
Vascular Calcification: In Vitro Models under the Magnifying Glass

 ,
,
Abstract
:1. Introduction
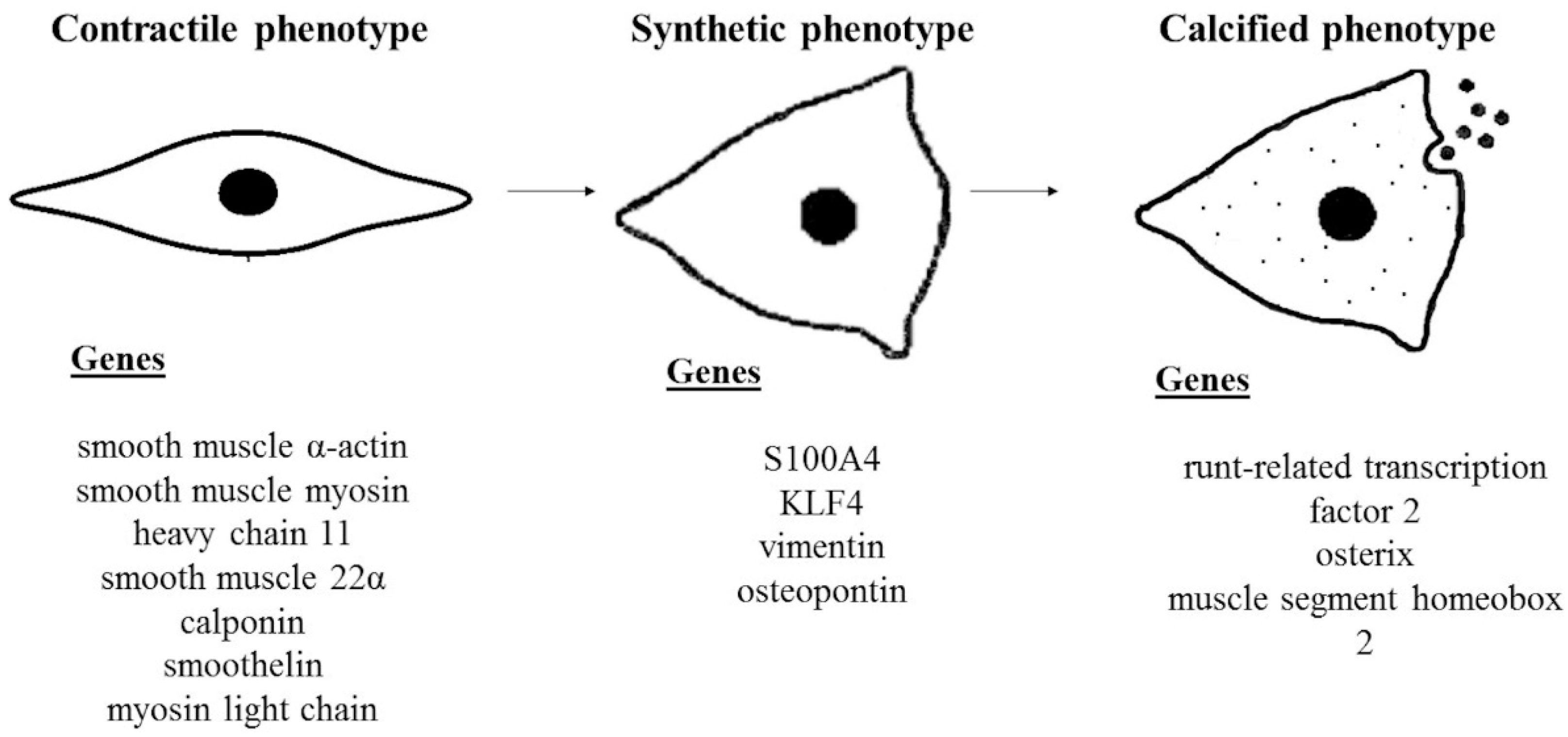
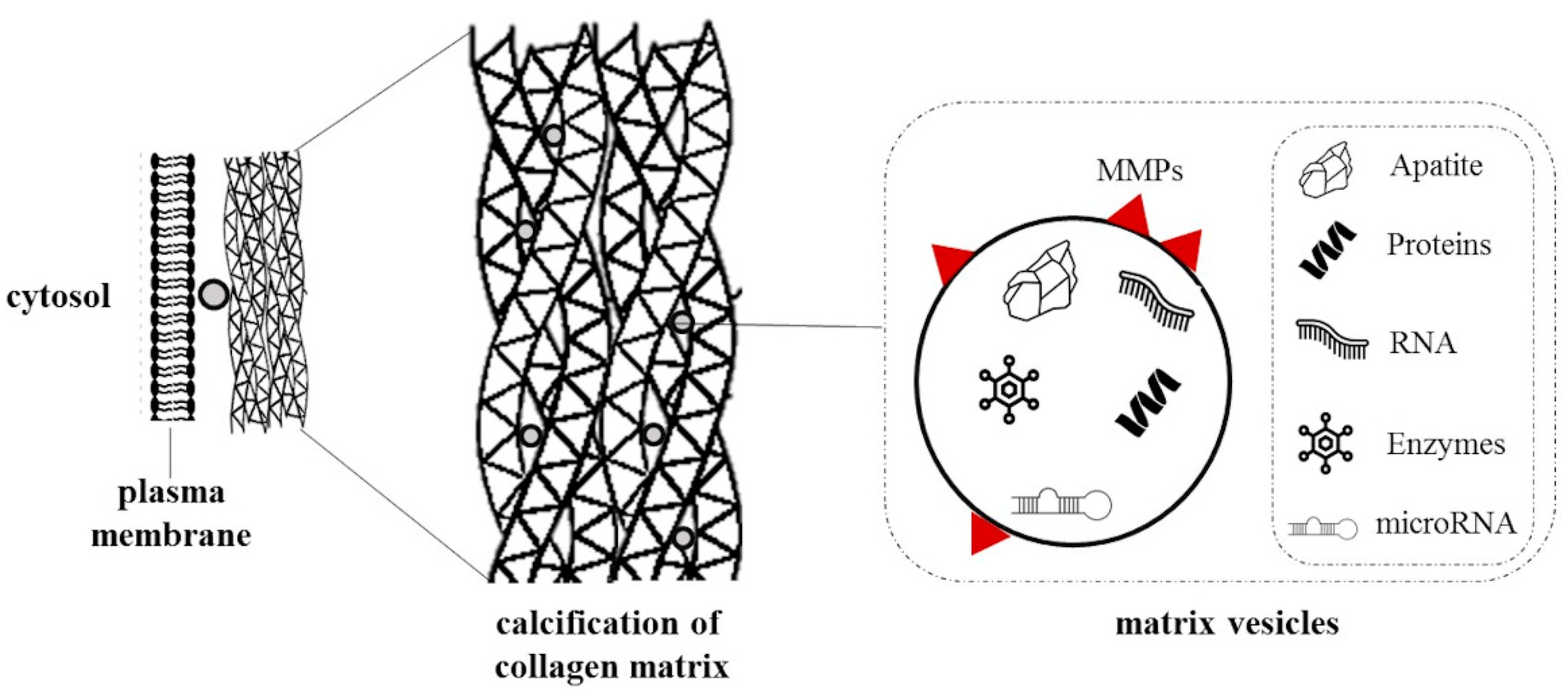
2. Relevant Mechanisms in Vascular Calcification
3. How to Investigate Vascular Calcification? From Old to Modern In Vitro Models
3.1. Monolayer Cell Cultures
3.2. ECs and VSMCs Co-Cultures
3.3. MSCs and VSMCs Co-Cultures
3.4. Extracellular Vesicles and Particles and VSMC Cultures
4. Anticalcifying Agents
4.1. Natural Compounds
4.2. Synthetic Compounds
5. Conclusions
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
References
- Xu, F.; Zhong, J.; Lin, X.; Shan, S.; Guo, B.; Zheng, M.; Wang, Y.; Li, F.; Cui, R.; Wu, F.; et al. Melatonin Alleviates Vascular Calcification and Ageing through Exosomal MiR-204/MiR-211 Cluster in a Paracrine Manner. J. Pineal Res. 2020, 68, e12631. [Google Scholar] [CrossRef] [Green Version]
- Leoncini, G.; Ratto, E.; Viazzi, F.; Vaccaro, V.; Parodi, A.; Falqui, V.; Conti, N.; Tomolillo, C.; Deferrari, G.; Pontremoli, R. Increased Ambulatory Arterial Stiffness Index Is Associated With Target Organ Damage in Primary Hypertension. Hypertension 2006, 48, 397–403. [Google Scholar] [CrossRef] [PubMed]
- Furmanik, M.; Chatrou, M.; van Gorp, R.; Akbulut, A.; Willems, B.; Schmidt, H.; van Eys, G.; Bochaton-Piallat, M.-L.; Proudfoot, D.; Biessen, E.; et al. Reactive Oxygen-Forming Nox5 Links Vascular Smooth Muscle Cell Phenotypic Switching and Extracellular Vesicle-Mediated Vascular Calcification. Circ. Res. 2020, 127, 911–927. [Google Scholar] [CrossRef] [PubMed]
- Qi, Y.; Dai, F.; Gu, J.; Yao, W. Biomarkers in VSMC Phenotypic Modulation and Vascular Remodeling. Pharmazie 2019, 74, 711–714. [Google Scholar] [CrossRef] [PubMed]
- Voelkl, J.; Luong, T.T.D.; Tuffaha, R.; Musculus, K.; Auer, T.; Lian, X.; Daniel, C.; Zickler, D.; Boehme, B.; Sacherer, M.; et al. SGK1 Induces Vascular Smooth Muscle Cell Calcification through NF-ΚB Signaling. J. Clin. Investig. 2018, 128, 3024–3040. [Google Scholar] [CrossRef]
- Albiero, M.; Avogaro, A.; Fadini, G.P. Circulating Cellular Players in Vascular Calcification. Curr. Pharm. Des. 2014, 20, 5889–5896. [Google Scholar] [CrossRef] [PubMed]
- Cianciolo, G.; Capelli, I.; Cappuccilli, M.; Schillaci, R.; Cozzolino, M.; La Manna, G. Calcifying Circulating Cells: An Uncharted Area in the Setting of Vascular Calcification in CKD Patients. Clin. Kidney J. 2016, 9, 280–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dube, P.; DeRiso, A.; Patel, M.; Battepati, D.; Khatib-Shahidi, B.; Sharma, H.; Gupta, R.; Malhotra, D.; Dworkin, L.; Haller, S.; et al. Vascular Calcification in Chronic Kidney Disease: Diversity in the Vessel Wall. Biomedicines 2021, 9, 404. [Google Scholar] [CrossRef]
- Boström, K.I.; Yao, J.; Guihard, P.J.; Blazquez-Medela, A.M.; Yao, Y. Endothelial-Mesenchymal Transition in Atherosclerotic Lesion Calcification. Atherosclerosis 2016, 253, 124–127. [Google Scholar] [CrossRef] [Green Version]
- Libby, P.; Ridker, P.M.; Hansson, G.K. Progress and Challenges in Translating the Biology of Atherosclerosis. Nature 2011, 473, 317–325. [Google Scholar] [CrossRef]
- Rogers, M.A.; Aikawa, E. Cardiovascular Calcification: Artificial Intelligence and Big Data Accelerate Mechanistic Discovery. Nat. Rev. Cardiol. 2019, 16, 261–274. [Google Scholar] [CrossRef] [PubMed]
- Yang, W.; Zou, B.; Hou, Y.; Yan, W.; Chen, T.; Qu, S. Extracellular Vesicles in Vascular Calcification. Clin. Chim. Acta 2019, 499, 118–122. [Google Scholar] [CrossRef] [PubMed]
- Alam, M.; Kirton, J.P.; Wilkinson, F.L.; Towers, E.; Sinha, S.; Rouhi, M.; Vizard, T.N.; Sage, A.P.; Martin, D.; Ward, D.T.; et al. Calcification Is Associated with Loss of Functional Calcium-Sensing Receptor in Vascular Smooth Muscle Cells. Cardiovasc. Res. 2009, 81, 260–268. [Google Scholar] [CrossRef] [Green Version]
- Nguyen, N.T.; Nguyen, T.T.; Da Ly, D.; Xia, J.-B.; Qi, X.-F.; Lee, I.-K.; Cha, S.-K.; Park, K.-S. Oxidative Stress by Ca2+ Overload Is Critical for Phosphate-Induced Vascular Calcification. Am. J. Physiol.-Heart Circ. Physiol. 2020, 319, H1302–H1312. [Google Scholar] [CrossRef] [PubMed]
- Sutra, T.; Morena, M.; Bargnoux, A.-S.; Caporiccio, B.; Canaud, B.; Cristol, J.-P. Superoxide Production: A Procalcifying Cell Signalling Event in Osteoblastic Differentiation of Vascular Smooth Muscle Cells Exposed to Calcification Media. Free Radic. Res. 2008, 42, 789–797. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.-M.; Xu, M.-J.; Cai, Y.; Zhao, G.; Guan, Y.; Kong, W.; Tang, C.; Wang, X. Mitochondrial Reactive Oxygen Species Promote P65 Nuclear Translocation Mediating High-Phosphate-Induced Vascular Calcification in Vitro and in Vivo. Kidney Int. 2011, 79, 1071–1079. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Navarro-González, J.F.; Mora-Fernández, C.; Muros, M.; Herrera, H.; García, J. Mineral Metabolism and Inflammation in Chronic Kidney Disease Patients: A Cross-Sectional Study. Clin. J. Am. Soc. Nephrol. 2009, 4, 1646–1654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shao, J.-S.; Cheng, S.-L.; Sadhu, J.; Towler, D.A. Inflammation and the Osteogenic Regulation of Vascular Calcification: A Review and Perspective. Hypertension 2010, 55, 579–592. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jin, C.; Wang, H.; Chen, Y.; Tang, M.; Fan, G.; Wang, Z.; Li, L.; Zhang, Y.; Zhang, W.; Zhong, M. Gas6 Delays Senescence in Vascular Smooth Muscle Cells through the PI3K/Akt/FoxO Signaling Pathway. Cell. Physiol. Biochem. 2015, 35, 1151–1166. [Google Scholar] [CrossRef] [PubMed]
- Freise, C.; Querfeld, U.; Ludwig, A.; Hamm, B.; Schnorr, J.; Taupitz, M. Uraemic Extracellular Vesicles Augment Osteogenic Transdifferentiation of Vascular Smooth Muscle Cells via Enhanced AKT Signalling and PiT-1 Expression. J. Cell. Mol. Med. 2021, 25, 5602–5614. [Google Scholar] [CrossRef]
- Phadwal, K.; Feng, D.; Zhu, D.; MacRae, V.E. Autophagy as a Novel Therapeutic Target in Vascular Calcification. Pharmacol. Ther. 2020, 206, 107430. [Google Scholar] [CrossRef] [PubMed]
- Dai, X.-Y.; Zhao, M.-M.; Cai, Y.; Guan, Q.-C.; Zhao, Y.; Guan, Y.; Kong, W.; Zhu, W.-G.; Xu, M.-J.; Wang, X. Phosphate-Induced Autophagy Counteracts Vascular Calcification by Reducing Matrix Vesicle Release. Kidney Int. 2013, 83, 1042–1051. [Google Scholar] [CrossRef] [Green Version]
- Hegner, B.; Lange, M.; Kusch, A.; Essin, K.; Sezer, O.; Schulze-Lohoff, E.; Luft, F.C.; Gollasch, M.; Dragun, D. MTOR Regulates Vascular Smooth Muscle Cell Differentiation From Human Bone Marrow–Derived Mesenchymal Progenitors. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 232–238. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martin, K.A.; Merenick, B.L.; Ding, M.; Fetalvero, K.M.; Rzucidlo, E.M.; Kozul, C.D.; Brown, D.J.; Chiu, H.Y.; Shyu, M.; Drapeau, B.L.; et al. Rapamycin Promotes Vascular Smooth Muscle Cell Differentiation through Insulin Receptor Substrate-1/Phosphatidylinositol 3-Kinase/Akt2 Feedback Signaling. J. Biol. Chem. 2007, 282, 36112–36120. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poon, M.; Marx, S.O.; Gallo, R.; Badimon, J.J.; Taubman, M.B.; Marks, A.R. Rapamycin Inhibits Vascular Smooth Muscle Cell Migration. J. Clin. Investig. 1996, 98, 2277–2283. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sun, J.; Marx, S.O.; Chen, H.J.; Poon, M.; Marks, A.R.; Rabbani, L.E. Role for P27(Kip1) in Vascular Smooth Muscle Cell Migration. Circulation 2001, 103, 2967–2972. [Google Scholar] [CrossRef] [Green Version]
- Feng, T.; Liu, P.; Wang, X.; Luo, J.; Zuo, X.; Jiang, X.; Liu, C.; Li, Y.; Li, N.; Chen, M.; et al. SIRT1 Activator E1231 Protects from Experimental Atherosclerosis and Lowers Plasma Cholesterol and Triglycerides by Enhancing ABCA1 Expression. Atherosclerosis 2018, 274, 172–181. [Google Scholar] [CrossRef]
- Badi, I.; Mancinelli, L.; Polizzotto, A.; Ferri, D.; Zeni, F.; Burba, I.; Milano, G.; Brambilla, F.; Saccu, C.; Bianchi, M.E.; et al. MiR-34a Promotes Vascular Smooth Muscle Cell Calcification by Downregulating SIRT1 (Sirtuin 1) and Axl (AXL Receptor Tyrosine Kinase). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 2079–2090. [Google Scholar] [CrossRef]
- Kida, Y.; Goligorsky, M.S. Sirtuins, Cell Senescence, and Vascular Aging. Can. J. Cardiol. 2016, 32, 634–641. [Google Scholar] [CrossRef] [Green Version]
- Akiyoshi, T.; Ota, H.; Iijima, K.; Son, B.-K.; Kahyo, T.; Setou, M.; Ogawa, S.; Ouchi, Y.; Akishita, M. A Novel Organ Culture Model of Aorta for Vascular Calcification. Atherosclerosis 2016, 244, 51–58. [Google Scholar] [CrossRef] [PubMed]
- Kanemaru, K.; Seya, K.; Miki, I.; Motomura, S.; Furukawa, K.-I. Calcification of Aortic Smooth Muscle Cells Isolated from Spontaneously Hypertensive Rats. J. Pharmacol. Sci. 2008, 106, 280–286. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shioi, A.; Nishizawa, Y.; Jono, S.; Koyama, H.; Hosoi, M.; Morii, H. Beta-Glycerophosphate Accelerates Calcification in Cultured Bovine Vascular Smooth Muscle Cells. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 2003–2009. [Google Scholar] [CrossRef]
- Schuchardt, M.; Tölle, M.; Prüfer, J.; Prüfer, N.; Huang, T.; Jankowski, V.; Jankowski, J.; Zidek, W.; van der Giet, M. Uridine Adenosine Tetraphosphate Activation of the Purinergic Receptor P2Y Enhances in Vitro Vascular Calcification. Kidney Int. 2012, 81, 256–265. [Google Scholar] [CrossRef] [Green Version]
- Prüfer, J.; Schuchardt, M.; Tölle, M.; Prüfer, N.; Höhne, M.; Zidek, W.; van der Giet, M. Harmful Effects of the Azathioprine Metabolite 6-Mercaptopurine in Vascular Cells: Induction of Mineralization. PLoS ONE 2014, 9, e101709. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.X.; Duan, D.; O’Neill, K.D.; Moe, S.M. High Glucose Increases the Expression of Cbfa1 and BMP-2 and Enhances the Calcification of Vascular Smooth Muscle Cells. Nephrol. Dial. Transplant. 2006, 21, 3435–3442. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beazley, K.E.; Deasey, S.; Lima, F.; Nurminskaya, M.V. Transglutaminase 2–Mediated Activation of β-Catenin Signaling Has a Critical Role in Warfarin-Induced Vascular Calcification. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 123–130. [Google Scholar] [CrossRef] [Green Version]
- Trion, A.; Schutte-Bart, C.; Bax, W.H.; Jukema, J.W.; van der Laarse, A. Modulation of Calcification of Vascular Smooth Muscle Cells in Culture by Calcium Antagonists, Statins, and Their Combination. Mol. Cell. Biochem. 2008, 308, 25–33. [Google Scholar] [CrossRef] [Green Version]
- Li, N.; Cheng, W.; Huang, T.; Yuan, J.; Wang, X.; Song, M. Vascular Adventitia Calcification and Its Underlying Mechanism. PLoS ONE 2015, 10, e0132506. [Google Scholar] [CrossRef] [PubMed]
- Frauscher, B.; Kirsch, A.H.; Schabhüttl, C.; Schweighofer, K.; Kétszeri, M.; Pollheimer, M.; Dragun, D.; Schröder, K.; Rosenkranz, A.R.; Eller, K.; et al. Autophagy Protects From Uremic Vascular Media Calcification. Front. Immunol. 2018, 9, 1866. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gayrard, N.; Muyor, K.; Notarnicola, C.; Duranton, F.; Jover, B.; Argilés, À. Optimisation of Cell and Ex Vivo Culture Conditions to Study Vascular Calcification. PLoS ONE 2020, 15, e0230201. [Google Scholar] [CrossRef] [PubMed]
- Holmar, J.; Noels, H.; Böhm, M.; Bhargava, S.; Jankowski, J.; Orth-Alampour, S. Development, Establishment and Validation of in Vitro and Ex Vivo Assays of Vascular Calcification. Biochem. Biophys. Res. Commun. 2020, 530, 462–470. [Google Scholar] [CrossRef]
- Méndez-Barbero, N.; Gutiérrez-Muñoz, C.; Blanco-Colio, L.M. Cellular Crosstalk between Endothelial and Smooth Muscle Cells in Vascular Wall Remodeling. Int. J. Mol. Sci. 2021, 22, 7284. [Google Scholar] [CrossRef] [PubMed]
- Powell, R.J.; Bhargava, J.; Basson, M.D.; Sumpio, B.E. Coculture Conditions Alter Endothelial Modulation of TGF-Β1 Activation and Smooth Muscle Growth Morphology. Am. J. Physiol.-Heart Circ. Physiol. 1998, 274, H642–H649. [Google Scholar] [CrossRef] [PubMed]
- Meng, F.; Zhao, Y.; Wang, B.; Li, B.; Sheng, Y.; Liu, M.; Li, H.; Xiu, R. Endothelial Cells Promote Calcification in Aortic Smooth Muscle Cells from Spontaneously Hypertensive Rats. Cell. Physiol. Biochem. 2018, 49, 2371–2381. [Google Scholar] [CrossRef] [PubMed]
- Evensen, L.; Micklem, D.R.; Blois, A.; Berge, S.V.; Aarsæther, N.; Littlewood-Evans, A.; Wood, J.; Lorens, J.B. Mural Cell Associated VEGF Is Required for Organotypic Vessel Formation. PLoS ONE 2009, 4, e5798. [Google Scholar] [CrossRef]
- Wang, Y.-H.; Yan, Z.-Q.; Shen, B.-R.; Zhang, L.; Zhang, P.; Jiang, Z.-L. Vascular Smooth Muscle Cells Promote Endothelial Cell Adhesion via Microtubule Dynamics and Activation of Paxillin and the Extracellular Signal-Regulated Kinase (ERK) Pathway in a Co-Culture System. Eur. J. Cell Biol. 2009, 88, 701–709. [Google Scholar] [CrossRef]
- Heydarkhan-Hagvall, S.; Helenius, G.; Johansson, B.R.; Li, J.Y.; Mattsson, E.; Risberg, B. Co-Culture of Endothelial Cells and Smooth Muscle Cells Affects Gene Expression of Angiogenic Factors. J. Cell. Biochem. 2003, 89, 1250–1259. [Google Scholar] [CrossRef]
- Bouabdallah, J.; Zibara, K.; Issa, H.; Lenglet, G.; Kchour, G.; Caus, T.; Six, I.; Choukroun, G.; Kamel, S.; Bennis, Y. Endothelial Cells Exposed to Phosphate and Indoxyl Sulphate Promote Vascular Calcification through Interleukin-8 Secretion. Nephrol. Dial. Transplant. 2019, 34, 1125–1134. [Google Scholar] [CrossRef]
- Fadini, G.P.; Rattazzi, M.; Matsumoto, T.; Asahara, T.; Khosla, S. Emerging Role of Circulating Calcifying Cells in the Bone-Vascular Axis. Circulation 2012, 125, 2772–2781. [Google Scholar] [CrossRef] [Green Version]
- Zhu, M.; Fang, X.; Zhou, S.; Li, W.; Guan, S. Indirect Co-Culture of Vascular Smooth Muscle Cells with Bone Marrow Mesenchymal Stem Cells Inhibits Vascular Calcification and Downregulates the Wnt Signaling Pathways. Mol. Med. Rep. 2016, 13, 5141–5148. [Google Scholar] [CrossRef] [PubMed]
- Proudfoot, D.; Skepper, J.N.; Hegyi, L.; Bennett, M.R.; Shanahan, C.M.; Weissberg, P.L. Apoptosis Regulates Human Vascular Calcification In Vitro: Evidence for Initiation of Vascular Calcification by Apoptotic Bodies. Circ. Res. 2000, 87, 1055–1062. [Google Scholar] [CrossRef] [Green Version]
- Pan, W.; Liang, J.; Tang, H.; Fang, X.; Wang, F.; Ding, Y.; Huang, H.; Zhang, H. Differentially expressed microRNA profiles in exosomes from vascular smooth muscle cells associated with coronary artery calcification. Int. J. Biochem. Cell Biol. 2020, 118, 105645. [Google Scholar] [CrossRef] [PubMed]
- Li, S.; Zhan, J.K.; Wang, Y.J.; Lin, X.; Zhong, J.Y.; Wang, Y.; Tan, P.; He, J.Y.; Cui, X.J.; Chen, Y.Y.; et al. Exosomes from hyperglycemia-stimulated vascular endothelial cells contain versican that regulate calcification/senescence in vascular smooth muscle cells. Cell Biosci. 2019, 9, 1. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kapustin, A.N.; Davies, J.D.; Reynolds, J.L.; McNair, R.; Jones, G.T.; Sidibe, A.; Schurgers, L.J.; Skepper, J.N.; Proudfoot, D.; Mayr, M.; et al. Calcium Regulates Key Components of Vascular Smooth Muscle Cell–Derived Matrix Vesicles to Enhance Mineralization. Circ. Res. 2011, 109, e1–e12. [Google Scholar] [CrossRef] [Green Version]
- Kapustin, A.N.; Chatrou, M.L.L.; Drozdov, I.; Zheng, Y.; Davidson, S.M.; Soong, D.; Furmanik, M.; Sanchis, P.; De Rosales, R.T.M.; Alvarez-Hernandez, D.; et al. Vascular smooth muscle cell calcification is mediated by regulated exosome secretion. Circ. Res. 2015, 116, 1312–1323. [Google Scholar] [CrossRef] [Green Version]
- Kapustin, A.N.; Shanahan, C.M. Emerging roles for vascular smooth muscle cell exosomes in calcification and coagulation. J. Physiol. 2016, 594, 2905–2914. [Google Scholar] [CrossRef]
- Cavallari, C.; Dellepiane, S.; Fonsato, V.; Medica, D.; Marengo, M.; Migliori, M.; Quercia, A.D.; Pitino, A.; Formica, M.; Panichi, V.; et al. Online Hemodiafiltration Inhibits Inflammation-Related Endothelial Dysfunction and Vascular Calcification of Uremic Patients Modulating MiR-223 Expression in Plasma Extracellular Vesicles. J. Immunol. 2019, 202, 2372–2383. [Google Scholar] [CrossRef] [Green Version]
- Alique, M.; Bodega, G.; Corchete, E.; García-Menéndez, E.; de Sequera, P.; Luque, R.; Rodríguez-Padrón, D.; Marqués, M.; Portolés, J.; Carracedo, J.; et al. Microvesicles from Indoxyl Sulfate-Treated Endothelial Cells Induce Vascular Calcification in Vitro. Comput. Struct. Biotechnol. J. 2020, 18, 953–966. [Google Scholar] [CrossRef] [PubMed]
- Leopold, J.A. Vascular Calcification: Mechanisms of Vascular Smooth Muscle Cell Calcification. Trends Cardiovasc. Med. 2015, 25, 267–274. [Google Scholar] [CrossRef] [Green Version]
- Schmidt, J.R.; Kliemt, S.; Preissler, C.; Moeller, S.; von Bergen, M.; Hempel, U.; Kalkhof, S. Osteoblast-Released Matrix Vesicles, Regulation of Activity and Composition by Sulfated and Non-Sulfated Glycosaminoglycans. Mol. Cell. Proteom. 2016, 15, 558–572. [Google Scholar] [CrossRef]
- Shapiro, I.M.; Landis, W.J.; Risbud, M.V. Matrix Vesicles: Are They Anchored Exosomes? Bone 2015, 79, 29–36. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, N.X.; O’Neill, K.D.; Moe, S.M. Matrix Vesicles Induce Calcification of Recipient Vascular Smooth Muscle Cells through Multiple Signaling Pathways. Kidney Int. 2018, 93, 343–354. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heiss, A.; DuChesne, A.; Denecke, B.; Grötzinger, J.; Yamamoto, K.; Renné, T.; Jahnen-Dechent, W. Structural Basis of Calcification Inhibition by A2-HS Glycoprotein/Fetuin-A. J. Biol. Chem. 2003, 278, 13333–13341. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Viegas, C.S.B.; Santos, L.; Macedo, A.L.; Matos, A.A.; Silva, A.P.; Neves, P.L.; Staes, A.; Gevaert, K.; Morais, R.; Vermeer, C.; et al. Chronic Kidney Disease Circulating Calciprotein Particles and Extracellular Vesicles Promote Vascular Calcification: A Role for GRP (Gla-Rich Protein). Arterioscler. Thromb. Vasc. Biol. 2018, 38, 575–587. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Smith, E.R.; Cai, M.M.; McMahon, L.P.; Pedagogos, E.; Toussaint, N.D.; Brumby, C.; Holt, S.G. Serum Fetuin-A Concentration and Fetuin-A-Containing Calciprotein Particles in Patients with Chronic Inflammatory Disease and Renal Failure: Serum Calciprotein Particles and Inflammation. Nephrology 2013, 18, 215–221. [Google Scholar] [CrossRef]
- Smith, E.R.; Ford, M.L.; Tomlinson, L.A.; Rajkumar, C.; McMahon, L.P.; Holt, S.G. Phosphorylated Fetuin-A-Containing Calciprotein Particles Are Associated with Aortic Stiffness and a Procalcific Milieu in Patients with Pre-Dialysis CKD. Nephrol. Dial. Transplant. 2012, 27, 1957–1966. [Google Scholar] [CrossRef] [Green Version]
- Aghagolzadeh, P.; Bachtler, M.; Bijarnia, R.; Jackson, C.; Smith, E.R.; Odermatt, A.; Radpour, R.; Pasch, A. Calcification of Vascular Smooth Muscle Cells Is Induced by Secondary Calciprotein Particles and Enhanced by Tumor Necrosis Factor-α. Atherosclerosis 2016, 251, 404–414. [Google Scholar] [CrossRef] [Green Version]
- Cai, M.M.X.; Smith, E.R.; Tan, S.-J.; Hewitson, T.D.; Holt, S.G. The Role of Secondary Calciprotein Particles in the Mineralisation Paradox of Chronic Kidney Disease. Calcif. Tissue Int. 2017, 101, 570–580. [Google Scholar] [CrossRef]
- ter Braake, A.D.; Eelderink, C.; Zeper, L.W.; Pasch, A.; Bakker, S.J.L.; de Borst, M.H.; Hoenderop, J.G.J.; de Baaij, J.H.F. Calciprotein Particle Inhibition Explains Magnesium-Mediated Protection against Vascular Calcification. Nephrol. Dial. Transplant. 2020, 35, 765–773. [Google Scholar] [CrossRef]
- Herrera, E.; Barbas, C. Vitamin E: Action, Metabolism and Perspectives. J. Physiol. Biochem. 2001, 57, 43–56. [Google Scholar] [CrossRef]
- You, H.; Yang, H.; Zhu, Q.; Li, M.; Xue, J.; Gu, Y.; Lin, S.; Ding, F. Advanced Oxidation Protein Products Induce Vascular Calcification by Promoting Osteoblastic Trans-Differentiation of Smooth Muscle Cells via Oxidative Stress and ERK Pathway. Ren. Fail. 2009, 31, 313–319. [Google Scholar] [CrossRef] [PubMed]
- Qiu, C.; Zheng, H.; Tao, H.; Yu, W.; Jiang, X.; Li, A.; Jin, H.; Lv, A.; Li, H. Vitamin K2 Inhibits Rat Vascular Smooth Muscle Cell Calcification by Restoring the Gas6/Axl/Akt Anti-Apoptotic Pathway. Mol. Cell. Biochem. 2017, 433, 149–159. [Google Scholar] [CrossRef]
- Andres, S.; Pevny, S.; Ziegenhagen, R.; Bakhiya, N.; Schäfer, B.; Hirsch-Ernst, K.I.; Lampen, A. Safety Aspects of the Use of Quercetin as a Dietary Supplement. Mol. Nutr. Food Res. 2018, 62, 1700447. [Google Scholar] [CrossRef] [PubMed]
- Beazley, K.E.; Eghtesad, S.; Nurminskaya, M.V. Quercetin Attenuates Warfarin-Induced Vascular Calcification in Vitro Independently from Matrix Gla Protein. J. Biol. Chem. 2013, 288, 2632–2640. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cui, L.; Li, Z.; Chang, X.; Cong, G.; Hao, L. Quercetin Attenuates Vascular Calcification by Inhibiting Oxidative Stress and Mitochondrial Fission. Vasc. Pharmacol. 2017, 88, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Liang, Q.; Chen, Y.; Li, C.; Lu, L. Quercetin attenuates Ox-LDL-induced calcification in vascular smooth muscle cells by regulating ROS-TLR4 signaling pathway. Nan Fang Yi Ke Da Xue Xue Bao 2018, 38, 980–985. [Google Scholar] [CrossRef] [PubMed]
- Son, D.O.; Satsu, H.; Kiso, Y.; Totsuka, M.; Shimizu, M. Inhibitory Effect of Carnosine on Interleukin-8 Production in Intestinal Epithelial Cells through Translational Regulation. Cytokine 2008, 42, 265–276. [Google Scholar] [CrossRef]
- Gallant, S.; Semyonova, M.; Yuneva, M. Carnosine as a Potential Anti-Senescence Drug. Biochem. Mosc. 2000, 65, 866–868. [Google Scholar]
- Huang, Y.; Wang, J.; Luo, M.; Yan, D.; Zhang, C. Carnosine Attenuates Vascular Smooth Muscle Cells Calcification through MTOR Signaling Pathway. Aging Med. 2020, 3, 153–158. [Google Scholar] [CrossRef]
- Zhang, Z.; Miao, L.; Wu, X.; Liu, G.; Peng, Y.; Xin, X.; Jiao, B.; Kong, X. Carnosine Inhibits the Proliferation of Human Gastric Carcinoma Cells by Retarding Akt/MTOR/P70S6K Signaling. J. Cancer 2014, 5, 382–389. [Google Scholar] [CrossRef] [Green Version]
- Ter Braake, A.D.; Tinnemans, P.T.; Shanahan, C.M.; Hoenderop, J.G.J.; de Baaij, J.H.F. Magnesium Prevents Vascular Calcification in Vitro by Inhibition of Hydroxyapatite Crystal Formation. Sci. Rep. 2018, 8, 2069. [Google Scholar] [CrossRef] [PubMed]
- Roh, J.; Chang, C.L.; Bhalla, A.; Klein, C.; Hsu, S.Y.T. Intermedin Is a Calcitonin/Calcitonin Gene-Related Peptide Family Peptide Acting through the Calcitonin Receptor-like Receptor/Receptor Activity-Modifying Protein Receptor Complexes. J. Biol. Chem. 2004, 279, 7264–7274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ni, X.; Zhang, J.; Tang, C.; Qi, Y. Intermedin/Adrenomedullin2: An Autocrine/Paracrine Factor in Vascular Homeostasis and Disease. Sci. China Life Sci. 2014, 57, 781–789. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chang, J.-R.; Duan, X.-H.; Zhang, B.-H.; Teng, X.; Zhou, Y.-B.; Liu, Y.; Yu, Y.-R.; Zhu, Y.; Tang, C.-S.; Qi, Y.-F. Intermedin1-53 Attenuates Vascular Smooth Muscle Cell Calcification by Inhibiting Endoplasmic Reticulum Stress via Cyclic Adenosine Monophosphate/Protein Kinase A Pathway. Exp. Biol. Med. 2013, 238, 1136–1146. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zhang, L.-S.; Ren, J.-L.; Zhang, Y.-R.; Wu, N.; Jia, M.-Z.; Yu, Y.-R.; Ning, Z.-P.; Tang, C.-S.; Qi, Y.-F. Intermedin1-53 Attenuates Aging-Associated Vascular Calcification in Rats by Upregulating Sirtuin 1. Aging 2020, 12, 5651–5674. [Google Scholar] [CrossRef]
- Rushworth, G.F.; Megson, I.L. Existing and Potential Therapeutic Uses for N-Acetylcysteine: The Need for Conversion to Intracellular Glutathione for Antioxidant Benefits. Pharmacol. Ther. 2014, 141, 150–159. [Google Scholar] [CrossRef]
- Bourne, L.E.; Patel, J.J.; Davies, B.K.; Neven, E.; Verhulst, A.; D’Haese, P.C.; Wheeler-Jones, C.P.D.; Orriss, I.R. N-Acetylcysteine (NAC) Differentially Affects Arterial Medial Calcification and Bone Formation: The Role of l-Cysteine and Hydrogen Sulphide. J. Cell. Physiol. 2022, 237, 1070–1086. [Google Scholar] [CrossRef]
- Son, B.-K.; Kozaki, K.; Iijima, K.; Eto, M.; Kojima, T.; Ota, H.; Senda, Y.; Maemura, K.; Nakano, T.; Akishita, M.; et al. Statins Protect Human Aortic Smooth Muscle Cells From Inorganic Phosphate-Induced Calcification by Restoring Gas6-Axl Survival Pathway. Circ. Res. 2006, 98, 1024–1031. [Google Scholar] [CrossRef] [Green Version]
- Liu, D.; Cui, W.; Liu, B.; Hu, H.; Liu, J.; Xie, R.; Yang, X.; Gu, G.; Zhang, J.; Zheng, H. Atorvastatin Protects Vascular Smooth Muscle Cells from TGF-Β1-Stimulated Calcification by Inducing Autophagy via Suppression of the β-Catenin Pathway. Cell. Physiol. Biochem. 2014, 33, 129–141. [Google Scholar] [CrossRef]
- Zhou, J.; Zhou, H.; Liu, C.; Huang, L.; Lu, D.; Gao, C. HDAC1-Mediated Deacetylation of LSD1 Regulates Vascular Calcification by Promoting Autophagy in Chronic Renal Failure. J. Cell. Mol. Med. 2020, 24, 8636–8649. [Google Scholar] [CrossRef]
- Shearer, M.J.; Okano, T. Key Pathways and Regulators of Vitamin K Function and Intermediary Metabolism. Annu. Rev. Nutr. 2018, 38, 127–151. [Google Scholar] [CrossRef] [PubMed]
- Hasanbasic, I.; Rajotte, I.; Blostein, M. The Role of Gamma-Carboxylation in the Anti-Apoptotic Function of Gas6. J. Thromb. Haemost. 2005, 3, 2790–2797. [Google Scholar] [CrossRef] [PubMed]
- Kim, H.; Kim, H.-J.; Lee, K.; Kim, J.-M.; Kim, H.S.; Kim, J.-R.; Ha, C.-M.; Choi, Y.-K.; Lee, S.J.; Kim, J.-Y.; et al. α-Lipoic Acid Attenuates Vascular Calcification via Reversal of Mitochondrial Function and Restoration of Gas6/Axl/Akt Survival Pathway. J. Cell. Mol. Med. 2012, 16, 273–286. [Google Scholar] [CrossRef] [PubMed]
- Kaesler, N.; Immendorf, S.; Ouyang, C.; Herfs, M.; Drummen, N.; Carmeliet, P.; Vermeer, C.; Floege, J.; Krüger, T.; Schlieper, G. Gas6 Protein: Its Role in Cardiovascular Calcification. BMC Nephrol. 2016, 17, 52. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danziger, J. Vitamin K-Dependent Proteins, Warfarin, and Vascular Calcification. Clin. J. Am. Soc. Nephrol. 2008, 3, 1504–1510. [Google Scholar] [CrossRef] [Green Version]
- Zhan, J.-K.; Wang, Y.-J.; Wang, Y.; Wang, S.; Tan, P.; Huang, W.; Liu, Y.-S. The Mammalian Target of Rapamycin Signalling Pathway Is Involved in Osteoblastic Differentiation of Vascular Smooth Muscle Cells. Can. J. Cardiol. 2014, 30, 568–575. [Google Scholar] [CrossRef]


{kind=link}
{kind=link}
{kind=link}
| Cell Type | Calcifying Medium | References |
|---|---|---|
| Bovine VSMC | DMEM high glucose, 15% FBS, 10 mmol/L sodium pyruvate, 10 mmol/L β-glycerophosphate, 10−7 mol/L insulin, 50 μg/mL of ascorbic acid | [32] |
| Mouse VSMC | DMEM, 15% FBS, 10 mmol/l sodium pyruvate, 50 mg/mL ascorbic acid, 10 mmol/l β-glycerophosphate | [33] |
| Mouse VSMC | DMEM high glucose, 15% FBS, 10 mmol/L β-glycerophosphate, 284 µmol/L ascorbic acid, 10 mmol/L sodium pyruvate | [34] |
| Bovine VSMC | DMEM high glucose, 10% FBS, 10 mM β-glycerophosphate, 50 μg/mL ascorbic acid, 25 mM glucose | [35] |
| Mouse VSMC | DMEM, 1% FBS, 1.6 mM inorganic phosphate, 10 μM warfarin | [36] |
| Mouse VSMC | DMEM high glucose, 15% FBS, 8 mmol/l CaCl2, 10 mmol/l sodium pyruvate, 1 μmol/l insulin, 50 μg/mL ascorbic acid, 10 mmol/l β-glycerophosphate, and 100 nmol/l dexamethasone | [37] |
| Mouse VSMC | DMEM, 10% FBS, 2.5 mmol/L CaCl2, 5 mmol/L β-glycerophosphate | [38] |
| Mouse VSMC | DMEM high glucose, 10% FBS, 1.25/2.5 mM β-glycerophosphate, 25/50 µg/mL ascorbic acid | [39] |
| Natural Compounds | References |
|---|---|
| Vitamin E | [70,71] |
| Vitamin K | [72] |
| Quercetin | [73,74,75,76] |
| Carnosine | [77,78,79,80] |
| Magnesium | [81] |
| Intermedin | [82,83,84,85] |
| Synthetic Compounds | |
| Acetylcysteine | [86,87] |
| Statins | [37,88,89] |
| Valproic acid | [90] |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ceccherini, E.; Cecchettini, A.; Gisone, I.; Persiani, E.; Morales, M.A.; Vozzi, F. Vascular Calcification: In Vitro Models under the Magnifying Glass. Biomedicines 2022, 10, 2491. https://doi.org/10.3390/biomedicines10102491
Ceccherini E, Cecchettini A, Gisone I, Persiani E, Morales MA, Vozzi F. Vascular Calcification: In Vitro Models under the Magnifying Glass. Biomedicines. 2022; 10(10):2491. https://doi.org/10.3390/biomedicines10102491
Chicago/Turabian StyleCeccherini, Elisa, Antonella Cecchettini, Ilaria Gisone, Elisa Persiani, Maria Aurora Morales, and Federico Vozzi. 2022. "Vascular Calcification: In Vitro Models under the Magnifying Glass" Biomedicines 10, no. 10: 2491. https://doi.org/10.3390/biomedicines10102491
APA StyleCeccherini, E., Cecchettini, A., Gisone, I., Persiani, E., Morales, M. A., & Vozzi, F. (2022). Vascular Calcification: In Vitro Models under the Magnifying Glass. Biomedicines, 10(10), 2491. https://doi.org/10.3390/biomedicines10102491