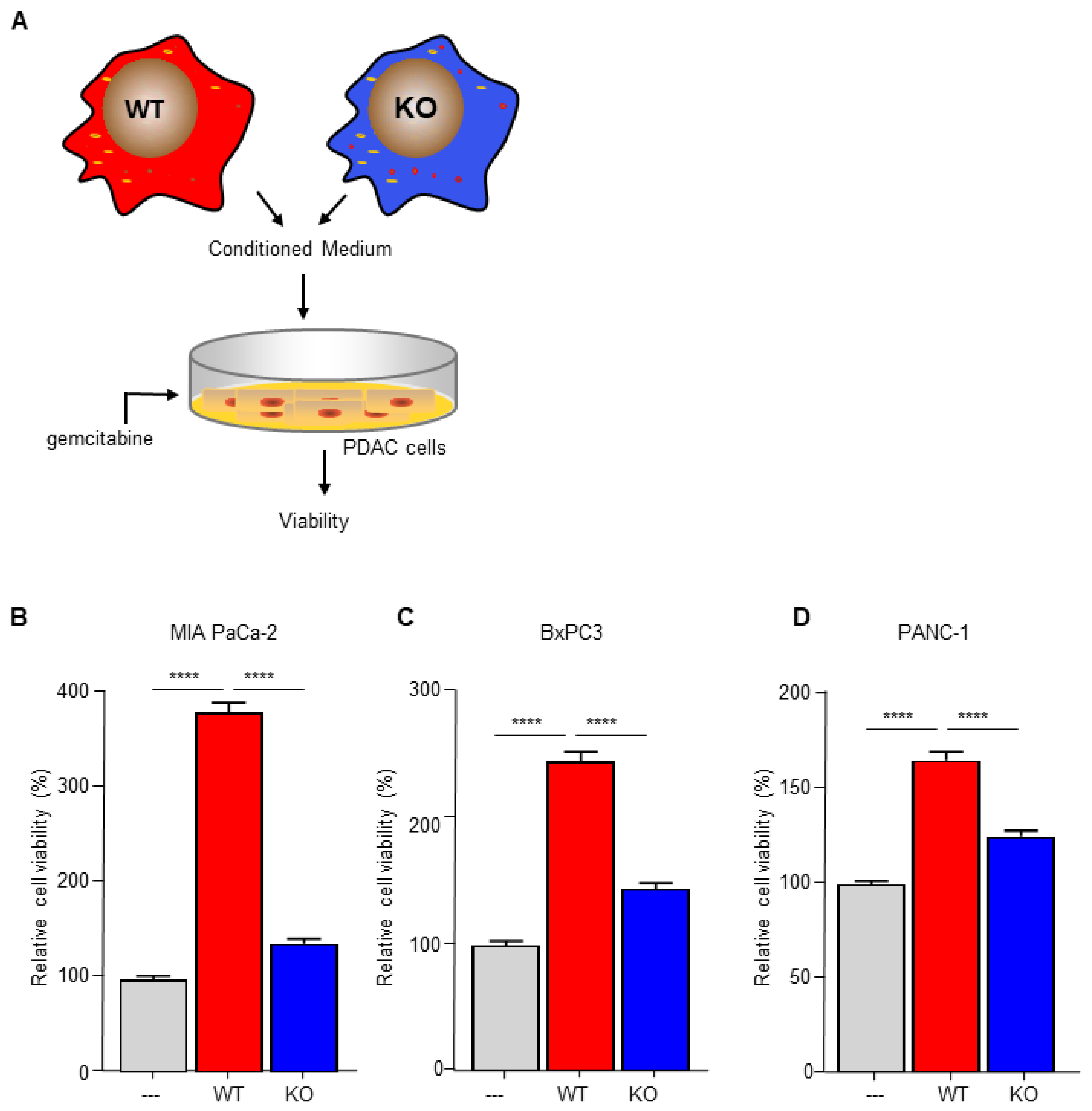
To investigate the importance of C/EBPδ in macrophage-dependent resistance to gemcitabine, we employed wild type and C/EBPδ-deficient RAW264.7 macrophages. Conditioned medium of these cells was compared to control medium in gemcitabine-induced pancreatic cancer cell cytotoxicity assays (Figure 1A). As shown in Figure 1B–D, conditioned medium obtained from wild type macrophages induced gemcitabine resistance in MIA PaCa-2 (Figure 1B), BxPc3 (Figure 1C) and PANC-1 (Figure 1D) cells. Of note, conditioned medium obtained from C/EBPδ-deficient macrophages did not affect gemcitabine-induced cytotoxicity of any of the pancreatic cancer cells tested. To exclude a direct effect of conditioned medium on cell viability, MIA PaCa-2 cells were incubated in conditioned medium in the absence of gemcitabine, showing that conditioned medium indeed did not affect cell viability by itself (Figure S2).
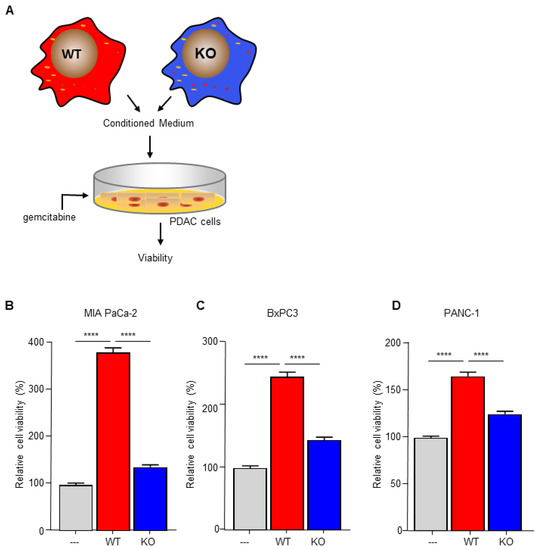
Figure 1.
Macrophage C/EBPδ drives gemcitabine resistance of pancreatic cancer cells. (A) Schematic overview of the experimental set up. (B–D) Viability of Mia PaCa-2 (B), BxPc3 (C) and PANC-1 (D) pancreatic cancer cells in control medium (---), conditioned medium obtained from wild type macrophages (WT) and conditioned medium obtained from C/EBPδ-deficient macrophages (KO) in the presence of gemcitabine. Shown is the mean ± SEM of two to three independent experiments performed in sixplo. **** p < 0.0001.
3.2. C/EBPδ Deficiency Inhibits Pyrimidine Syntheses in Macrophages
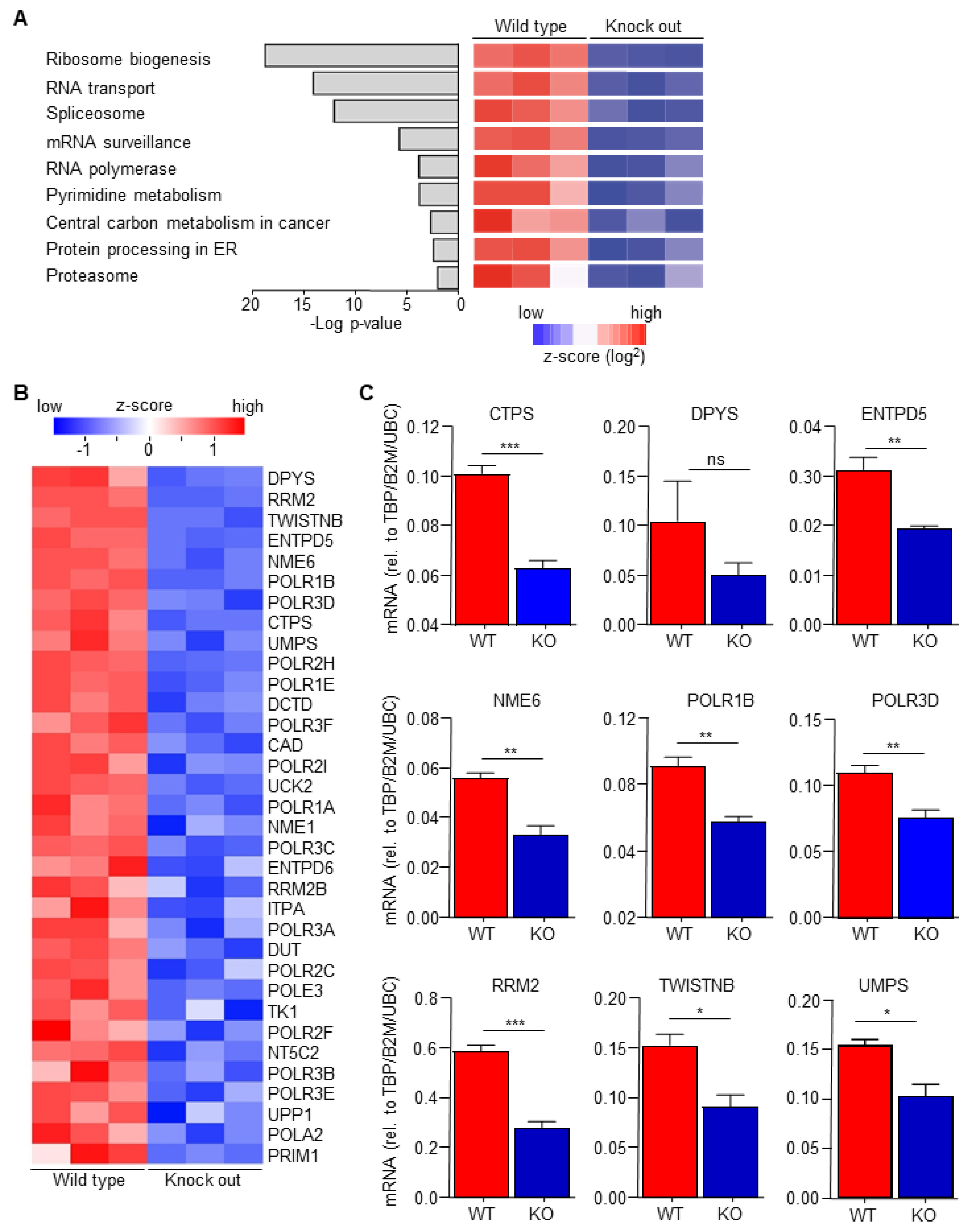
Macrophage-secreted chemokines and growth factors are suggested to induce drug resistance [18], suggesting that macrophage C/EBPδ potentiates gemcitabine resistance by enhancing chemokine/cytokine secretion. Analysis of recently obtained RNA-Seq data to determine transcriptional changes induced by C/EBPδ deletion in macrophages (GSE173552 [31]) revealed, however, no apparent changes in the expression of chemokines/cytokines. KEGG pathway enrichment analyses of differentially expressed genes between wild type and C/EBPδ-deficient macrophages, in fact, identified the pyrimidine metabolism pathway amongst the most significant pathways down-regulated by C/EBPδ deficiency (Figure 2A). Detailed analysis of the pyrimidine metabolism pathway showed that 34 genes were significantly decreased in C/EBPδ-deficient macrophages (Figure 2B) of which the 9 most down-regulated genes were confirmed by RT-PCR analysis (Figure 2C).
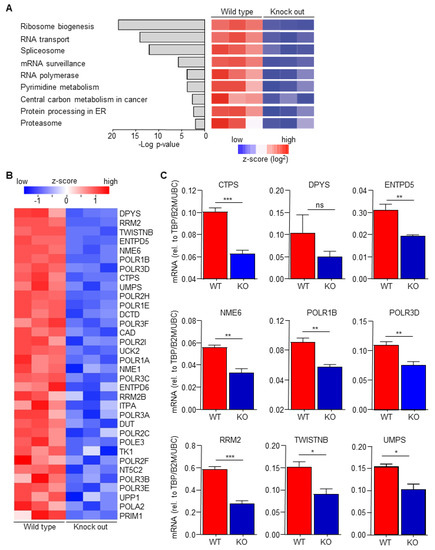
Figure 2.
C/EBPδ deficiency inhibits pyrimidine syntheses. (A) The p-values and names of the most over-represented KEGG pathways, calculated on the basis of all the differentially expressed genes between wild type and C/EBPδ-deficient macrophages. (B) Heatmap of all the differentially expressed genes from the pyrimidine metabolism pathway. (C) RT-PCR validation of the top differentially expressed genes of the pyrimidine metabolism pathway. * p < 0.05; ** p < 0.01; *** p < 0.005; ns: not significant.
3.3. Macrophage C/EBPδ Drives Gemcitabine Resistance in a Deoxycytidine-Dependent Manner
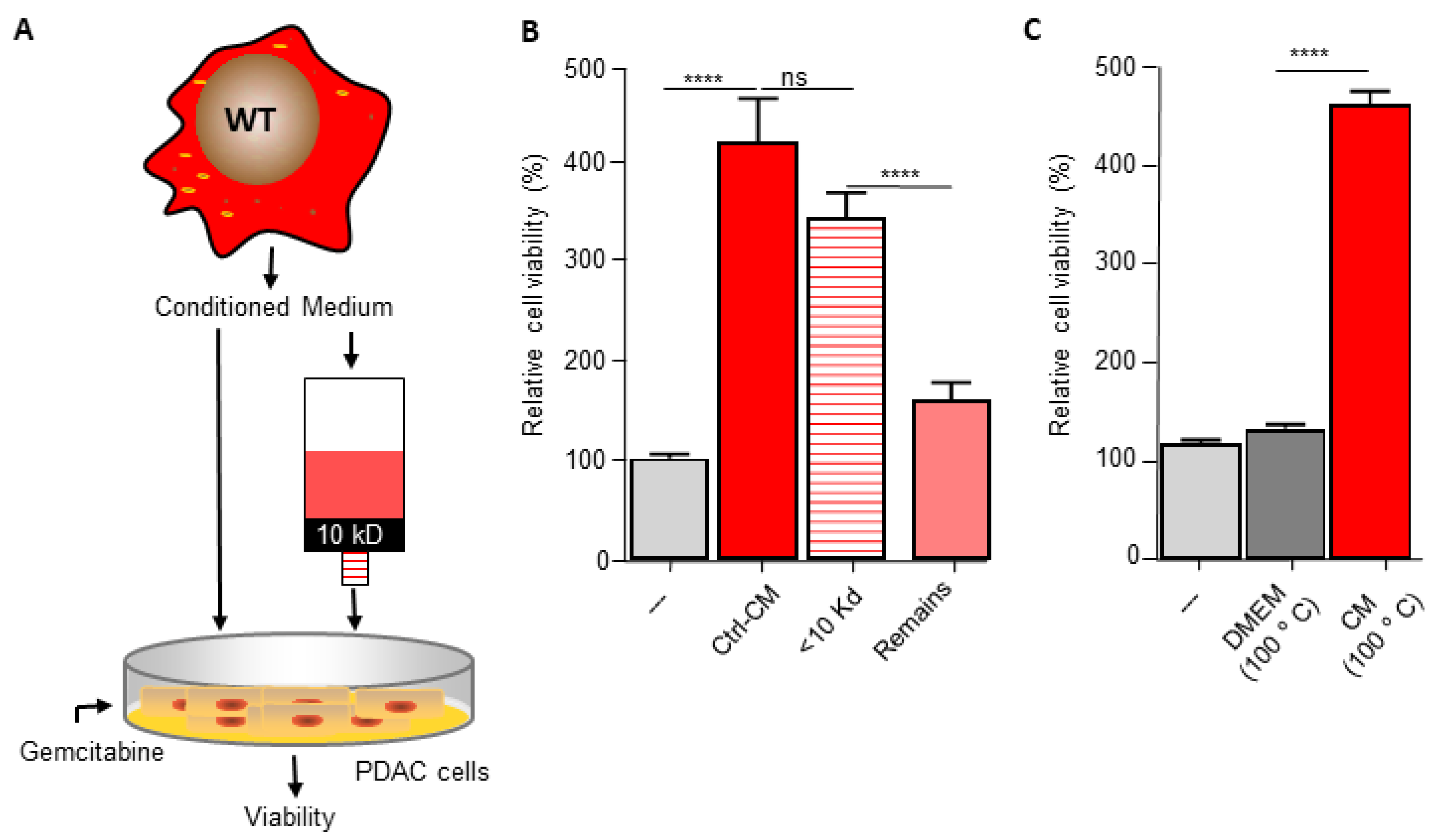
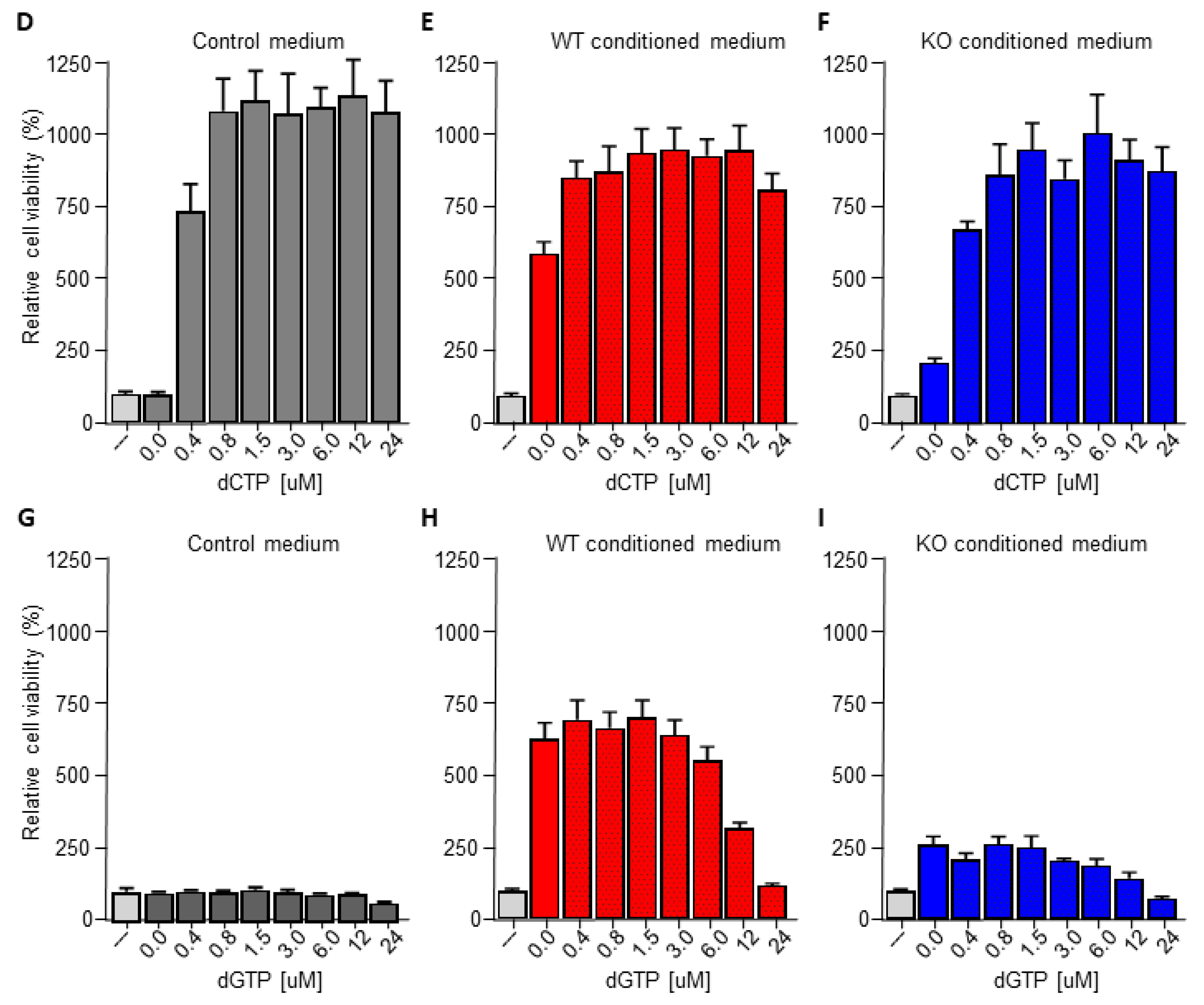
The pyrimidine metabolism pathway has previously been shown to affect gemcitabine resistance [33,34]. Indeed, deoxycytidine directly competes with gemcitabine thereby inhibiting its uptake and activation, leading to diminished intracellular gemcitabine activity and reduced gemcitabine-induced cytotoxicity [33]. It is thus tempting to speculate that macrophage C/EBPδ drives gemcitabine resistance in a deoxycytidine-dependent manner. To prove or refute this hypothesis, we first subjected conditioned medium obtained from wild type macrophages to a size filtration spin column with a 10 kD cutoff to separate proteins from free metabolites (Figure 3A). As shown in Figure 3B, the flow through the fraction containing free metabolites was almost as effective in inducing gemcitabine resistance as control, non-filtrated, conditioned medium. The fraction that retained on the filter (containing proteins but also residual metabolites) was clearly less effective in inducing gemcitabine resistance of pancreatic cancer cells. In line with these findings that suggest that the resistance-inducing factor is not a protein, heat denaturation of the conditioned medium (15 min 100 °C) prior to transfer to pancreatic cancer cells did not diminish gemcitabine resistance (Figure 3C). Based on a model in which C/EBPδ-dependent deoxycytidine biosynthesis in macrophages induces gemcitabine resistance of pancreatic cancer cells, exogenous administration of deoxycytidine should overrule the effect of macrophage C/EBPδ. As shown in Figure 3D–F, deoxycytidine co-treatment indeed limits gemcitabine-induced cytotoxicity to a similar extent in control, wild type and C/EBPδ-deficient conditioned medium. Of note, supplementing control or C/EBPδ-deficient conditioned medium with low dose deoxycytidine (0.4 uM) already results in similar gemcitabine resistance as induced by wild type conditioned medium (compare the 0.4 uM bars of panels D and F with the 0 uM bar of panel E). Supplementing control or conditioned medium with the pyrimidine deoxyguanosine did not induce gemcitabine resistance (Figure 3G–I), in line with the notion that deoxyguanosine does not compete with gemcitabine uptake and activation.
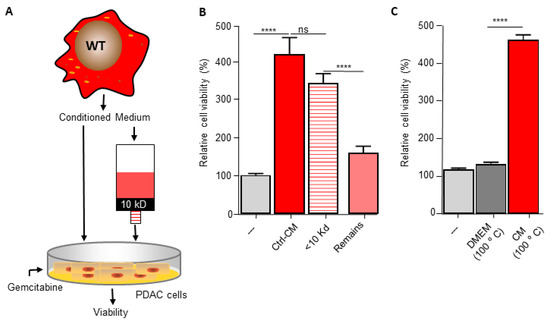
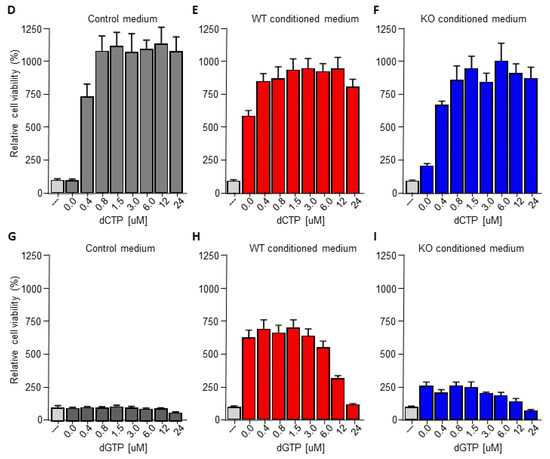
Figure 3.
Macrophage C/EBPδ drives gemcitabine resistance in a deoxycytidine-dependent manner. (A) Schematic overview of the experimental set up. (B) Viability of Mia PaCa-2 pancreatic cancer cells in control medium (---), unfractionated conditioned medium obtained from wild type macrophages (Ctrl-CM), in the flow through fraction containing free metabolites (<10 kD) and in the fraction that retained on the filter (remains) in the presence of gemcitabine. **** p < 0.0001; ns: not significant. (C) Viability of Mia PaCa-2 pancreatic cancer cells in control medium (---), boiled DMEM (DMEM 100 °C) or boiled conditioned medium obtained from wild type macrophages (CM 100 °C) in the presence of gemcitabine. **** p < 0.0001. (D–F) Viability of Mia PaCa-2 pancreatic cancer cells in control medium (D) or conditioned medium obtained from wild type (E) or C/EBPδ-deficient macrophages (F) in the presence of gemcitabine and the indicated concentration of deoxycytidine (dCTP). (G–I) Viability of Mia PaCa-2 pancreatic cancer cells in control medium (G) or conditioned medium obtained from wild type (H) or C/EBPδ-deficient macrophages (I) in the presence of gemcitabine and the indicated concentration of deoxycytidine (dCTP). Shown is the mean ± SEM of experiments performed in sixplo. Note that the light grey bars in panels (D–I) (---) represent cell viability in control conditions (i.e., control medium supplemented with gemcitabine).
3.4. Macrophage C/EBPδ-Dependent Resistance of Pancreatic Cancer Cells Is Specific for Deoxycytidine Analogs
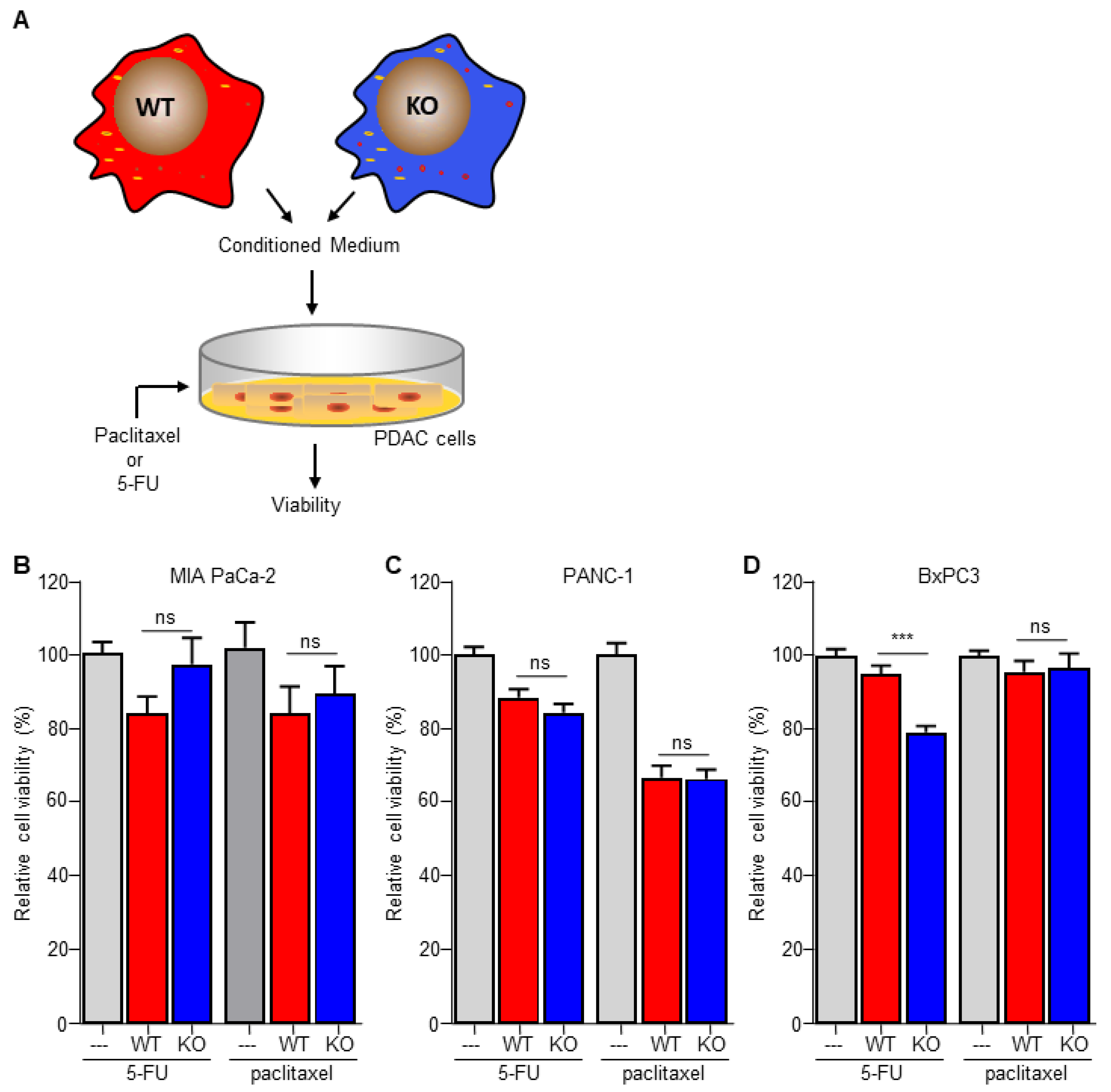
The data above suggest that macrophage C/EBPδ induces gemcitabine resistance by potentiating deoxycytidine biosynthesis and secretion. For this hypothesis to be true, macrophage C/EBPδ-induced resistance should be limited to gemcitabine. Consequently, we next assessed the effect of macrophage C/EBPδ on paclitaxel-, a cytotoxic agent routinely used in pancreatic cancer treatment that acts independent of DNA replication, and 5-FU-, a pyrimidine nucleoside-based cytotoxic agent with different transport and activation properties as deoxycytidine [33], induced cell death (Figure 4A). In line with our hypothesis, conditioned medium obtained from both wild type and C/EBPδ-deficient macrophages did not induce resistance to 5-FU or paclitaxel in MIA PaCa-2 (Figure 4B), PANC-1 (Figure 4C) or BxPc3 (Figure 4D) cells. Of note, conditioned medium did slightly enhance 5-FU- and paclitaxel-induced cytotoxicity in PANC1 and BxPc3 cells. Except for 5-FU-treated BxPc3 cells, this effect was similar for conditioned medium obtained from wild type and C/EBPδ-deficient macrophages.
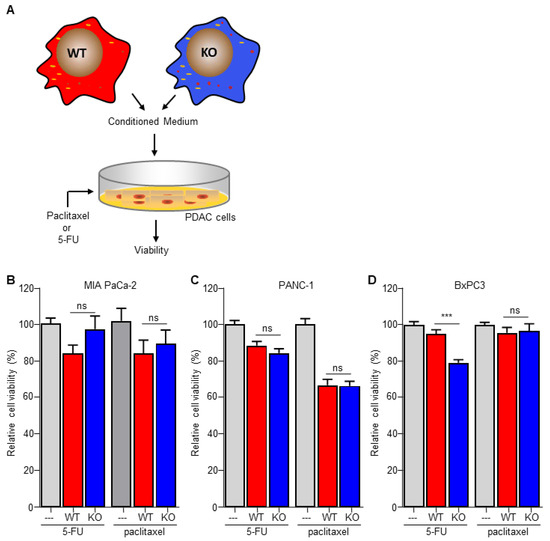
Figure 4.
Macrophage C/EBPδ selectively induces resistance to gemcitabine. (A) Schematic overview of the experimental set up. (B–D) Viability of Mia PaCa-2 (B), BxPc3 (C) and PANC-1 (D) pancreatic cancer cells in control medium (---), conditioned medium obtained from wild type macrophages (WT) and conditioned medium obtained from C/EBPδ-deficient macrophages (KO) in the presence of 5-FU or paclitaxel. Shown is the mean ± SEM of an experiment performed in sixplo. *** p < 0.0001; ns: not significant.
4. Discussion
In the current study, we aimed to elucidate the importance of macrophage C/EBPδ in drug resistance of pancreatic cancer cells. We confirm that macrophage-derived conditioned medium induces gemcitabine resistance of pancreatic cancer cells and show that conditioned medium obtained from C/EBPδ-deficient macrophages no longer induces gemcitabine resistance. The fact that resistance, which occurs in most PDAC patients within weeks of treatment initiation, profoundly limits the efficacy of gemcitabine treatment [35], underscores the potential importance of our findings and suggests that C/EBPδ is a key factor underlying the poor prognosis of PDAC patients.
Single agent gemcitabine chemotherapy has been the backbone of PDAC treatment for many years [36,37,38,39], but combination therapy with folinic acid, fluorouracil, irinotecan and oxaliplatin (FOLFIRINOX) recently replaced gemcitabine as first-line treatment modality [40]. The increased clinical benefit of FOLFIRINOX comes, however, at the expense of increased toxicity [41]. In view of the substantial toxicity of FOLFIRINOX, understanding the underlying mechanisms by which PDAC patients become resistant to gemcitabine is not just of scientific importance. The identification of C/EBPδ as a driver of macrophage-dependent gemcitabine resistance suggests that patients with low C/EBPδ expression in their macrophages may be particularly eligible for gemcitabine treatment. On the contrary, patients with high macrophage C/EBPδ levels should preferably not be treated with gemcitabine, as they are prone to be resistant and experience limited benefit of treatment. Future studies should prove or refute the validity of this tantalizing hypothesis.
Macrophage numbers in or around tumors are typically associated with poor prognosis and reduced overall survival [11,12,13]. In line with this, pharmacological macrophage depletion was recently shown to enhance the therapeutic response to gemcitabine in a preclinical murine PDAC model [20]. These data not only confirm the general concept that macrophages, despite their cytotoxic nature, exert a pro-tumorigenic role [42] but also suggest that macrophage depletion could be an attractive option to increase the clinical efficacy of gemcitabine. Of note, however, is that specific macrophage subsets may possess tumor-suppressive functions based upon which it has been suggested that macrophage reprogramming rather than depletion may be the most promising strategy to pursue [20,43]. We identify C/EBPδ as an attractive candidate to reprogram macrophages in the setting of gemcitabine therapy in PDAC patients. Our data suggest that combination therapy of gemcitabine and a C/EBPδ inhibitor will enhance gemcitabine efficacy and will improve treatment response in PDAC patients. In particular, patients with high C/EBPδ positive macrophage numbers are likely to benefit from combination therapy, but this hypothesis needs to be addressed in preclinical PDAC models before one could pursue to clinical studies. Although no C/EBPδ inhibitor has yet been clinically approved, a recent study showed that two histone deacetylase (HDAC) inhibitors, suberoylanilide hydroxamic acid (SAHA) and trichostatin A (TSA), abolished endogenous C/EBPδ mRNA expression levels in THP-1 macrophages [44]. Whether these HDAC inhibitors do reduce C/EBPδ levels in tumor associated macrophages and subsequently potentiate gemcitabine efficacy in PDAC remains to be established in future experiments.
C/EBPδ plays a pleiotropic role in cancer biology and may either potentiate or inhibit cancer progression in a context-dependent manner. In PDAC, tumor cell C/EBPδ expression is lost and re-expression of C/EBPδ in PDAC cell lines slowed down proliferation and decreased the clonogenic capacity [26]. In contrast, however, primary tumor growth was not affected in C/EBPδ-deficient mice but metastases were observed in numerous organs of tumor cell grafted wild type mice but not in C/EBPδ-deficient mice [27]. Here, we extend the notion that C/EBPδ seems to exert opposing effects in tumor and non-tumor cells by showing that macrophage C/EBPδ limits the cytotoxicity of gemcitabine towards pancreatic cancer cells. Of note, a previous study suggested that macrophage C/EBPδ induced chemoresistance of breast cancer cells to both cisplatin and 5-FU [45]. The latter is especially interesting as we here show that wild type and C/EBPδ-deficient macrophages both do not induce resistance of pancreatic cancer cells towards 5-FU, further underscoring the context-dependent role of C/EBPδ in tumor biology.
Gemcitabine resistance of pancreatic cancer cells is multifactorial [46] and may result from pancreatic cancer cell autonomous processes, for instance mitochondria-mediated apoptosis [47,48] or nucleoside transporter down-regulation [49], and from interactions between pancreatic cancer cells and stromal cells. Tumor associated macrophages may induce gemcitabine resistance of pancreatic cancer cells by several mechanisms (excellently reviewed by Yang and colleagues [50]). Most importantly for the current study, macrophages release a spectrum of pyrimidine species of which deoxycytidine molecularly competes with gemcitabine uptake and metabolism thereby hindering its efficacy as a chemotherapy in PDAC [34]. We here confirm the importance of macrophage-derived deoxycytidine in gemcitabine resistance and identify C/EBPδ as a key transcription factor driving deoxycytidine biosynthesis in macrophages. In line with its specific effect on deoxycytidine biosynthesis, macrophage C/EBPδ did not impact resistance to either 5-FU or paclitaxel. Indeed, conditioned medium obtained from both wild type and C/EBPδ-deficient macrophages did not induce resistance to 5-FU and paclitaxel, but actually slightly enhanced 5-FU- and paclitaxel-induced cytotoxicity in PANC-1 and BxPc3 cells. At this moment, we do not have an explanation for the small additive effect of macrophage conditioned medium on 5-FU- and paclitaxel-induced cytotoxicity, but most importantly our data highlight that C/EBPδ specifically acts upon gemcitabine resistance of pancreatic cancer cells and that C/EBPδ is not a generic player in PDAC chemoresistance. Interestingly, however, pancreatic cancer cells are more resistant to gemcitabine than other chemotherapeutic drugs [46], making C/EBPδ an attractive transcription factor in the setting of chemoresistance in PDAC.
5. Conclusions
In conclusion, we here show that macrophage C/EBPδ drives gemcitabine resistance of pancreatic cancer cells in a deoxycytidine-dependent manner. Our data contribute to a better understanding of gemcitabine resistance in PDAC which may ultimately aid in improved prognosis of this dismal disorder.
Supplementary Materials
The following supporting information can be downloaded at: www.mdpi.com/article/10.3390/biomedicines10020219/s1, Figure S1: Dose–response curves of gemcitabine treatment, Figure S2: Conditioned medium does not affect cell viability, Figure S3: Dose–response curves of paclitaxel and 5-FU treatment, Table S1: Primers for LC validation.
Author Contributions
Conceptualization, C.A.S. and J.D.; methodology, C.A.S. and J.D.; validation, C.A.S. and J.D.; investigation, H.L.A. and J.D.; data curation, C.A.S., H.L.A. and J.D; writing—original draft preparation, C.A.S.; writing—review and editing, C.A.S. and J.D.; visualization, J.D. and C.A.S.; supervision, C.A.S. and J.D.; project administration, C.A.S. and J.D.; funding acquisition, J.D. All authors have read and agreed to the published version of the manuscript.
Funding
This research was supported by NWO (Netherlands Organization for Scientific Research), VENI grant (016.186.046). The APC was funded by the University of Amsterdam. The funders did not participate in the study design, data collection, data analysis, interpretation or writing of the report.
Institutional Review Board Statement
Not applicable.
Informed Consent Statement
Not applicable.
Data Availability Statement
The data presented in this study are available from the corresponding author upon reasonable request.
Conflicts of Interest
The authors declare no conflict of interest.
References
- Ferlay, J.; Ervik, M.; Lam, F.; Colombet, M.; Mery, L.; Piñeros, M.; Znaor, A.; Soerjomataram, I.; Bray, F. Global Cancer Observatory: Cancer Today. Lyon: International Agency for Research on Cancer; 2020. Available online: https://gco.iarc.fr/today (accessed on 11 January 2022).
- Rawla, P.; Sunkara, T.; Gaduputi, V. Epidemiology of Pancreatic Cancer: Global Trends, Etiology and Risk Factors. World J. Oncol. 2019, 10, 10–27. [Google Scholar] [CrossRef] [PubMed]
- Siegel, R.L.; Miller, K.D.; Jemal, A. Cancer statistics, 2020. CA Cancer J. Clin. 2020, 70, 7–30. [Google Scholar] [CrossRef] [PubMed]
- Kamisawa, T.; Wood, L.D.; Itoi, T.; Takaori, K. Pancreatic cancer. Lancet 2016, 388, 73–85. [Google Scholar] [CrossRef]
- Principe, D.R.; Underwood, P.W.; Korc, M.; Trevino, J.G.; Munshi, H.G.; Rana, A. The Current Treatment Paradigm for Pancreatic Ductal Adenocarcinoma and Barriers to Therapeutic Efficacy. Front. Oncol. 2021, 11, 688377. [Google Scholar] [CrossRef]
- Von Hoff, D.D.; Ervin, T.; Arena, F.P.; Chiorean, E.G.; Infante, J.; Moore, M.; Seay, T.; Tjulandin, S.A.; Ma, W.W.; Saleh, M.N.; et al. Increased Survival in Pancreatic Cancer with Nab-Paclitaxel Plus Gemcitabine. N. Engl. J. Med. 2013, 369, 1691–1703. [Google Scholar] [CrossRef] [Green Version]
- Conroy, T.; Hammel, P.; Hebbar, M.; Ben Abdelghani, M.; Wei, A.C.; Raoul, J.L.; Choné, L.; Francois, E.; Artru, P.; Biagi, J.J.; et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. N. Engl. J. Med. 2018, 379, 2395–2406. [Google Scholar] [CrossRef]
- Porta, C.; Riboldi, E.; Totaro, M.G.; Strauss, L.; Sica, A.; Mantovani, A. Macrophages in cancer and infectious diseases: The ‘good’ and the ‘bad’. Immunotherapy 2011, 3, 1185–1202. [Google Scholar] [CrossRef]
- Jadus, M.R.; Irwin, M.C.; Irwin, M.R.; Horansky, R.D.; Sekhon, S.; Pepper, K.A.; Kohn, D.B.; Wepsic, H.T. Macrophages can recognize and kill tumor cells bearing the membrane isoform of macrophage colony-stimulating factor. Blood 1996, 87, 5232–5241. [Google Scholar] [CrossRef] [Green Version]
- Goswami, K.K.; Ghosh, T.; Ghosh, S.; Sarkar, M.; Bose, A.; Baral, R. Tumor promoting role of anti-tumor macrophages in tumor microenvironment. Cell Immunol. 2017, 316, 1–10. [Google Scholar] [CrossRef]
- Jung, K.Y.; Cho, S.W.; Kim, Y.A.; Kim, D.; Oh, B.C.; Park, D.J.; Park, Y.J. Cancers with Higher Density of Tumor-Associated Macrophages Were Associated with Poor Survival Rates. J. Pathol. Transl. Med. 2015, 49, 318–324. [Google Scholar] [CrossRef] [Green Version]
- Lindsten, T.; Hedbrant, A.; Ramberg, A.; Wijkander, J.; Solterbeck, A.; Eriksson, M.; Delbro, D.; Erlandsson, A. Effect of macrophages on breast cancer cell proliferation.; and on expression of hormone receptors.; uPAR and HER-2. Int. J. Oncol. 2017, 51, 104–114. [Google Scholar] [CrossRef] [Green Version]
- Zhang, Q.W.; Liu, L.; Gong, C.Y.; Shi, H.S.; Zeng, Y.H.; Wang, X.Z.; Zhao, Y.W.; Wei, Y.Q. Prognostic significance of tumor-associated macrophages in solid tumor: A meta-analysis of the literature. PLoS ONE 2012, 7, e50946. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Su, S.; Liu, Q.; Chen, J.; Chen, J.; Chen, F.; He, C.; Huang, D.; Wu, W.; Lin, L.; Huang, W.; et al. A positive feedback loop between mesenchymal-like cancer cells and macrophages is essential to breast cancer metastasis. Cancer Cell 2014, 25, 605–620. [Google Scholar] [CrossRef] [Green Version]
- Fan, Q.M.; Jing, Y.Y.; Yu, G.F.; Kou, X.R.; Ye, F.; Gao, L.; Li, R.; Zhao, Q.D.; Yang, Y.; Lu, Z.H.; et al. Tumor-associated macrophages promote cancer stem cell-like properties via transforming growth factor-beta1-induced epithelial-mesenchymal transition in hepatocellular carcinoma. Cancer Lett. 2014, 352, 160–168. [Google Scholar] [CrossRef]
- Liu, C.Y.; Xu, J.Y.; Shi, X.Y.; Huang, W.; Ruan, T.Y.; Xie, P.; Ding, J.L. M2-polarized tumor-associated macrophages promoted epithelial-mesenchymal transition in pancreatic cancer cells; partially through TLR4/IL-10 signaling pathway. Lab. Investig. 2013, 93, 844–854. [Google Scholar] [CrossRef] [Green Version]
- Mitchem, J.B.; Brennan, D.J.; Knolhoff, B.L.; Belt, B.A.; Zhu, Y.; Sanford, D.E.; Belaygorod, L.; Carpenter, D.; Collins, L.; Piwnica-Worms, D.; et al. Targeting tumor-infiltrating macrophages decreases tumor-initiating cells.; relieves immunosuppression.; and improves chemotherapeutic responses. Cancer Res. 2013, 73, 1128–1141. [Google Scholar] [CrossRef] [Green Version]
- Weizman, N.; Krelin, Y.; Shabtay-Orbach, A.; Amit, M.; Binenbaum, Y.; Wong, R.J.; Gil, Z. Macrophages mediate gemcitabine resistance of pancreatic adenocarcinoma by upregulating cytidine deaminase. Oncogene 2014, 33, 3812–3819. [Google Scholar] [CrossRef] [Green Version]
- Adamska, A.; Domenichini, A.; Falasca, M. Pancreatic Ductal Adenocarcinoma: Current and Evolving Therapies. Int. J. Mol. Sci. 2017, 18, 1338. [Google Scholar] [CrossRef]
- Buchholz, S.M.; Goetze, R.G.; Singh, S.K.; Ammer-Herrmenau, C.; Richards, F.M.; Jodrell, D.I.; Buchholz, M.; Michl, P.; Ellenrieder, V.; Hessmann, E.; et al. Depletion of Macrophages Improves Therapeutic Response to Gemcitabine in Murine Pancreas Cancer. Cancers 2020, 12, 1978. [Google Scholar] [CrossRef] [PubMed]
- Lekstrom-Himes, J.; Xanthopoulos, K.G. Biological role of the CCAAT/enhancer-binding protein family of transcription factors. J. Biol. Chem. 1998, 273, 28545–28548. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gery, S.; Tanosaki, S.; Hofmann, W.K.; Koppel, A.; Koeffler, H.P. C/EBPdelta expression in a BCR-ABL-positive cell line induces growth arrest and myeloid differentiation. Oncogene 2005, 24, 1589–1597. [Google Scholar] [CrossRef] [Green Version]
- Stein, T.; Salomonis, N.; Nuyten, D.S.; van de Vijver, M.J.; Gusterson, B.A. A mouse mammary gland involution mRNA signature identifies biological pathways potentially associated with breast cancer metastasis. J. Mammary Gland Biol. Neoplasia 2009, 14, 99–116. [Google Scholar] [CrossRef]
- Ikezoe, T.; Gery, S.; Yin, D.; O’Kelly, J.; Binderup, L.; Lemp, N.; Taguchi, H.; Koeffler, H.P. CCAAT/enhancer-binding protein delta: A molecular target of 1.;25-dihydroxyvitamin D3 in androgen-responsive prostate cancer LNCaP cells. Cancer Res. 2005, 65, 4762–4768. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sivko, G.S.; Sanford, D.C.; Dearth, L.D.; Tang, D.; DeWille, J.W. CCAAT/Enhancer binding protein delta (c/EBPdelta) regulation and expression in human mammary epithelial cells: II. Analysis of activating signal transduction pathways; transcriptional; post-transcriptional; and post-translational control. J. Cell Biochem. 2004, 93, 844–856. [Google Scholar] [CrossRef] [PubMed]
- Hartl, L.; Duitman, J.; Aberson, H.L.; Chen, K.; Dijk, F.; Roelofs, J.J.T.H.; Dings, M.P.G.; Hooijer, G.K.J.; Hernanda, P.Y.; Pan, Q.; et al. CCAAT/Enhancer-Binding Protein Delta (C/EBPδ): A Previously Unrecognized Tumor Suppressor that Limits the Oncogenic Potential of Pancreatic Ductal Adenocarcinoma Cells. Cancers 2020, 12, 2546. [Google Scholar] [CrossRef] [PubMed]
- Duitman, J.; Hartl, L.; Roelofs, J.J.T.H.; Bijlsma, M.F.; Spek, C.A. Non-Tumor CCAAT/Enhancer-Binding Protein Delta Potentiates Tumor Cell Extravasation and Pancreatic Cancer Metastasis Formation. Biomolecules 2021, 11, 1079. [Google Scholar] [CrossRef]
- Chang, L.H.; Huang, H.S.; Wu, P.T.; Jou, I.M.; Pan, M.H.; Chang, W.C.; Wang, D.D.; Wang, J.M. Role of macrophage CCAAT/enhancer binding protein delta in the pathogenesis of rheumatoid arthritis in collagen-induced arthritic mice. PLoS ONE 2012, 7, e45378. [Google Scholar] [CrossRef]
- Banerjee, S.; Xie, N.; Cui, H.; Tan, Z.; Yang, S.; Icyuz, M.; Abraham, E.; Liu, G. MicroRNA let-7c regulates macrophage polarization. J. Immunol. 2013, 190, 6542–6549. [Google Scholar] [CrossRef]
- Jones, V.S.; Huang, R.Y.; Chen, L.P.; Chen, Z.S.; Fu, L.; Huang, R.P. Cytokines in cancer drug resistance: Cues to new therapeutic strategies. Biochim. Biophys. Acta 2016, 1865, 255–265. [Google Scholar] [CrossRef] [Green Version]
- Spek, C.A.; Aberson, H.L.; Butler, J.M.; de Vos, A.F.; Duitman, J. CEBPD Potentiates the Macrophage Inflammatory Response but CEBPD Knock-Out Macrophages Fail to Identify CEBPD-Dependent Pro-Inflammatory Transcriptional Programs. Cells 2021, 10, 2233. [Google Scholar] [CrossRef]
- Slapak, E.J.; Kong, L.; El Mandili, M.; Nieuwland, R.; Kros, A.; Bijlsma, M.F.; Spek, C.A. ADAM9-Responsive Mesoporous Silica Nanoparticles for Targeted Drug Delivery in Pancreatic Cancer. Cancers 2021, 13, 3321. [Google Scholar] [CrossRef] [PubMed]
- Halbrook, C.J.; Pontious, C.; Kovalenko, I.; Lapienyte, L.; Dreyer, S.; Lee, H.J.; Thurston, G.; Zhang, Y.; Lazarus, J.; Sajjakulnukit, P.; et al. Macrophage-Released Pyrimidines Inhibit Gemcitabine Therapy in Pancreatic Cancer. Cell Metab. 2019, 29, 1390–1399.e6. [Google Scholar] [CrossRef]
- Shukla, S.K.; Purohit, V.; Mehla, K.; Gunda, V.; Chaika, N.V.; Vernucci, E.; King, R.J.; Abrego, J.; Goode, G.D.; Dasgupta, A.; et al. MUC1 and HIF-1alpha Signaling Crosstalk Induces Anabolic Glucose Metabolism to Impart Gemcitabine Resistance to Pancreatic Cancer. Cancer Cell 2017, 32, 71–87.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Amrutkar, M.; Gladhaug, I.P. Pancreatic Cancer Chemoresistance to Gemcitabine. Cancers 2017, 9, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burris, H.A., 3rd; Moore, M.J.; Andersen, J.; Green, M.R.; Rothenberg, M.L.; Modiano, M.R.; Cripps, M.C.; Portenoy, R.K.; Storniolo, A.M.; Tarassoff, P.; et al. Improvements in survival and clinical benefit with gemcitabine as first-line therapy for patients with advanced pancreas cancer: A randomized trial. J. Clin. Oncol. 1997, 15, 2403–2413. [Google Scholar]
- Cunningham, D.; Chau, I.; Stocken, D.D.; Valle, J.W.; Smith, D.; Steward, W.; Harper, P.G.; Dunn, J.; Tudur-Smith, C.; West, J.; et al. Phase III randomized comparison of gemcitabine versus gemcitabine plus capecitabine in patients with advanced pancreatic cancer. J. Clin. Oncol. 2009, 27, 5513–5518. [Google Scholar] [CrossRef]
- Heinemann, V.; Quietzsch, D.; Gieseler, F.; Gonnermann, M.; Schönekäs, H.; Rost, A.; Neuhaus, H.; Haag, C.; Clemens, M.; Heinrich, B.; et al. Randomized phase III trial of gemcitabine plus cisplatin compared with gemcitabine alone in advanced pancreatic cancer. J. Clin. Oncol. 2006, 24, 3946–3952. [Google Scholar] [CrossRef]
- Louvet, C.; Labianca, R.; Hammel, P.; Lledo, G.; Zampino, M.G.; André, T.; Zaniboni, A.; Ducreux, M.; Aitini, E.; Taïeb, J.; et al. Gemcitabine in combination with oxaliplatin compared with gemcitabine alone in locally advanced or metastatic pancreatic cancer: Results of a GERCOR and GISCAD phase III trial. J. Clin. Oncol. 2005, 23, 3509–3516. [Google Scholar] [CrossRef]
- Conroy, T.; Desseigne, F.; Ychou, M.; Bouché, O.; Guimbaud, R.; Bécouarn, Y.; Adenis, A.; Raoul, J.L.; Gourgou-Bourgade, S.; de la Fouchardière, C.; et al. FOLFIRINOX versus gemcitabine for metastatic pancreatic cancer. N. Engl. J. Med. 2011, 364, 1817–1825. [Google Scholar] [CrossRef] [Green Version]
- de Jesus, V.H.F.; Camandaroba, M.P.G.; Donadio, M.D.S.; Cabral, A.; Muniz, T.P.; de Moura Leite, L.; Sant’Ana, L.F. Retrospective comparison of the efficacy and the toxicity of standard and modified FOLFIRINOX regimens in patients with metastatic pancreatic adenocarcinoma. J. Gastrointest. Oncol. 2018, 9, 694–707. [Google Scholar] [CrossRef]
- Noy, R.; Pollard, J.W. Tumor-associated macrophages: From mechanisms to therapy. Immunity 2014, 41, 49–61. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Clappaert, E.J.; Murgaski, A.; Van Damme, H.; Kiss, M.; Laoui, D. Diamonds in the Rough: Harnessing Tumor-Associated Myeloid Cells for Cancer Therapy. Front. Immunol. 2018, 9, 2250. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ullmann, T.; Luckhardt, S.; Wolf, M.; Parnham, M.J.; Resch, E. High-Throughput Screening for CEBPD-Modulating Compounds in THP-1-Derived Reporter Macrophages Identifies Anti-Inflammatory HDAC and BET Inhibitors. Int. J. Mol. Sci. 2021, 22, 3022. [Google Scholar] [CrossRef] [PubMed]
- Chi, J.Y.; Hsiao, Y.W.; Li, C.F.; Lo, Y.C.; Lin, Z.Y.; Hong, J.Y.; Liu, Y.M.; Han, X.; Wang, S.M.; Chen, B.K.; et al. Targeting chemotherapy-induced PTX3 in tumor stroma to prevent the progression of drug-resistant cancers. Oncotarget 2015, 6, 23987–24001. [Google Scholar] [CrossRef] [Green Version]
- Zeng, S.; Pöttler, M.; Lan, B.; Grützmann, R.; Pilarsky, C.; Yang, H. Chemoresistance in Pancreatic Cancer. Int. J. Mol. Sci. 2019, 20, 4504. [Google Scholar] [CrossRef] [Green Version]
- Schniewind, B.; Christgen, M.; Kurdow, R.; Haye, S.; Kremer, B.; Kalthoff, H.; Ungefroren, H. Resistance of pancreatic cancer to gemcitabine treatment is dependent on mitochondria-mediated apoptosis. Int. J. Cancer 2004, 109, 182–188. [Google Scholar] [CrossRef]
- Fu, Y.; Ricciardiello, F.; Yang, G.; Qiu, J.; Huang, H.; Xiao, J.; Cao, Z.; Zhao, F.; Liu, Y.; Luo, W.; et al. The Role of Mitochondria in the Chemoresistance of Pancreatic Cancer Cells. Cells 2021, 10, 497. [Google Scholar] [CrossRef]
- Rauchwerger, D.R.; Firby, P.S.; Hedley, D.W.; Moore, M.J. Equilibrative-sensitive nucleoside transporter and its role in gemcitabine sensitivity. Cancer Res. 2000, 60, 6075–6079. [Google Scholar]
- Yang, S.; Liu, Q.; Liao, Q. Tumor-Associated Macrophages in Pancreatic Ductal Adenocarcinoma: Origin, Polarization, Function, and Reprogramming. Front. Cell Dev. Biol. 2021, 8, 607209. [Google Scholar] [CrossRef]
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).