
Characterization of the Urinary Metagenome and Virome in Healthy Children


 , ,
, ,
Abstract
1. Introduction
2. Material and Methods
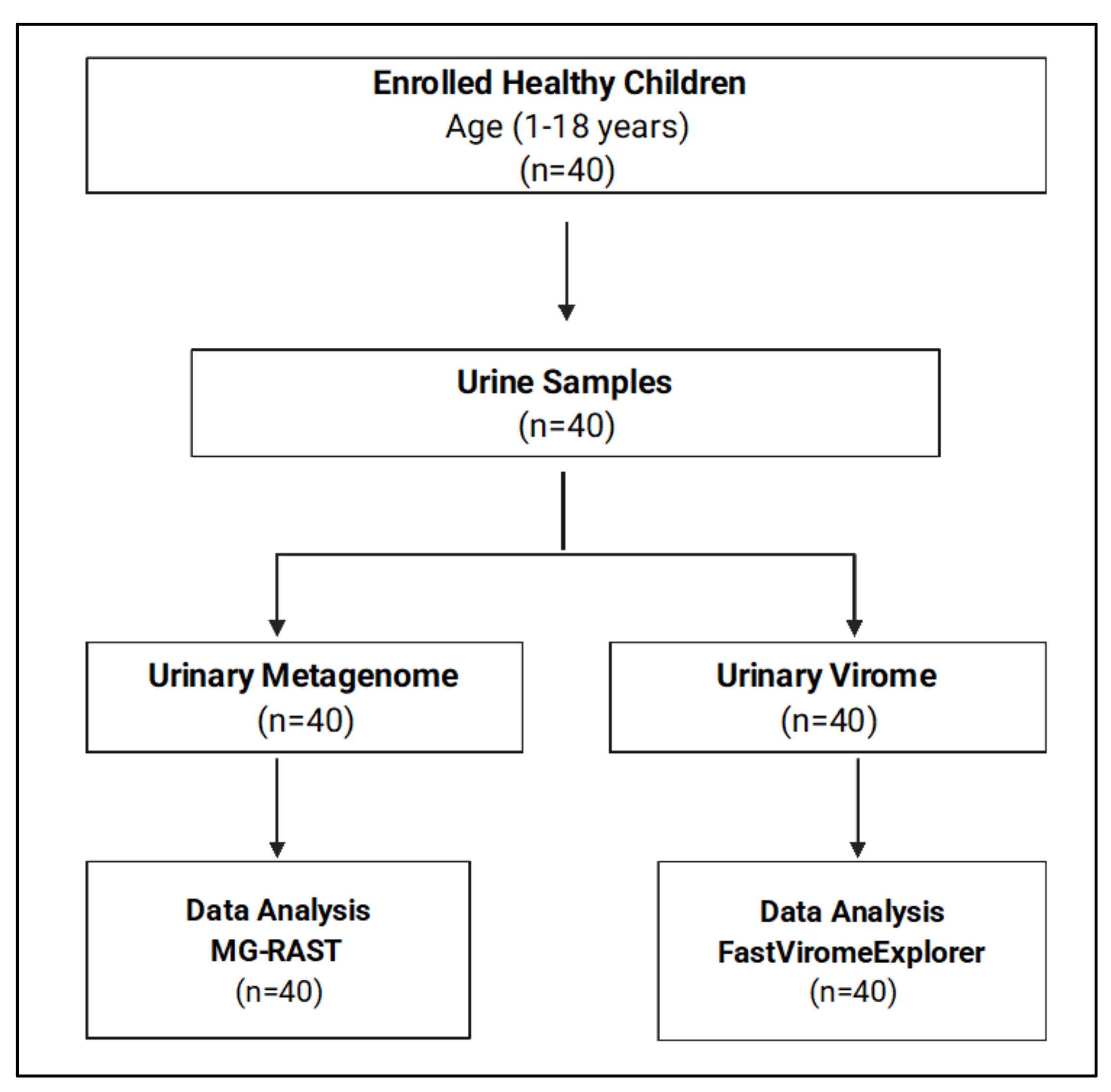
2.1. Study Design
2.2. DNA Extraction for Urinary Metagenome
2.3. RNA Extraction for Urinary Virome
2.4. cDNA Synthesis and Virus Amplification
2.5. Library Preparation and Shotgun Sequencing
2.6. Data Analysis and Statistical Analysis
3. Results
3.1. Demographic Data
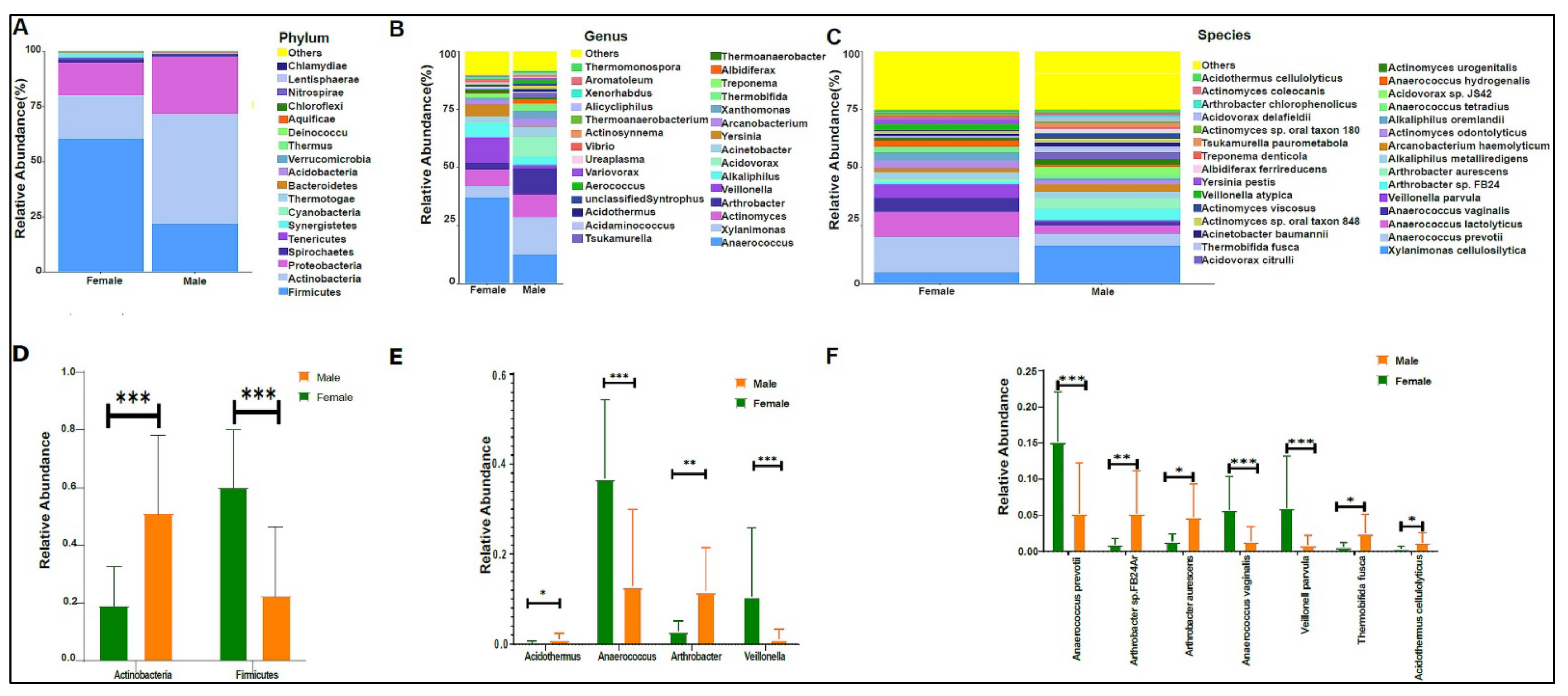
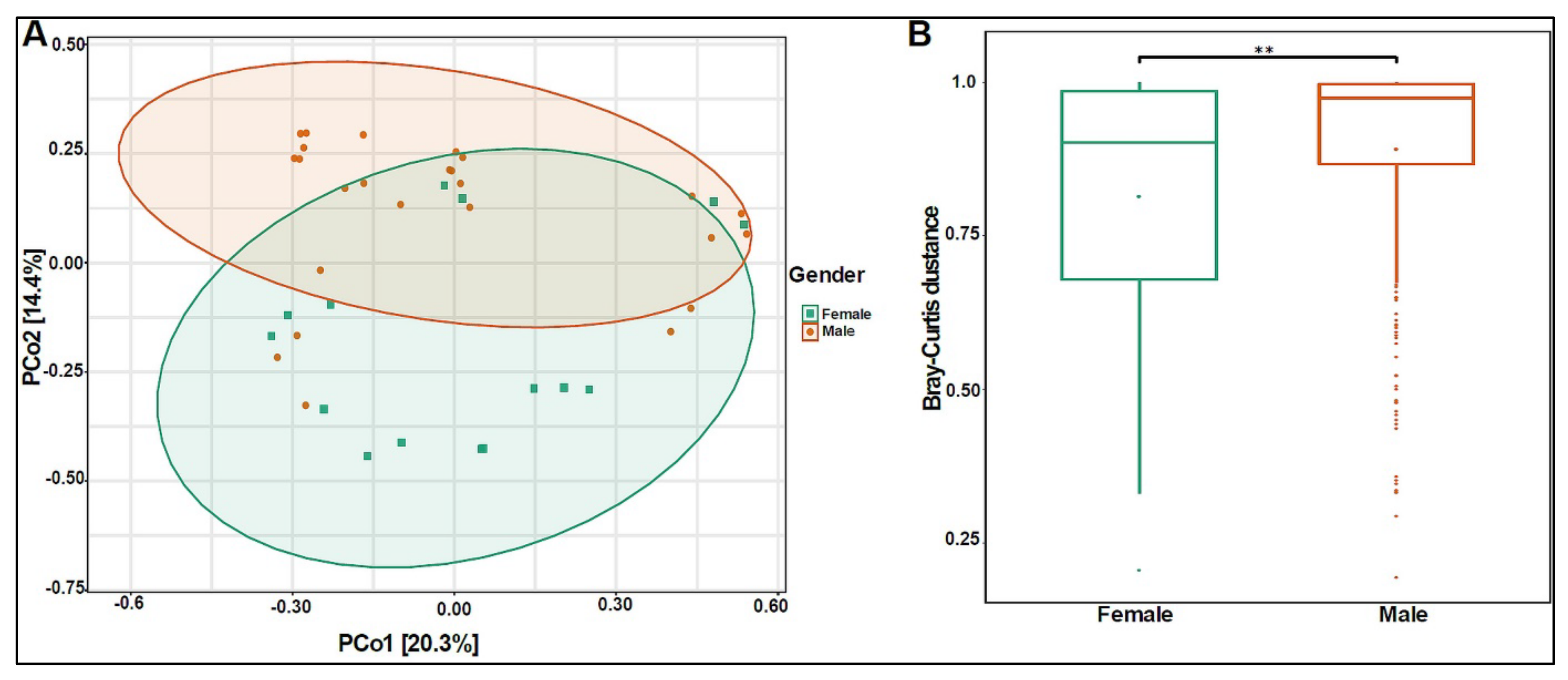
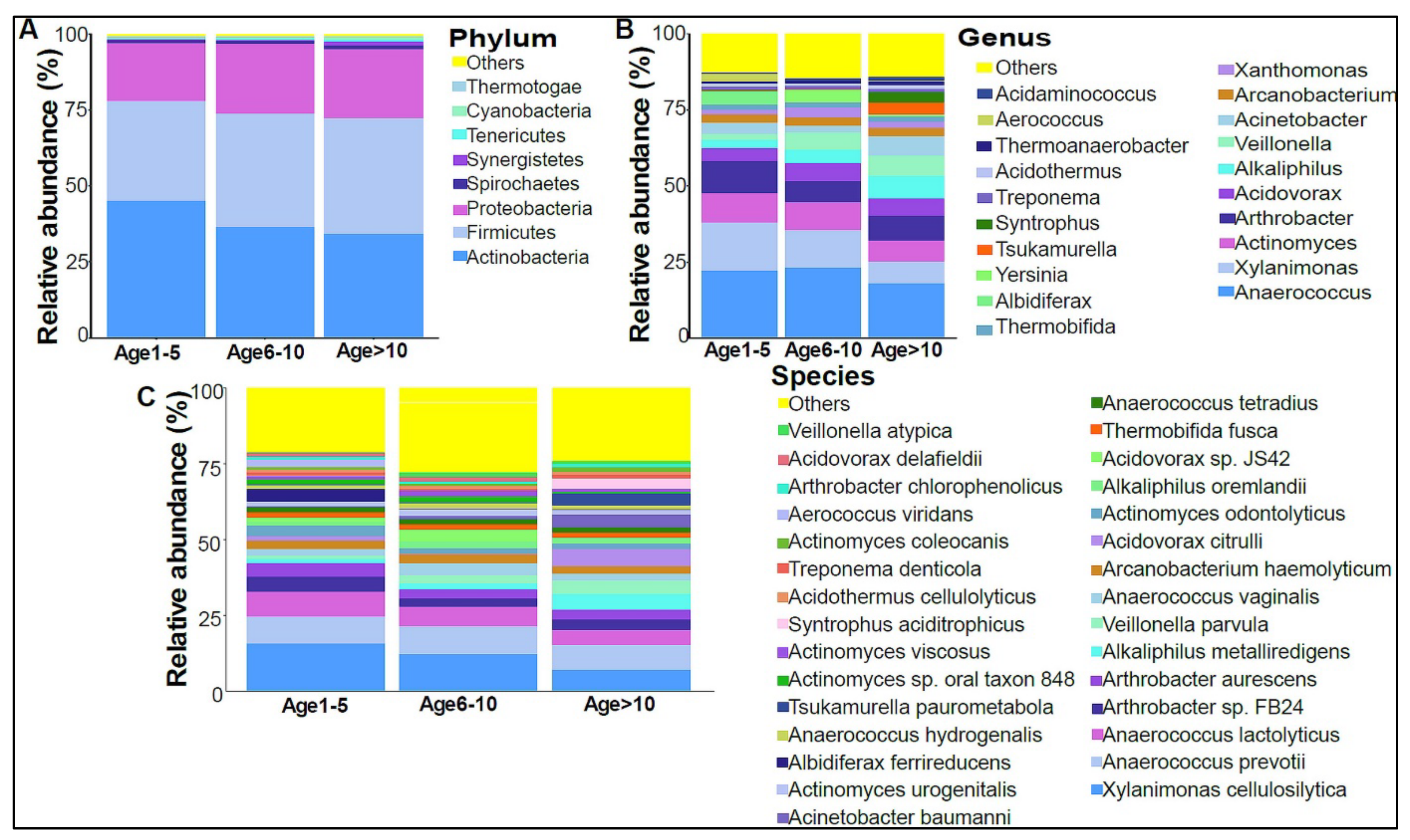
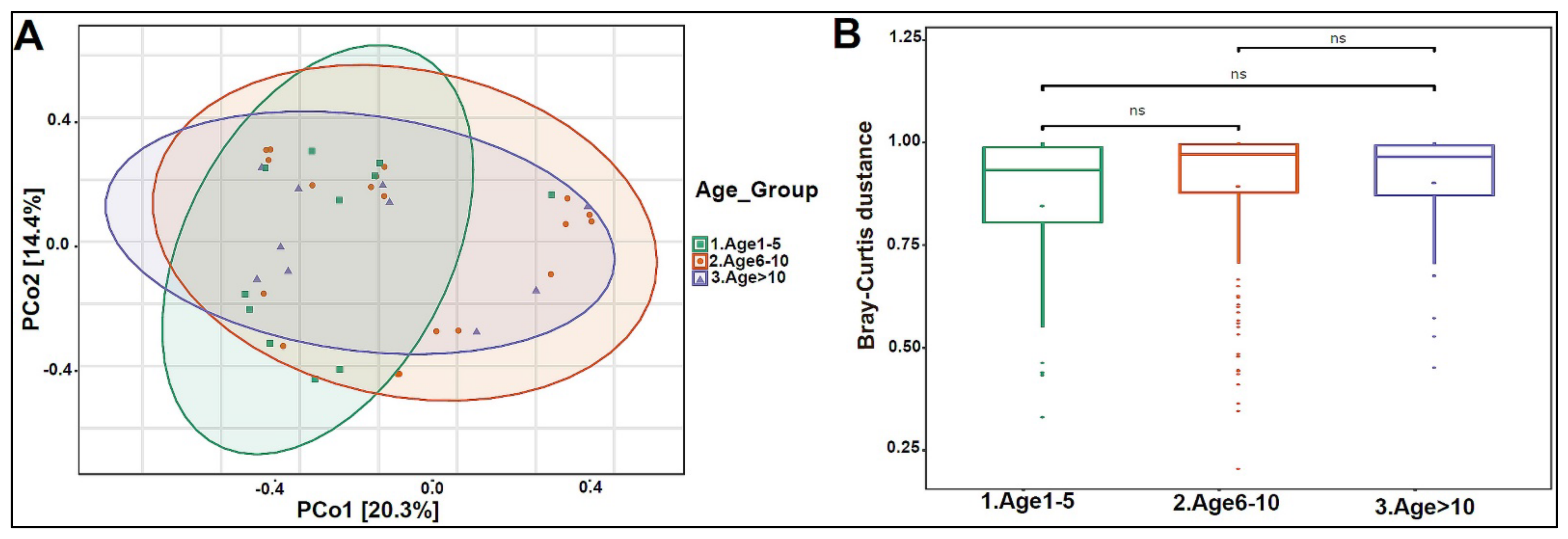
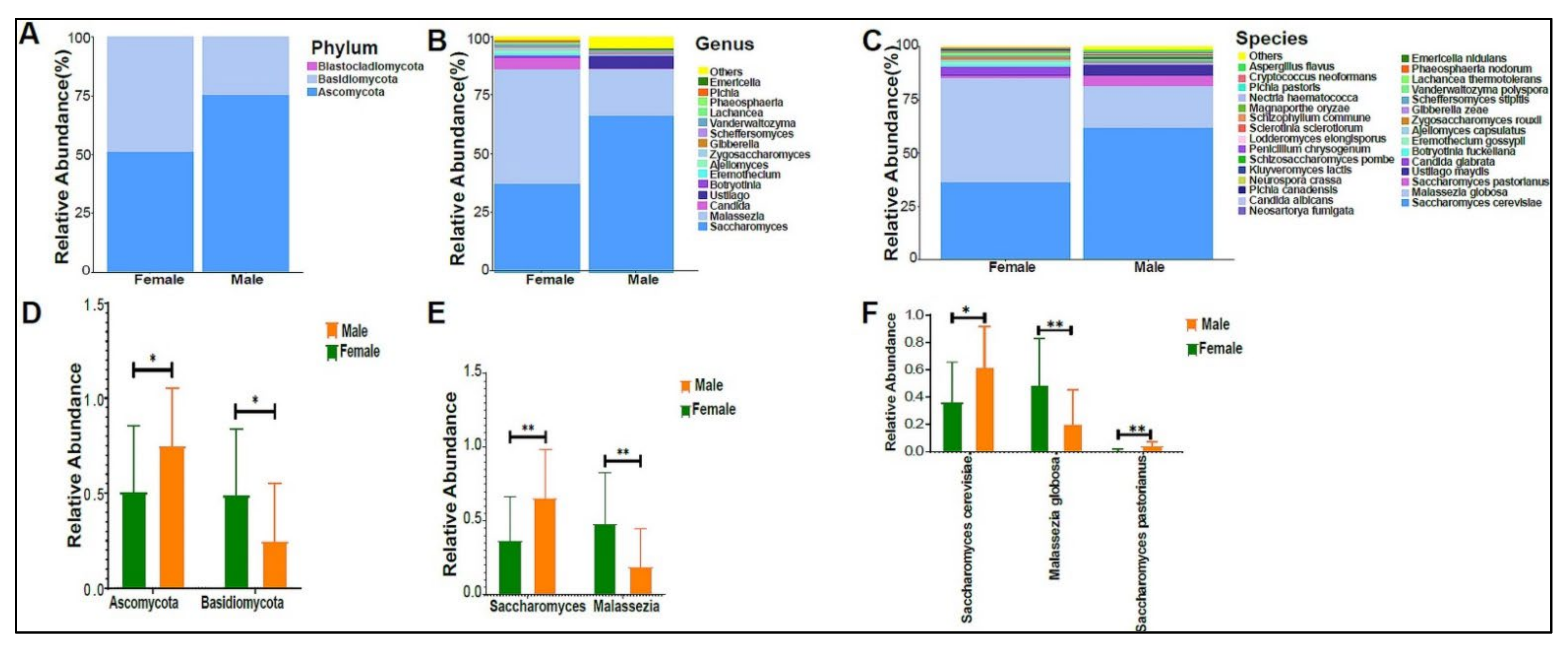
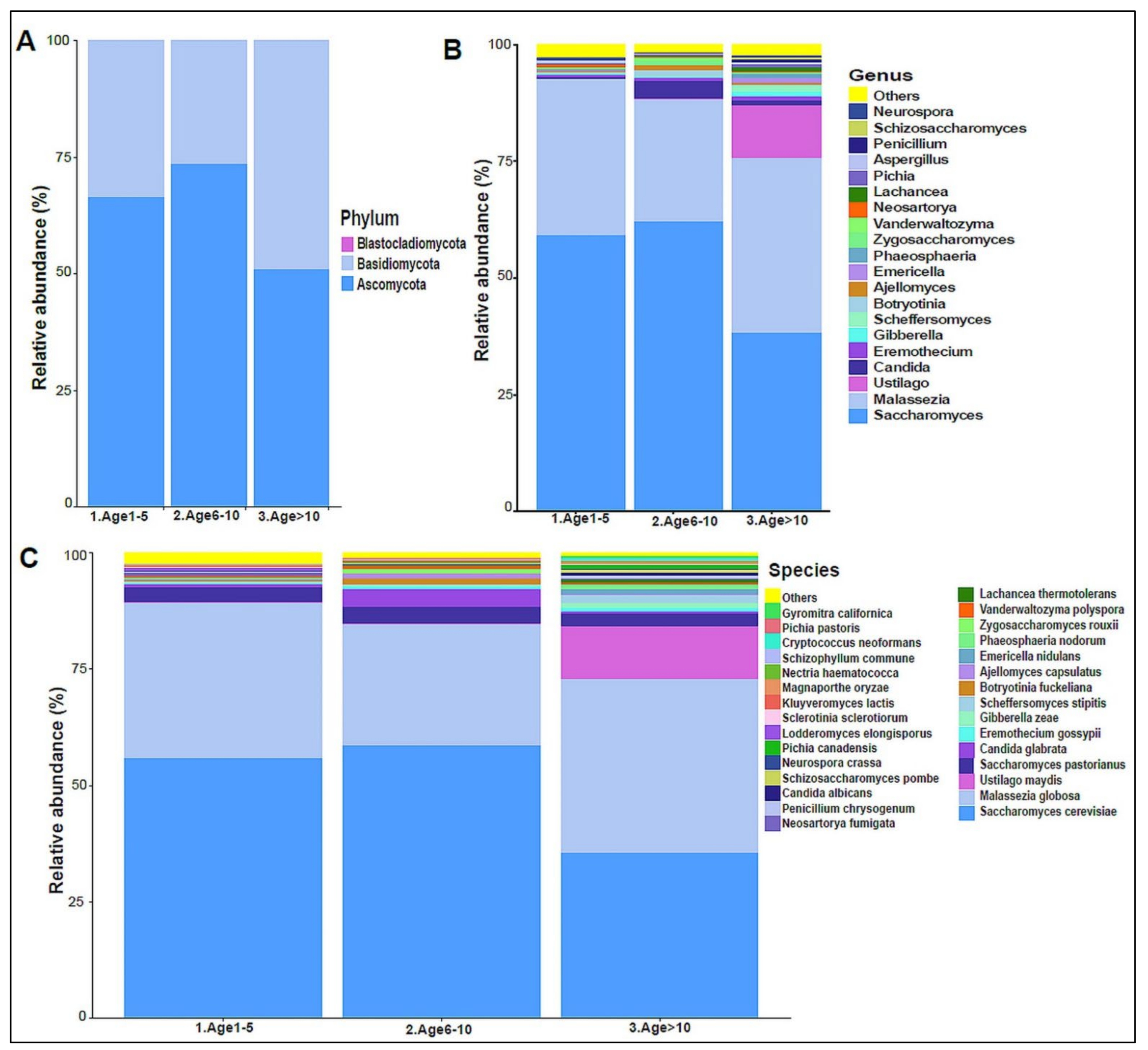
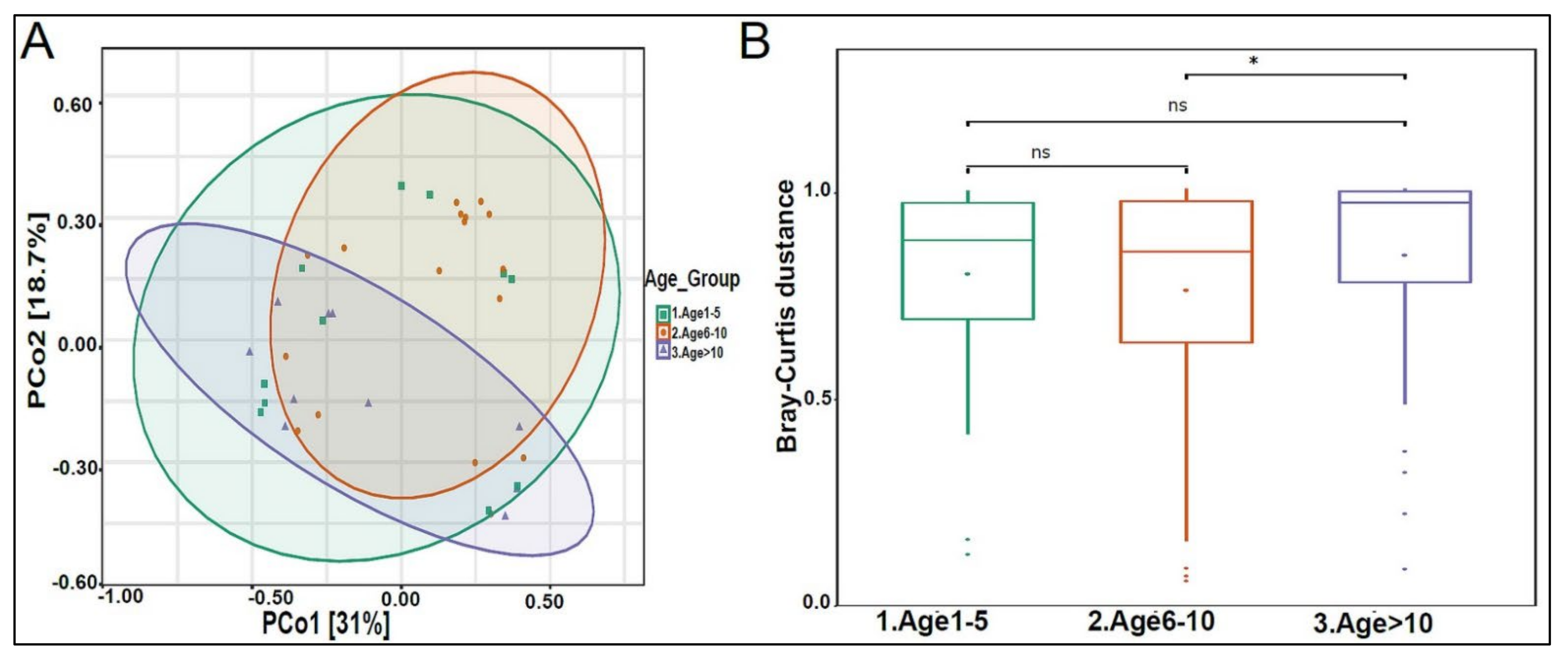
3.2. Urinary Metagenome Composition in Healthy Children
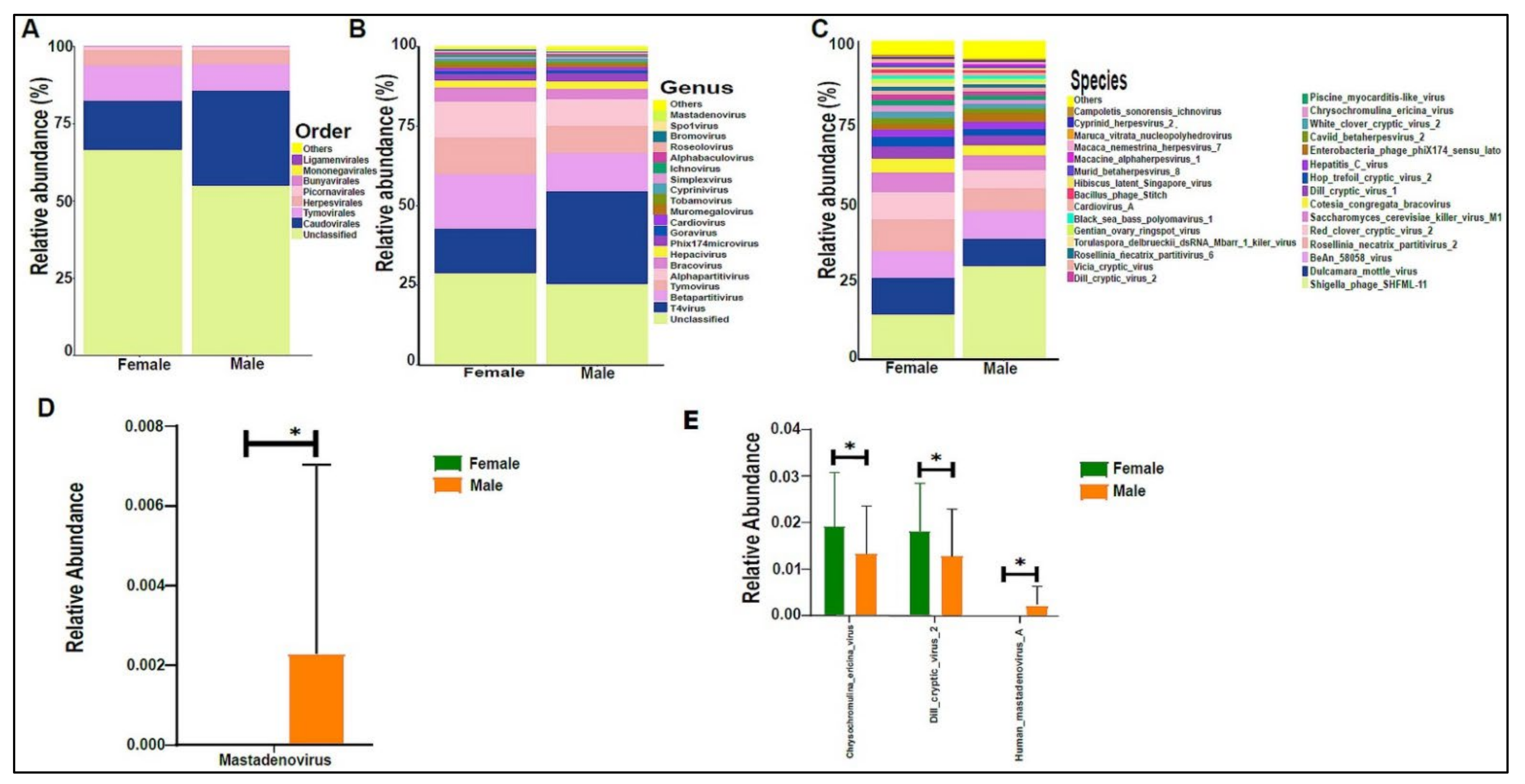
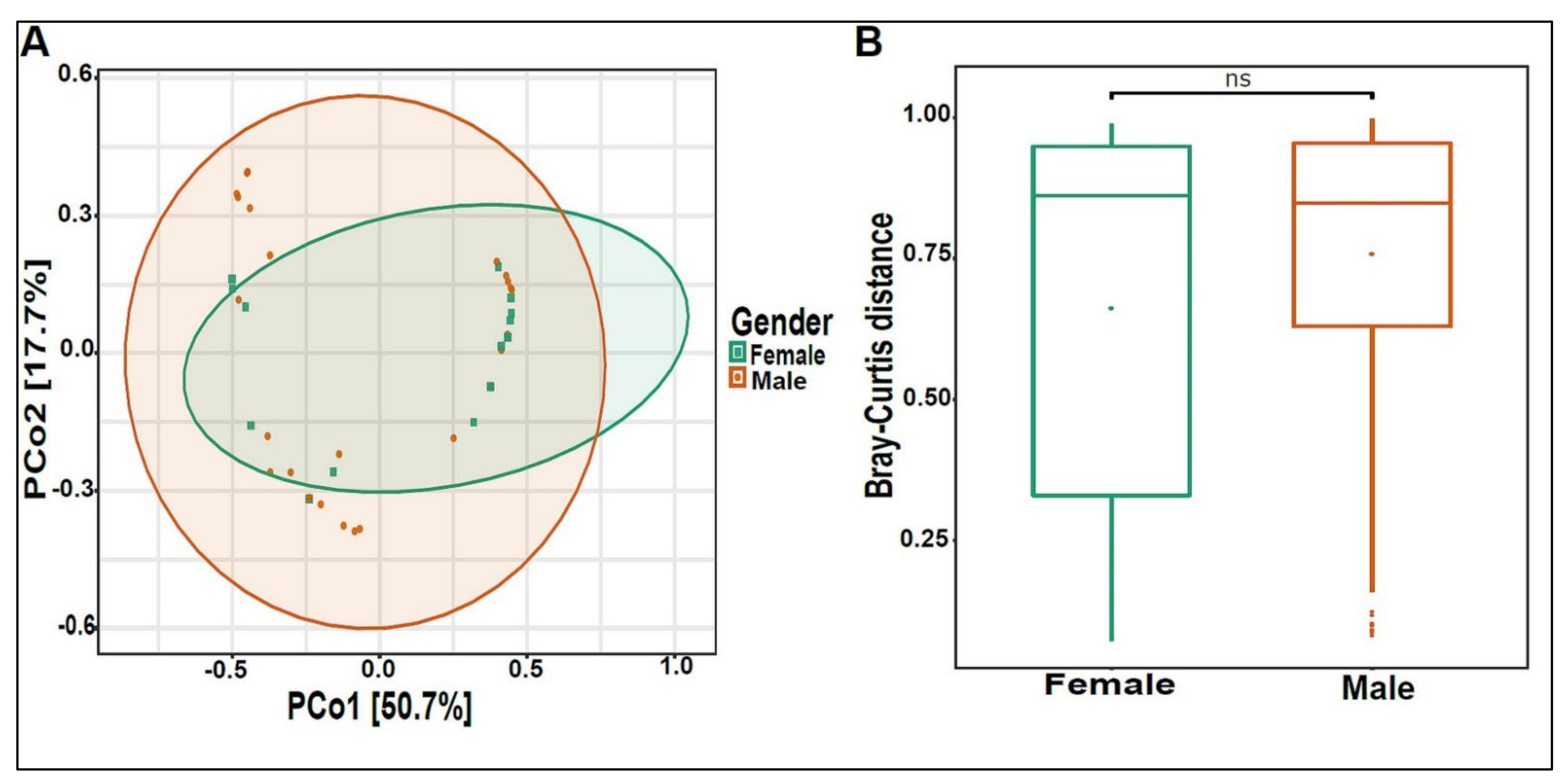
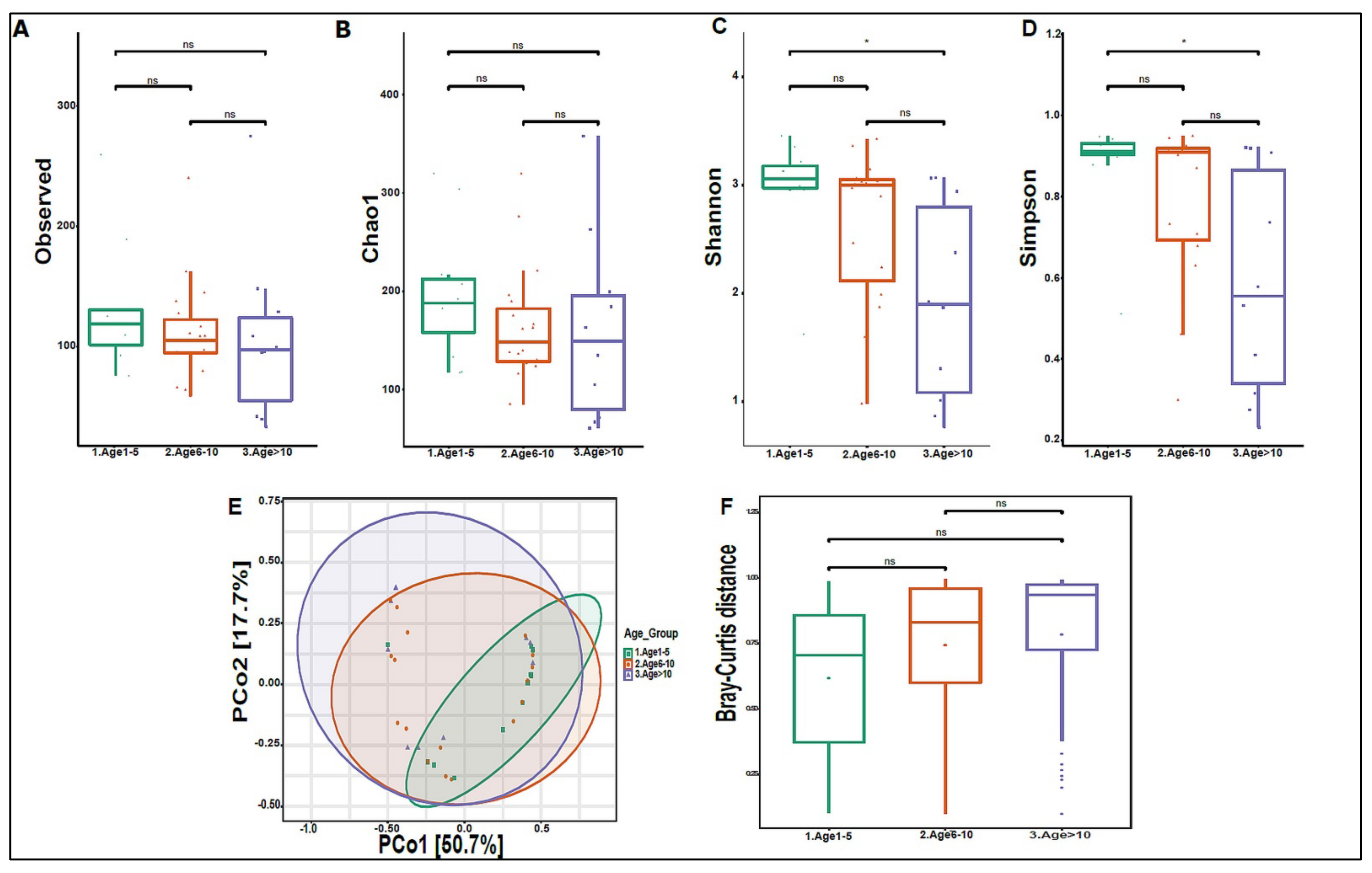
3.3. Urinary Virome Profile of Healthy Children
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Institutional Review Board Statement
Informed Consent Statement
Data Availability Statement
Conflicts of Interest
Abbreviations
| IRB | Institutional review board |
| OUT | Operational taxonomic unit |
| REDCap | Research Electronic Data Capture |
| RT | Reverse transcription |
| SM | Sulfate magnesium |
| UTIs | Urinary tract infections |
| VLP | Viral-like particles |
References
- The Human Microbiome Project Consortium. A framework for human microbiome research. Nature 2012, 486, 215–221. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Price, J.; Abu-Ali, G.; Huttenhower, C. The healthy human microbiome. Genome. Med. 2016, 8, 51. [Google Scholar] [CrossRef] [PubMed]
- Thursby, E.; Juge, N. Introduction to the human gut microbiota. Biochem. J. 2017, 474, 1823–1836. [Google Scholar] [CrossRef] [PubMed]
- Lynch, S.V.; Pedersen, O. The human intestinal microbiome in health and disease. N. Engl. J. Med. 2016, 375, 2369–2379. [Google Scholar] [CrossRef]
- Morand, A.; Cornu, F.; Dufour, J.C.; Tsimaratos, M.; Lagier, J.C.; Raoult, D. Human Bacterial Repertoire of the Urinary Tract: A Potential Paradigm Shift. J. Clin. Microbiol. 2019, 57, e00675-18. [Google Scholar] [CrossRef]
- Whiteside, S.A.; Razvi, H.; Dave, S.; Reid, G.; Burton, J.P. The microbiome of the urinary tract--a role beyond infection. Nat. Rev. Urol. 2015, 12, 81–90. [Google Scholar] [CrossRef]
- Magistro, G.; Stief, C.G. The Urinary Tract Microbiome: The Answer to All Our Open Questions? Eur. Urol. Focus 2019, 5, 36–38. [Google Scholar] [CrossRef]
- Foddai, A.C.G.; Grant, I.R. Methods for detection of viable foodborne pathogens: Current state-of-art and future prospects. Appl. Microbiol. Biotechnol. 2020, 104, 4281–4288. [Google Scholar] [CrossRef]
- Lagier, J.C.; Edouard, S.; Pagnier, I.; Mediannikov, O.; Drancourt, M.; Raoult, D. Current and past strategies for bacterial culture in clinical microbiology. Clin. Microbiol. Rev. 2015, 28, 208–236. [Google Scholar] [CrossRef]
- Brubaker, L.; Wolfe, A.J. The new world of the urinary microbiota in women. Am. J. Obstet. Gynecol. 2015, 213, 644–649. [Google Scholar] [CrossRef]
- Garcia, L.S.; Isenberg, H.D. Aerobic Bacteriology. In Clinical Microbiology Procedures Handbook; ASM Press: Washingon, DC, USA, 2010. [Google Scholar] [CrossRef]
- Gerges-Knafl, D.; Pichler, P.; Zimprich, A.; Hotzy, C.; Barousch, W.; Lang, R.M.; Lobmeyr, E.; Baumgartner-Parzer, S.; Wagner, L.; Winnicki, W. The urinary microbiome shows different bacterial genera in renal transplant recipients and non-transplant patients at time of acute kidney injury—A pilot study. BMC Nephrol. 2020, 21, 117. [Google Scholar] [CrossRef]
- Wolfe, A.J.; Toh, E.; Shibata, N.; Rong, R.; Kenton, K.; Fitzgerald, M.; Mueller, E.R.; Schreckenberger, P.; Dong, Q.; Nelson, D.E.; et al. Evidence of uncultivated bacteria in the adult female bladder. J. Clin. Microbiol. 2012, 50, 1376–1383. [Google Scholar] [CrossRef]
- Dutilh, B.E.; Cassman, N.; McNair, K.; Sanchez, S.E.; Silva, G.G.; Boling, L.; Barr, J.J.; Speth, D.R.; Seguritan, V.; Aziz, R.K.; et al. A highly abundant bacteriophage discovered in the unknown sequences of human faecal metagenomes. Nat. Commun. 2014, 5, 4498. [Google Scholar] [CrossRef]
- Siddiqui, H.; Nederbragt, A.J.; Lagesen, K.; Jeansson, S.L.; Jakobsen, K.S. Assessing diversity of the female urine microbiota by high throughput sequencing of 16S rDNA amplicons. BMC Microbiol. 2011, 11, 244. [Google Scholar] [CrossRef]
- Fouts, D.E.; Pieper, R.; Szpakowski, S.; Pohl, H.; Knoblach, S.; Suh, M.J.; Huang, S.T.; Ljungberg, I.; Sprague, B.M.; Lucas, S.K.; et al. Integrated next-generation sequencing of 16S rDNA and metaproteomics differentiate the healthy urine microbiome from asymptomatic bacteriuria in neuropathic bladder associated with spinal cord injury. J. Transl. Med. 2012, 10, 174. [Google Scholar] [CrossRef]
- Abelson, B.; Sun, D.; Que, L.; Nebel, R.A.; Baker, D.; Popiel, P.; Amundsen, C.L.; Chai, T.; Close, C.; DiSanto, M.; et al. Sex differences in lower urinary tract biology and physiology. Biol. Sex. Differ. 2018, 9, 45. [Google Scholar] [CrossRef]
- Brubaker, L.; Wolfe, A.J. The female urinary microbiota, urinary health and common urinary disorders. Ann. Transl. Med. 2017, 5, 34. [Google Scholar] [CrossRef]
- Rani, A.; Ranjan, R.; McGee, H.S.; Andropolis, K.E.; Panchal, D.V.; Hajjiri, Z.; Brennan, D.C.; Finn, P.W.; Perkins, D.L. Urinary microbiome of kidney transplant patients reveals dysbiosis with potential for antibiotic resistance. Transl. Res. 2017, 181, 59–70. [Google Scholar] [CrossRef]
- Hallen-Adams, H.E.; Suhr, M.J. Fungi in the healthy human gastrointestinal tract. Virulence 2017, 8, 352–358. [Google Scholar] [CrossRef]
- Ackerman, A.L.; Underhill, D.M. The mycobiome of the human urinary tract: Potential roles for fungi in urology. Ann. Transl. Med. 2017, 5, 31. [Google Scholar] [CrossRef]
- Brandt, M.E.; Park, B.J. Think fungus-prevention and control of fungal infections. Emerg. Infect. Dis. 2013, 19, 1688–1689. [Google Scholar] [CrossRef]
- Seifi, Z.; Azish, M.; Salehi, Z.; Zarei Mahmoudabadi, A.; Shamsizadeh, A. Candiduria in children and susceptibility patterns of recovered Candida species to antifungal drugs in Ahvaz. J. Nephropathol. 2013, 2, 122–128. [Google Scholar] [CrossRef]
- Delwart, E. A roadmap to the human virome. PLoS Pathog. 2013, 9, e1003146. [Google Scholar] [CrossRef]
- Santiago-Rodriguez, T.M.; Hollister, E.B. Human Virome and Disease: High-Throughput Sequencing for Virus Discovery, Identification of Phage-Bacteria Dysbiosis and Development of Therapeutic Approaches with Emphasis on the Human Gut. Viruses 2019, 11, 656. [Google Scholar] [CrossRef]
- Navarro, F.; Muniesa, M. Phages in the Human Body. Front. Microbiol. 2017, 8, 566. [Google Scholar] [CrossRef]
- Salabura, A.; Łuniewski, A.; Kucharska, M.; Myszak, D.; Dołęgowska, B.; Ciechanowski, K.; Kędzierska-Kapuza, K.; Wojciuk, B. Urinary Tract Virome as an Urgent Target for Metagenomics. Life 2021, 11, 1264. [Google Scholar] [CrossRef]
- Yumoto, Y. The procedures for sampling specimens in pediatrics. Rinsho Byori 2014, 62, 766–774. [Google Scholar]
- Soliman, N.A.; Rizvi, S.A.H. Endemic bladder calculi in children. Pediatr. Nephrol. 2017, 32, 1489–1499. [Google Scholar] [CrossRef]
- Song, L.; Maalouf, N.M. Nephrolithiasis. In Endotext; Feingold, K.R., Anawalt, B., Boyce, A., Chrousos, G., de Herder, W.W., Dhatariya, K., Dungan, K., Hershman, J.M., Hofland, J., Kalra, S., et al., Eds.; MDText.com, Inc.: South Dartmouth, MA, USA, 2000. [Google Scholar]
- Moustafa, A.; Li, W.; Singh, H.; Moncera, K.J.; Torralba, M.G.; Yu, Y.; Manuel, O.; Biggs, W.; Venter, J.C.; Nelson, K.E.; et al. Microbial metagenome of urinary tract infection. Sci. Rep. 2018, 8, 4333. [Google Scholar] [CrossRef]
- Norman, J.M.; Handley, S.A.; Baldridge, M.T.; Droit, L.; Liu, C.Y.; Keller, B.C.; Kambal, A.; Monaco, C.L.; Zhao, G.; Fleshner, P.; et al. Disease-specific alterations in the enteric virome in inflammatory bowel disease. Cell 2015, 160, 447–460. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Ryan, F.J.; Draper, L.A.; Forde, A.; Stockdale, S.R.; Daly, K.M.; McDonnell, S.A.; Nolan, J.A.; Sutton, T.D.S.; Dalmasso, M.; et al. Reproducible protocols for metagenomic analysis of human faecal phageomes. Microbiome 2018, 6, 68. [Google Scholar] [CrossRef] [PubMed]
- Keegan, K.P.; Glass, E.M.; Meyer, F. MG-RAST, a Metagenomics Service for Analysis of Microbial Community Structure and Function. Methods Mol. Biol. 2016, 1399, 207–233. [Google Scholar] [CrossRef] [PubMed]
- Keegan, K.P.; Trimble, W.L.; Wilkening, J.; Wilke, A.; Harrison, T.; D’Souza, M.; Meyer, F. A platform-independent method for detecting errors in metagenomic sequencing data: DRISEE. PLoS Comput. Biol. 2012, 8, e1002541. [Google Scholar] [CrossRef] [PubMed]
- Marcais, G.; Kingsford, C. A fast, lock-free approach for efficient parallel counting of occurrences of k-mers. Bioinformatics 2011, 27, 764–770. [Google Scholar] [CrossRef] [PubMed]
- Jiang, H.; Lei, R.; Ding, S.W.; Zhu, S. Skewer: A fast and accurate adapter trimmer for next-generation sequencing paired-end reads. BMC Bioinform. 2014, 15, 182. [Google Scholar] [CrossRef] [PubMed]
- Aronesty, E. Comparison of Sequencing Utility Programs. Open Bioinform. J. 2013, 7, 1–8. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Kopylova, E.; Noe, L.; Touzet, H. SortMeRNA: Fast and accurate filtering of ribosomal RNAs in metatranscriptomic data. Bioinformatics 2012, 28, 3211–3217. [Google Scholar] [CrossRef]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef]
- Kent, W.J. BLAT--the BLAST-like alignment tool. Genome. Res. 2002, 12, 656–664. [Google Scholar] [CrossRef]
- Tithi, S.S.; Aylward, F.O.; Jensen, R.V.; Zhang, L. FastViromeExplorer: A pipeline for virus and phage identification and abundance profiling in metagenomics data. PeerJ 2018, 6, e4227. [Google Scholar] [CrossRef]
- Liu, C.; Cui, Y.; Li, X.; Yao, M. Microeco: An R package for data mining in microbial community ecology. FEMS Microbiol. Ecol. 2021, 97, fiaa255. [Google Scholar] [CrossRef]
- Prism, G. Unpaired Multiple t-test followed by Mann-Whiteney’s comparison of ranking test was performed using GraphPad Prism version 9.0.1 for Windows. Graphpad Prism 2021. [Google Scholar]
- Flores-Mireles, A.L.; Walker, J.N.; Caparon, M.; Hultgren, S.J. Urinary tract infections: Epidemiology, mechanisms of infection and treatment options. Nat. Rev. Microbiol. 2015, 13, 269–284. [Google Scholar] [CrossRef]
- Tamadonfar, K.O.; Omattage, N.S.; Spaulding, C.N.; Hultgren, S.J. Reaching the End of the Line: Urinary Tract Infections. Microbiol. Spectr. 2019, 7, 3. [Google Scholar] [CrossRef]
- Clark, D.J.; Hu, Y.; Schnaubelt, M.; Fu, Y.; Ponce, S.; Chen, S.Y.; Zhou, Y.; Shah, P.; Zhang, H. Simple Tip-Based Sample Processing Method for Urinary Proteomic Analysis. Anal. Chem. 2019, 91, 5517–5522. [Google Scholar] [CrossRef]
- Wu, J.; Chen, Y.D.; Gu, W. Urinary proteomics as a novel tool for biomarker discovery in kidney diseases. J. Zhejiang Univ. Sci. B 2010, 11, 227–237. [Google Scholar] [CrossRef]
- Ragland, S.A.; Criss, A.K. From bacterial killing to immune modulation: Recent insights into the functions of lysozyme. PLoS Pathog. 2017, 13, e1006512. [Google Scholar] [CrossRef]
- Simpson, R.J. Preparation of extracts from yeast. Cold Spring Harb. Protoc. 2011, 2011, pdb.prot55455. [Google Scholar] [CrossRef]
- Thomas-White, K.; Forster, S.C.; Kumar, N.; Van Kuiken, M.; Putonti, C.; Stares, M.D.; Hilt, E.E.; Price, T.K.; Wolfe, A.J.; Lawley, T.D. Culturing of female bladder bacteria reveals an interconnected urogenital microbiota. Nat. Commun. 2018, 9, 1557. [Google Scholar] [CrossRef]
- Tachedjian, G.; Aldunate, M.; Bradshaw, C.S.; Cone, R.A. The role of lactic acid production by probiotic Lactobacillus species in vaginal health. Res. Microbiol. 2017, 168, 782–792. [Google Scholar] [CrossRef]
- Borges, S.; Silva, J.; Teixeira, P. The role of lactobacilli and probiotics in maintaining vaginal health. Arch. Gynecol. Obstet. 2014, 289, 479–489. [Google Scholar] [CrossRef]
- Xiao, B.B.; Liao, Q.P. Analysis of diversity of vaginal microbiota in healthy Chinese women by using DNA-fingerprinting. J. Peking Univ. Health Sci. 2012, 44, 281–287. [Google Scholar]
- Storm, D.W.; Copp, H.L.; Halverson, T.M.; Du, J.; Juhr, D.; Wolfe, A.J. A Child’s urine is not sterile: A pilot study evaluating the Pediatric Urinary Microbiome. J. Pediatr. Urol. 2022, 18, 383–392. [Google Scholar] [CrossRef]
- Pederzoli, F.; Ferrarese, R.; Amato, V.; Locatelli, I.; Alchera, E.; Luciano, R.; Nebuloni, M.; Briganti, A.; Gallina, A.; Colombo, R.; et al. Sex-specific Alterations in the Urinary and Tissue Microbiome in Therapy-naive Urothelial Bladder Cancer Patients. Eur. Urol. Oncol. 2020, 3, 784–788. [Google Scholar] [CrossRef]
- Coyte, K.Z.; Schluter, J.; Foster, K.R. The ecology of the microbiome: Networks, competition, and stability. Science 2015, 350, 663–666. [Google Scholar] [CrossRef]
- Fassarella, M.; Blaak, E.E.; Penders, J.; Nauta, A.; Smidt, H.; Zoetendal, E.G. Gut microbiome stability and resilience: Elucidating the response to perturbations in order to modulate gut health. Gut 2021, 70, 595–605. [Google Scholar] [CrossRef]
- Backhed, F.; Roswall, J.; Peng, Y.; Feng, Q.; Jia, H.; Kovatcheva-Datchary, P.; Li, Y.; Xia, Y.; Xie, H.; Zhong, H.; et al. Dynamics and Stabilization of the Human Gut Microbiome during the First Year of Life. Cell Host Microbe 2015, 17, 690–703. [Google Scholar] [CrossRef]
- Oh, J.; Byrd, A.L.; Park, M.; Program, N.C.S.; Kong, H.H.; Segre, J.A. Temporal Stability of the Human Skin Microbiome. Cell 2016, 165, 854–866. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Wang, D.; Garmaeva, S.; Kurilshikov, A.; Vich Vila, A.; Gacesa, R.; Sinha, T.; Lifelines Cohort, S.; Segal, E.; Weersma, R.K.; et al. The long-term genetic stability and individual specificity of the human gut microbiome. Cell 2021, 184, 2302–2315.e2312. [Google Scholar] [CrossRef] [PubMed]
- Nash, A.K.; Auchtung, T.A.; Wong, M.C.; Smith, D.P.; Gesell, J.R.; Ross, M.C.; Stewart, C.J.; Metcalf, G.A.; Muzny, D.M.; Gibbs, R.A.; et al. The gut mycobiome of the Human Microbiome Project healthy cohort. Microbiome 2017, 5, 153. [Google Scholar] [CrossRef] [PubMed]
- Spatz, M.; Richard, M.L. Overview of the Potential Role of Malassezia in Gut Health and Disease. Front. Cell Infect. Microbiol. 2020, 10, 201. [Google Scholar] [CrossRef] [PubMed]
- Sparber, F.; Ruchti, F.; LeibundGut-Landmann, S. Host Immunity to Malassezia in Health and Disease. Front. Cell Infect. Microbiol. 2020, 10, 198. [Google Scholar] [CrossRef]
- Boix-Amoros, A.; Puente-Sanchez, F.; du Toit, E.; Linderborg, K.M.; Zhang, Y.; Yang, B.; Salminen, S.; Isolauri, E.; Tamames, J.; Mira, A.; et al. Mycobiome Profiles in Breast Milk from Healthy Women Depend on Mode of Delivery, Geographic Location, and Interaction with Bacteria. Appl. Environ. Microbiol. 2019, 85, e02994-18. [Google Scholar] [CrossRef]
- You, L.; Zhao, D.; Zhou, R.; Tan, Y.; Wang, T.; Zheng, J. Distribution and function of dominant yeast species in the fermentation of strong-flavor baijiu. World J. Microbiol. Biotechnol. 2021, 37, 26. [Google Scholar] [CrossRef]
- Chen, Y.; Nielsen, J. In vitro turnover numbers do not reflect in vivo activities of yeast enzymes. Proc. Natl. Acad. Sci. USA 2021, 118, e2108391118. [Google Scholar] [CrossRef]
- Zuo, T.; Lu, X.J.; Zhang, Y.; Cheung, C.P.; Lam, S.; Zhang, F.; Tang, W.; Ching, J.Y.L.; Zhao, R.; Chan, P.K.S.; et al. Gut mucosal virome alterations in ulcerative colitis. Gut 2019, 68, 1169–1179. [Google Scholar] [CrossRef]
- Fernandes, M.A.; Verstraete, S.G.; Phan, T.G.; Deng, X.; Stekol, E.; LaMere, B.; Lynch, S.V.; Heyman, M.B.; Delwart, E. Enteric Virome and Bacterial Microbiota in Children With Ulcerative Colitis and Crohn Disease. J. Pediatr. Gastroenterol. Nutr. 2019, 68, 30–36. [Google Scholar] [CrossRef]
- De Sordi, L.; Lourenco, M.; Debarbieux, L. The Battle Within: Interactions of Bacteriophages and Bacteria in the Gastrointestinal Tract. Cell Host. Microbe. 2019, 25, 210–218. [Google Scholar] [CrossRef]
- Letarov, A.; Kulikov, E. The bacteriophages in human- and animal body-associated microbial communities. J. Appl. Microbiol. 2009, 107, 1–13. [Google Scholar] [CrossRef]
- Balique, F.; Lecoq, H.; Raoult, D.; Colson, P. Can plant viruses cross the kingdom border and be pathogenic to humans? Viruses 2015, 7, 2074–2098. [Google Scholar] [CrossRef]
- Granato, M. Nanotechnology Frontiers in gamma-Herpesviruses Treatments. Int. J. Mol. Sci. 2021, 22, 1470. [Google Scholar] [CrossRef]
- Huff, J.L.; Barry, P.A. B-virus (Cercopithecine herpesvirus 1) infection in humans and macaques: Potential for zoonotic disease. Emerg. Infect. Dis. 2003, 9, 246–250. [Google Scholar] [CrossRef]
- Shkoporov, A.N.; Clooney, A.G.; Sutton, T.D.S.; Ryan, F.J.; Daly, K.M.; Nolan, J.A.; McDonnell, S.A.; Khokhlova, E.V.; Draper, L.A.; Forde, A.; et al. The Human Gut Virome Is Highly Diverse, Stable, and Individual Specific. Cell Host Microbe 2019, 26, 527–541.e525. [Google Scholar] [CrossRef]
- Autio, A.; Kettunen, J.; Nevalainen, T.; Kimura, B.; Hurme, M. Herpesviruses and their genetic diversity in the blood virome of healthy individuals: Effect of aging. Immun. Ageing 2022, 19, 15. [Google Scholar] [CrossRef]













{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Number of Participants | 40 |
|---|---|
| Sex n (%) | |
| Male | 25 (62.5) |
| Female | 15 (37.5) |
| Age | |
| Average in years (Standard deviation) | 8.275 (4.49) |
| Age 1–5 | n = 10 |
| Age 6–10 | n = 19 |
| Age 11–17 (Age > 10) | n = 11 |
| Nationalities n (%) | |
| Algerian | 2 (5) |
| Egyptian | 13 (32.5) |
| Indian | 11 (27.5) |
| Qatari | 1 (2.5) |
| Spanish | 5 (12.5) |
| Sri Lankan | 3 (7.5) |
| Syrian | 2 (5) |
| British | 3 (7.5) |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2022 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (https://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wehedy, E.; Murugesan, S.; George, C.R.; Shatat, I.F.; Al Khodor, S. Characterization of the Urinary Metagenome and Virome in Healthy Children. Biomedicines 2022, 10, 2412. https://doi.org/10.3390/biomedicines10102412
Wehedy E, Murugesan S, George CR, Shatat IF, Al Khodor S. Characterization of the Urinary Metagenome and Virome in Healthy Children. Biomedicines. 2022; 10(10):2412. https://doi.org/10.3390/biomedicines10102412
Chicago/Turabian StyleWehedy, Eman, Selvasankar Murugesan, Chinnu Reeba George, Ibrahim F. Shatat, and Souhaila Al Khodor. 2022. "Characterization of the Urinary Metagenome and Virome in Healthy Children" Biomedicines 10, no. 10: 2412. https://doi.org/10.3390/biomedicines10102412
APA StyleWehedy, E., Murugesan, S., George, C. R., Shatat, I. F., & Al Khodor, S. (2022). Characterization of the Urinary Metagenome and Virome in Healthy Children. Biomedicines, 10(10), 2412. https://doi.org/10.3390/biomedicines10102412