Reelin Mediates Hippocampal Cajal-Retzius Cell Positioning and Infrapyramidal Blade Morphogenesis

 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Mice
2.2. Immunohistochemistry and Histology
2.3. Antibody Characterization
2.4. BrdU Incorporation Assay
2.5. Statistical Analysis
3. Results
3.1. Cajal-Retzius Cells Are Absent in the IPB
3.2. Neurogenic Cluster Does Not Form at the Fimbriodentate Junction
3.3. Distribution of Tbr2-Positive Intermediate Progenitor Cells Is Abnormal
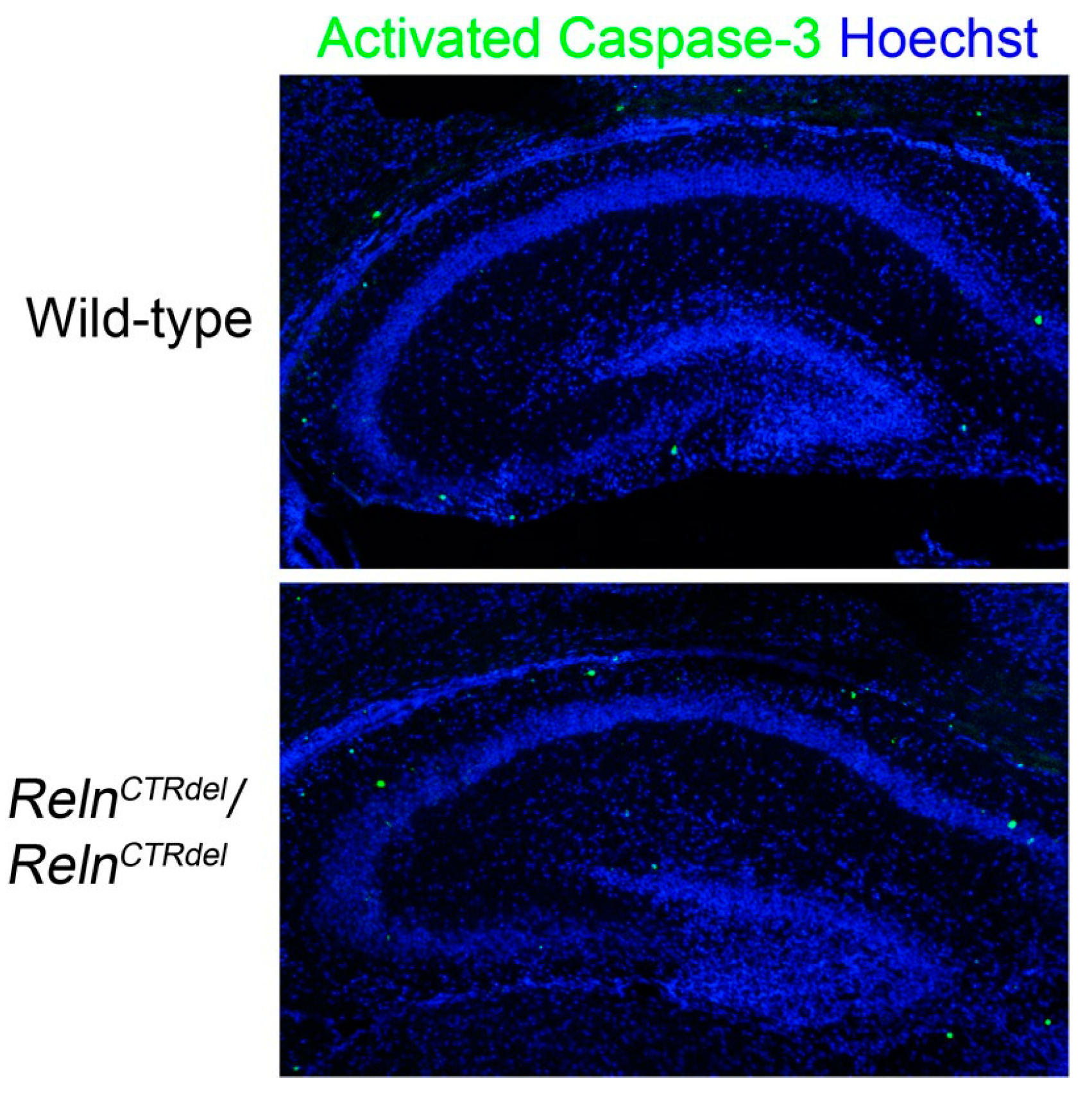
3.4. Granule Neuron Precursor Distribution in the Hilus Is Abnormal
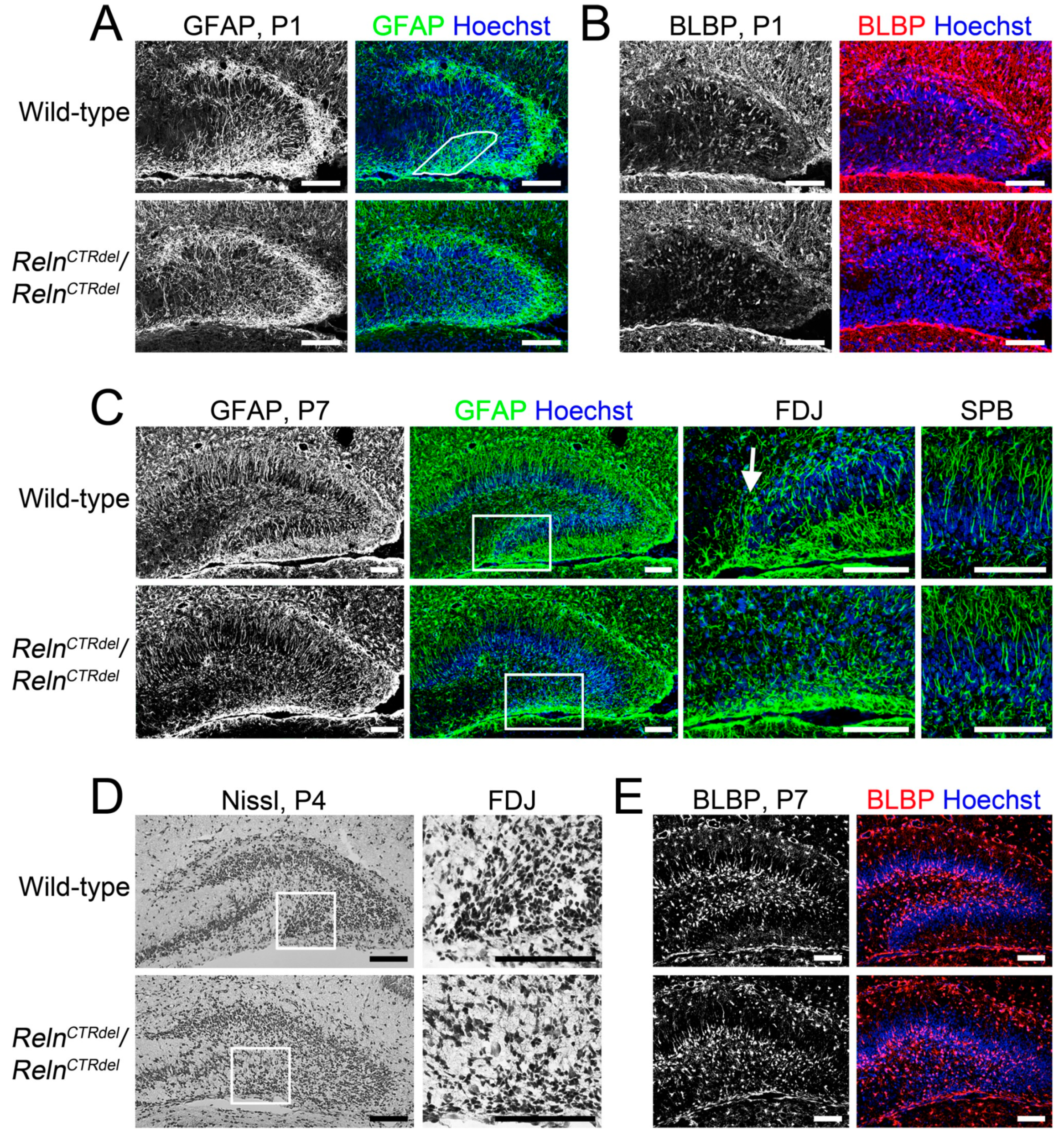
3.5. Radial Glial Scaffold Defect Is Specific to the IPB
4. Discussion
4.1. A Reln Mutation Can Cause Abnormal Positioning of Cajal-Retzius Cells
4.2. Defects in the Secondary Glial Scaffold and Neurogenic Niche at the Fimbriodentate Junction Result in Truncation of the Infrapyramidal Blade
4.3. Does Reelin Act as a Repulsive Signal at the Fimbriodentate Junction?
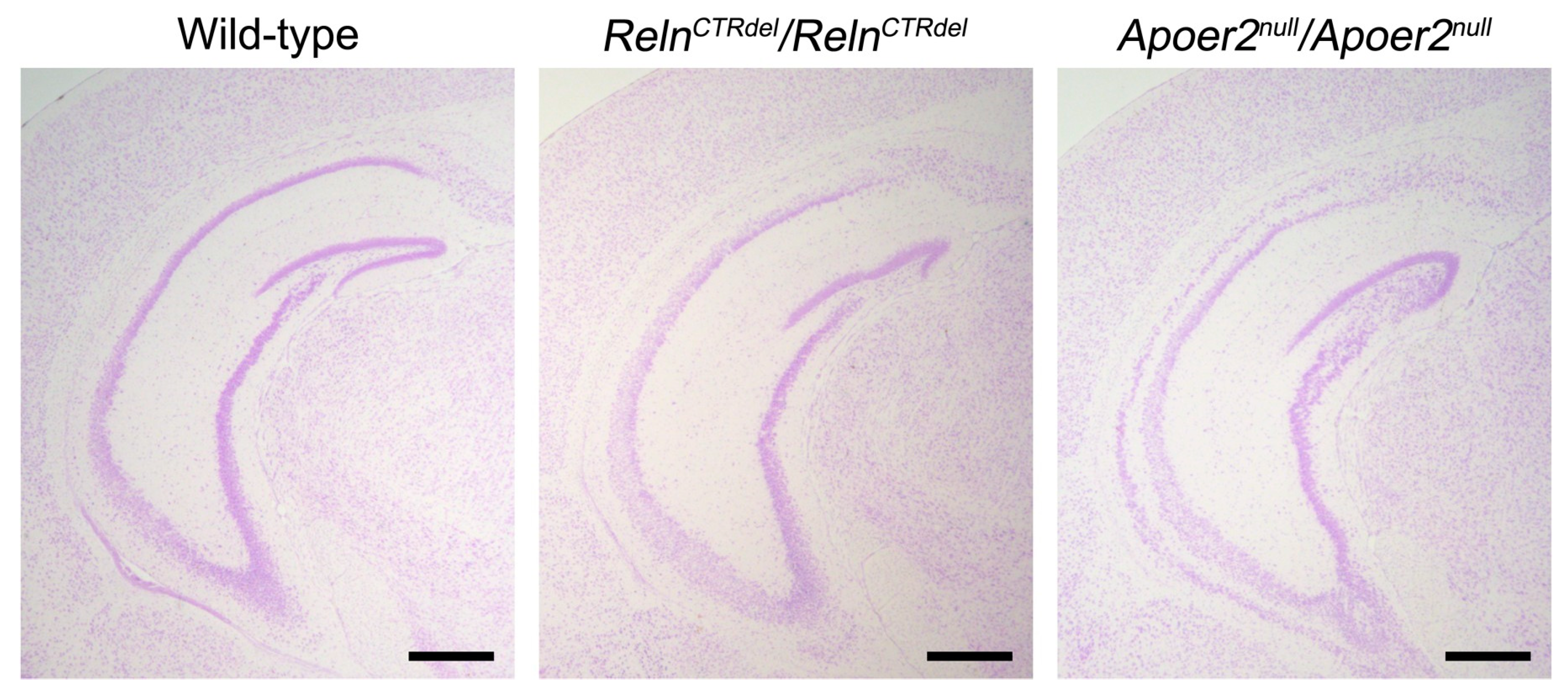
4.4. Can Differential Receptor Binding Account for the IPB Defect?
4.5. Potential Clinical Relevance
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Appendix A



References
- Förster, E.; Zhao, S.; Frotscher, M. Laminating the hippocampus. Nat. Rev. Neurosci. 2006, 7, 259–268. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.; Bayer, S.A. Migration and distribution of two populations of hippocampal granule cell precursors during the perinatal and postnatal periods. J. Comp. Neurol. 1990, 301, 365–381. [Google Scholar] [CrossRef] [PubMed]
- Li, G.; Pleasure, S.J. Morphogenesis of the Dentate Gyrus: What We Are Learning from Mouse Mutants. Dev. Neurosci. 2005, 27, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Altman, J.; Bayer, S.A. Mosaic organization of the hippocampal neuroepithelium and the multiple germinal sources of dentate granule cells. J. Comp. Neurol. 1990, 301, 325–342. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.D.; Garcia, A.J.; Elsen, G.E.; Nelson, B.R.; Mussar, K.E.; Reiner, S.L.; Ramirez, J.-M.; Hevner, R.F. Tbr2 Expression in Cajal-Retzius Cells and Intermediate Neuronal Progenitors Is Required for Morphogenesis of the Dentate Gyrus. J. Neurosci. 2013, 33, 4165–4180. [Google Scholar] [CrossRef]
- Hayashi, K.; Kubo, K.-I.; Kitazawa, A.; Nakajima, K. Cellular dynamics of neuronal migration in the hippocampus. Front. Mol. Neurosci. 2015, 9, 135. [Google Scholar] [CrossRef]
- Li, G.; Kataoka, H.; Coughlin, S.R.; Pleasure, S.J. Identification of a transient subpial neurogenic zone in the developing dentate gyrus and its regulation by Cxcl12 and reelin signaling. Development 2009, 136, 327–335. [Google Scholar] [CrossRef]
- Nicola, Z.; Fabel, K.; Kempermann, G. Development of the adult neurogenic niche in the hippocampus of mice. Front. Neuroanat. 2015, 9, 53. [Google Scholar] [CrossRef]
- Stanfield, B.B.; Cowan, W.M. The morphology of the hippocampus and dentate gyrus in normal and reeler mice. J. Comp. Neurol. 1979, 185, 393–422. [Google Scholar] [CrossRef]
- Frotscher, M.; Haas, C.A.; Förster, E. Reelin controls granule cell migration in the dentate gyrus by acting on the radial glial scaffold. Cereb. Cortex 2003, 13, 634–640. [Google Scholar] [CrossRef]
- Zhao, S.; Frotscher, M. Go or Stop? Divergent Roles of Reelin in Radial Neuronal Migration. Neuroscientist 2010, 16, 421–434. [Google Scholar] [CrossRef]
- Zhao, S.; Chai, X.; Förster, E.; Frotscher, M. Reelin is a positional signal for the lamination of dentate granule cells. Development 2004, 131, 5117–5125. [Google Scholar] [CrossRef] [PubMed]
- Förster, E.; Tielsch, A.; Saum, B.; Weiss, K.H.; Johanssen, C.; Graus-Porta, D.; Müller, U.; Frotscher, M. Reelin, Disabled 1, and 1 integrins are required for the formation of the radial glial scaffold in the hippocampus. Proc. Natl. Acad. Sci. USA 2002, 99, 13178–13183. [Google Scholar] [CrossRef] [PubMed]
- Weiss, K.H.; Johanssen, C.; Tielsch, A.; Herz, J.; Deller, T.; Frotscher, M.; Förster, E. Malformation of the radial glial scaffold in the dentate gyrus of reeler mice, scrambler mice, and ApoER2/VLDLR-deficient mice. J. Comp. Neurol. 2003, 460, 56–65. [Google Scholar] [CrossRef]
- Brunne, B.; Franco, S.; Bouché, E.; Herz, J.; Howell, B.W.; Pahle, J.; Müller, U.; May, P.; Frotscher, M.; Bock, H.H. Role of the postnatal radial glial scaffold for the development of the dentate gyrus as revealed by Reelin signaling mutant mice. Glia 2013, 61, 1347–1363. [Google Scholar] [CrossRef] [PubMed]
- Tian, C.; Gong, Y.; Yang, Y.; Shen, W.; Wang, K.; Liu, J.; Xu, B.; Zhao, J.; Zhao, C. Foxg1 Has an Essential Role in Postnatal Development of the Dentate Gyrus. J. Neurosci. 2012, 32, 2931–2949. [Google Scholar] [CrossRef]
- Yang, A.; Walker, N.; Bronson, R.; Kaghad, M.; Oosterwegel, M.; Bonnin, J.; Vagner, C.; Bonnet, H.; Dikkes, P.; Sharpe, A.; et al. p73-deficient mice have neurological, pheromonal and inflammatory defects but lack spontaneous tumours. Nature 2000, 404, 99–103. [Google Scholar] [CrossRef]
- Deller, T.; Drakew, A.; Frotscher, M. Different Primary Target Cells Are Important for Fiber Lamination in the Fascia Dentata: A Lesson from Reeler Mutant Mice. Exp. Neurol. 1999, 156, 239–253. [Google Scholar] [CrossRef]
- Ha, S.; Stottmann, R.W.; Furley, A.J.; Beier, D.R. A Forward Genetic Screen in Mice Identifies Mutants with Abnormal Cortical Patterning. Cereb. Cortex 2015, 25, 167–179. [Google Scholar] [CrossRef]
- De Bergeyck, V.; Nakajima, K.; De Rouvroit, C.L.; Naerhuyzen, B.; Goffinet, A.; Miyata, T.; Ogawa, M.; Mikoshiba, K. A truncated Reelin protein is produced but not secreted in the ‘Orleans’ reeler mutation (Relnrl-Orl). Mol. Brain Res. 1997, 50, 85–90. [Google Scholar] [CrossRef]
- D’Arcangelo, G.; Nakajima, K.; Miyata, T.; Ogawa, M.; Mikoshiba, K.; Curran, T. Reelin Is a Secreted Glycoprotein Recognized by the CR-50 Monoclonal Antibody. J. Neurosci. 1997, 17, 23–31. [Google Scholar] [CrossRef] [PubMed]
- Kohno, T.; Honda, T.; Kubo, K.-I.; Nakano, Y.; Tsuchiya, A.; Murakami, T.; Banno, H.; Nakajima, K.; Hattori, M. Importance of Reelin C-Terminal Region in the Development and Maintenance of the Postnatal Cerebral Cortex and Its Regulation by Specific Proteolysis. J. Neurosci. 2015, 35, 4776–4787. [Google Scholar] [CrossRef] [PubMed]
- Nakano, Y.; Kohno, T.; Hibi, T.; Kohno, S.; Baba, A.; Mikoshiba, K.; Nakajima, K.; Hattori, M. The Extremely Conserved C-terminal Region of Reelin Is Not Necessary for Secretion but Is Required for Efficient Activation of Downstream Signaling. J. Boil. Chem. 2007, 282, 20544–20552. [Google Scholar] [CrossRef] [PubMed]
- Sakai, K.; Shoji, H.; Kohno, T.; Miyakawa, T.; Hattori, M. Mice that lack the C-terminal region of Reelin exhibit behavioral abnormalities related to neuropsychiatric disorders. Sci. Rep. 2016, 6, 28636. [Google Scholar] [CrossRef]
- Nakamura, K.; Beppu, M.; Sakai, K.; Yagyu, H.; Matsumaru, S.; Kohno, T.; Hattori, M. The C-terminal region of Reelin is necessary for proper positioning of a subset of Purkinje cells in the postnatal cerebellum. Neuroscientist 2016, 336, 20–29. [Google Scholar] [CrossRef]
- Ha, S.; Tripathi, P.P.; Mihalas, A.B.; Hevner, R.F.; Beier, D. C-Terminal Region Truncation of RELN Disrupts an Interaction with VLDLR, Causing Abnormal Development of the Cerebral Cortex and Hippocampus. J. Neurosci. 2017, 37, 960–971. [Google Scholar] [CrossRef]
- Anstötz, M.; Karsak, M.; Rune, G.M. Integrity of Cajal-Retzius cells in the reeler-mouse hippocampus. Hippocampus 2019, 29, 550–565. [Google Scholar] [CrossRef]
- Derer, P. Comparative localization of Cajal-Retzius cells in the neocortex of normal and reeler mutant mice fetuses. Neurosci. Lett. 1985, 54, 1–6. [Google Scholar] [CrossRef]
- De Bergeyck, V.; Naerhuyzen, B.; Goffinet, A.M.; De Rouvroit, C.L. A panel of monoclonal antibodies against reelin, the extracellular matrix protein defective in reeler mutant mice. J. Neurosci. Methods 1998, 82, 17–24. [Google Scholar] [CrossRef]
- Pesold, C.; Impagnatiello, F.; Pisu, M.G.; Uzunov, D.P.; Costa, E.; Guidotti, A.; Caruncho, H.J. Reelin is preferentially expressed in neurons synthesizing gamma-aminobutyric acid in cortex and hippocampus of adult rats. Proc. Natl. Acad. Sci. USA 1998, 95, 3221–3226. [Google Scholar] [CrossRef]
- Gu, X.; Liu, B.; Wu, X.; Yan, Y.; Zhang, Y.; Wei, Y.; Pleasure, S.J.; Zhao, C. Inducible Genetic Lineage Tracing of Cortical Hem Derived Cajal-Retzius Cells Reveals Novel Properties. PLoS ONE 2011, 6, e28653. [Google Scholar] [CrossRef]
- Hodge, R.D.; Nelson, B.R.; Kahoud, R.J.; Yang, R.; Mussar, K.E.; Reiner, S.L.; Hevner, R.F. Tbr2 is essential for hippocampal lineage progression from neural stem cells to intermediate progenitors and neurons. J. Neurosci. 2012, 32, 6275–6287. [Google Scholar] [CrossRef] [PubMed]
- Brunne, B.; Zhao, S.; Derouiche, A.; Herz, J.; May, P.; Frotscher, M.; Bock, H.H. Origin, maturation, and astroglial transformation of secondary radial glial cells in the developing dentate gyrus. Glia 2010, 58, 1553–1569. [Google Scholar] [CrossRef] [PubMed]
- Pedroni, A.; Minh, D.D.; Mallamaci, A.; Cherubini, E. Electrophysiological characterization of granule cells in the dentate gyrus immediately after birth. Front. Cell. Neurosci. 2014, 8, 44. [Google Scholar] [CrossRef] [PubMed]
- Hodge, R.D.; Kowalczyk, T.D.; Wolf, S.; Encinas, J.M.; Rippey, C.; Enikolopov, G.; Kempermann, G.; Hevner, R.F. Intermediate Progenitors in Adult Hippocampal Neurogenesis: Tbr2 Expression and Coordinate Regulation of Neuronal Output. J. Neurosci. 2008, 28, 3707–3717. [Google Scholar] [CrossRef] [PubMed]
- Stanfield, B.B.; Cowan, W.M. The development of the hippocampus and dentate gyrus in normal and reeler mice. J. Comp. Neurol. 1979, 185, 423–459. [Google Scholar] [CrossRef] [PubMed]
- Rickmann, M.; Amaral, D.G.; Cowan, W.M. Organization of radial glial cells during the development of the rat dentate gyrus. J. Comp. Neurol. 1987, 264, 449–479. [Google Scholar] [CrossRef] [PubMed]
- Borrell, V.; Pujadas, L.; Simó, S.; Durà, D.; Solé, M.; Cooper, J.A.; Del Río, J.A.; Soriano, E. Reelin and mDab1 regulate the development of hippocampal connections. Mol. Cell. Neurosci. 2007, 36, 158–173. [Google Scholar] [CrossRef] [PubMed]
- Borrell, V.; Ruiz, M.; Del Río, J.A.; Soriano, E. Development of Commissural Connections in the Hippocampus of Reeler Mice: Evidence of an Inhibitory Influence of Cajal–Retzius Cells. Exp. Neurol. 1999, 156, 268–282. [Google Scholar] [CrossRef]
- Borrell, V.; Del Río, J.A.; Alcantara, S.; Dérer, M.; Martínez, A.; D’Arcangelo, G.; Nakajima, K.; Mikoshiba, K.; Derer, P.; Curran, T.; et al. Reelin Regulates the Development and Synaptogenesis of the Layer-Specific Entorhino-Hippocampal Connections. J. Neurosci. 1999, 19, 1345–1358. [Google Scholar] [CrossRef]
- Del Río, J.A.; Heimrich, B.; Borrell, V.; Förster, E.; Drakew, A.; Alcantara, S.; Nakajima, K.; Miyata, T.; Ogawa, M.; Mikoshiba, K.; et al. A role for Cajal–Retzius cells and reelin in the development of hippocampal connections. Nature 1997, 385, 70–74. [Google Scholar] [CrossRef] [PubMed]
- Supèr, H.; Martínez, A.; Del Río, J.A.; Soriano, E. Involvement of Distinct Pioneer Neurons in the Formation of Layer-Specific Connections in the Hippocampus. J. Neurosci. 1998, 18, 4616–4626. [Google Scholar] [CrossRef] [PubMed]
- Barber, M.; Pierani, A. Tangential migration of glutamatergic neurons and cortical patterning during development: Lessons from Cajal-Retzius cells. Dev. Neurobiol. 2016, 76, 847–881. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Perez-Garcia, C.G.; Abraham, H.; Caput, D. Expression of p73 and Reelin in the Developing Human Cortex. J. Neurosci. 2002, 22, 4973–4986. [Google Scholar] [CrossRef] [PubMed]
- Grove, E.A.; Tole, S.; Limon, J.; Yip, L.; Ragsdale, C.W. The hem of the embryonic cerebral cortex is defined by the expression of multiple Wnt genes and is compromised in Gli3-deficient mice. Development 1998, 125, 2315–2325. [Google Scholar]
- Yoshida, M.; Assimacopoulos, S.; Grove, E.A.; Jones, K.R. Massive loss of Cajal-Retzius cells does not disrupt neocortical layer order. Development 2006, 133, 537–545. [Google Scholar] [CrossRef]
- Takiguchi-Hayashi, K.; Sekiguchi, M.; Ashigaki, S.; Takamatsu, M.; Hasegawa, H.; Suzuki-Migishima, R.; Yokoyama, M.; Nakanishi, S.; Tanabe, Y. Generation of Reelin-Positive Marginal Zone Cells from the Caudomedial Wall of Telencephalic Vesicles. J. Neurosci. 2004, 24, 2286–2295. [Google Scholar] [CrossRef]
- Lu, M.; Grove, E.A.; Miller, R.J. Abnormal development of the hippocampal dentate gyrus in mice lacking the CXCR4 chemokine receptor. Proc. Natl. Acad. Sci. USA 2002, 99, 7090–7095. [Google Scholar] [CrossRef]
- Bagri, A.; Gurney, T.; He, X.; Zou, Y.-R.; Littman, D.R.; Tessier-Lavigne, M.; Pleasure, S.J. The chemokine SDF1 regulates migration of dentate granule cells. Development 2002, 129, 4249–4260. [Google Scholar]
- Parisot, J.; Flore, G.; Bertacchi, M.; Studer, M. COUP-TFI mitotically regulates production and migration of dentate granule cells and modulates hippocampal Cxcr4 expression. Development 2017, 144, 2045–2058. [Google Scholar] [CrossRef]
- Villar-Cerviño, V.; Molano-Mazón, M.; Catchpole, T.; Valdeolmillos, M.; Henkemeyer, M.; Martínez, L.; Borrell, V.; Marin, O. Contact repulsion controls the dispersion and final distribution of Cajal-Retzius cells. Neuron 2013, 77, 457–471. [Google Scholar] [CrossRef] [PubMed]
- Sibbe, M.; Förster, E.; Basak, O.; Taylor, V.; Frotscher, M. Reelin and Notch1 Cooperate in the Development of the Dentate Gyrus. J. Neurosci. 2009, 29, 8578–8585. [Google Scholar] [CrossRef] [PubMed]
- Abraham, H.; Perez-Garcia, C.G.; Meyer, G. p73 and Reelin in Cajal-Retzius Cells of the Developing Human Hippocampal Formation. Cereb. Cortex 2004, 14, 484–495. [Google Scholar] [CrossRef] [PubMed]
- Meyer, G.; Socorro, A.C.; Perez-Garcia, C.G.; Millan, L.M.; Walker, N.; Caput, D. Developmental Roles of p73 in Cajal-Retzius Cells and Cortical Patterning. J. Neurosci. 2004, 24, 9878–9887. [Google Scholar] [CrossRef] [PubMed]
- Dulabon, L.; Olson, E.C.; Taglienti, M.G.; Eisenhuth, S.; McGrath, B.; Walsh, C.A.; Kreidberg, J.A.; Anton, E.S. Reelin binds α3β1 integrin and inhibits neuronal migration. Neuron 2000, 27, 33–44. [Google Scholar] [CrossRef]
- Frotscher, M. Dual role of Cajal-Retzius cells and reelin in cortical development. Cell Tissue Res. 1997, 290, 315–322. [Google Scholar] [CrossRef]
- Hack, I.; Hellwig, S.; Junghans, D.; Brunne, B.; Bock, H.H.; Zhao, S.; Frotscher, M. Divergent roles of ApoER2 and Vldlr in the migration of cortical neurons. Development 2007, 134, 3883–3891. [Google Scholar] [CrossRef]
- Herrick, T.M.; Cooper, J.A. A hypomorphic allele of dab1 reveals regional differences in reelin-Dab1 signaling during brain development. Development 2002, 129, 787–796. [Google Scholar]
- Ogawa, M.; Miyata, T.; Nakajima, K.; Yagyu, K.; Seike, M.; Ikenaka, K.; Yamamoto, H.; Mikoshibat, K. The reeler gene-associated antigen on cajal-retzius neurons is a crucial molecule for laminar organization of cortical neurons. Neuron 1995, 14, 899–912. [Google Scholar] [CrossRef]
- Schiffmann, S.N.; Bernier, B.; Goffinet, A.M. Reelin mRNA expression during mouse brain development. Eur. J. Neurosci. 1997, 9, 1055–1071. [Google Scholar] [CrossRef]
- Sheppard, A.; Pearlman, A. Abnormal reorganization of preplate neurons and their associated extracellular matrix: An early manifestation of altered neocortical development in the reeler mutant mouse. J. Comp. Neurol. 1997, 378, 173–179. [Google Scholar] [CrossRef]
- Olson, E.C.; Kim, S.; Walsh, C.A. Impaired Neuronal Positioning and Dendritogenesis in the Neocortex after Cell-Autonomous Dab1 Suppression. J. Neurosci. 2006, 26, 1767–1775. [Google Scholar] [CrossRef] [PubMed]
- Fish, K.N.; Krucker, T. Functional consequences of hippocampal neuronal ectopia in the apolipoprotein E receptor-2 knockout mouse. Neurobiol. Dis. 2008, 32, 391–401. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Dazzo, E.; Fanciulli, M.; Serioli, E.; Minervini, G.; Pulitano, P.; Binelli, S.; Di Bonaventura, C.; Luisi, C.; Pasini, E.; Striano, S.; et al. Heterozygous Reelin Mutations Cause Autosomal-Dominant Lateral Temporal Epilepsy. Am. J. Hum. Genet. 2015, 96, 992–1000. [Google Scholar] [CrossRef]
- Michelucci, R.; Pulitano, P.; Di Bonaventura, C.; Binelli, S.; Luisi, C.; Pasini, E.; Striano, S.; Striano, P.; Coppola, G.; La Neve, A.; et al. The clinical phenotype of autosomal dominant lateral temporal lobe epilepsy related to reelin mutations. Epilepsy Behav. 2017, 68, 103–107. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Antigen | Description of Immunogen | Source, Host Species, Clonality, Catalog No., Clone No., RRID | Stock Concentration, Dilution Used |
|---|---|---|---|
| Reelin | Recombinant reelin amino acids 164–496 | Millipore, mouse, monoclonal, Cat# MAB5364, clone G10, RRID:AB_2179313 | 1 mg/mL, 1:1000 |
| p73 | Recombinant human p73α amino acids 1–80 | Santa Cruz, rabbit, polyclonal, Cat# sc-7957, clone H-79, RRID:AB_2207314 | 200 µg/mL, 1:200 |
| BrdU | BrdU | Accurate, rat, monoclonal, Cat# OBT0030, clone BU1/75 (ICR1), RRID:AB_2313756 | 0.5 mg/mL, 1:300 |
| Ki67 | Synthetic peptide derived from within amino acids 2300–2400 of human Ki67 | Vector Lab, rabbit, monoclonal, Cat# VP-Rm04, clone SP6, RRID:AB_2336545 | Unknown, 1:300 |
| Tbr2 | Mouse Tbr2/Eomes | eBioscience, rat, monoclonal, Cat# 14-4875-82, RRID:AB_11042577 | 0.5 mg/mL, 1:300 |
| GFAP | GFAP isolated from cow spinal cord | Dako, rabbit, polyclonal, Cat# Z0334, RRID:AB_10013382 | 2.9 mg/mL, 1:500 |
| BLBP | Synthetic peptide conjugated to KLH derived from within amino acids 1–100 of mouse BLBP | Abcam, rabbit, polyclonal, Cat# ab32423, RRID:AB_880078 | Unknown, 1:1000 |
| NeuroD | Peptide mapping at the N-terminus of mouse NeuroD | Santa Cruz, goat, polyclonal, Cat# sc-1084, clone N-19, RRID:AB_630922 | 100 μg/mL, 1:400 |
| Prox1 | Synthetic peptide EIFKSPNCLQELLHE, corresponding to amino acids 723–737 of mouse Prox1 | Abcam, rabbit, polyclonal, Cat# ab37128, RRID:AB_882189 | Unknown, 1:1000 |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ha, S.; Tripathi, P.P.; Daza, R.A.; Hevner, R.F.; Beier, D.R. Reelin Mediates Hippocampal Cajal-Retzius Cell Positioning and Infrapyramidal Blade Morphogenesis. J. Dev. Biol. 2020, 8, 20. https://doi.org/10.3390/jdb8030020
Ha S, Tripathi PP, Daza RA, Hevner RF, Beier DR. Reelin Mediates Hippocampal Cajal-Retzius Cell Positioning and Infrapyramidal Blade Morphogenesis. Journal of Developmental Biology. 2020; 8(3):20. https://doi.org/10.3390/jdb8030020
Chicago/Turabian StyleHa, Seungshin, Prem P. Tripathi, Ray A. Daza, Robert F. Hevner, and David R. Beier. 2020. "Reelin Mediates Hippocampal Cajal-Retzius Cell Positioning and Infrapyramidal Blade Morphogenesis" Journal of Developmental Biology 8, no. 3: 20. https://doi.org/10.3390/jdb8030020
APA StyleHa, S., Tripathi, P. P., Daza, R. A., Hevner, R. F., & Beier, D. R. (2020). Reelin Mediates Hippocampal Cajal-Retzius Cell Positioning and Infrapyramidal Blade Morphogenesis. Journal of Developmental Biology, 8(3), 20. https://doi.org/10.3390/jdb8030020