Transcriptional Control of Cell Lineage Development in Epicardium-Derived Cells


Abstract
:1. Epicardium-Derived Cells (EPDCs) in Heart Development and Disease
2. Overview of Epicardial Formation and Cell Lineage Diversification
3. Transcriptional Regulation of Epicardial EMT and EPDC Lineage Specification

3.1. Wt1

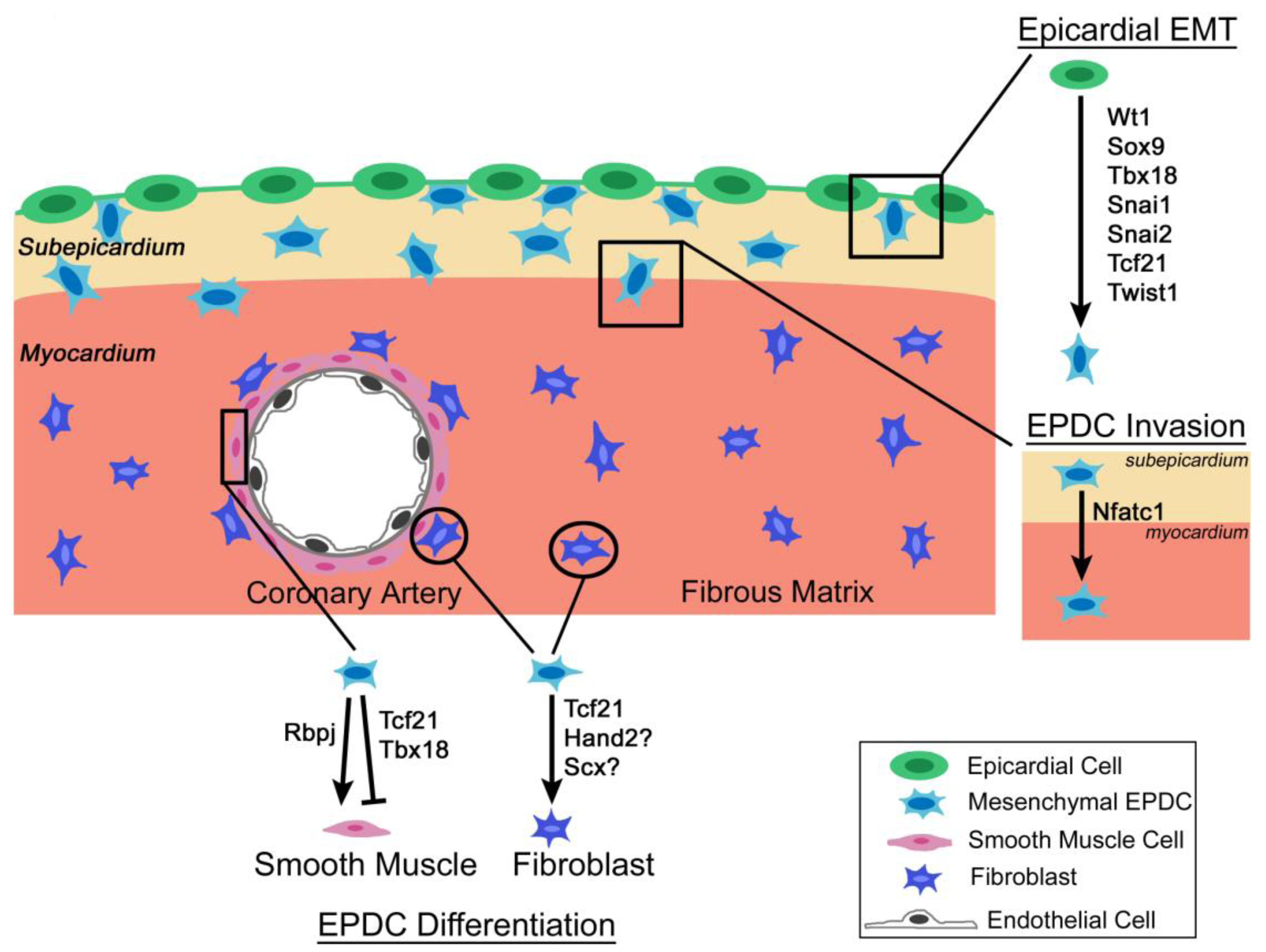{kind=link}
{kind=link}
| Gene | Loss-of-function cardiac phenotype a | Known downstream targets expressed in EPDCs | References |
|---|---|---|---|
| Wt1 | Ventricular non-compaction; impaired epicardial EMT; impaired coronary plexus formation; pericardial hemorrhaging; die by E13.5 | Itga4, Nestin, TrkB, Coronin1B, Raldh2, Snai1, Snai2 | [11,12,37,38,39,40,41,42,43] |
| Tbx18 | Caval vein defects; sinus horn myocardial hypoplasia; neonatal lethality | Snai2 | [40,74,75] |
| Tcf21 | Aberrant smooth muscle differentiation; loss of cardiac fibroblasts; pericardial hemorrhaging; neonatal lethality | None identified | [8,13,57] |
| Nfatc1 | bReduced cardiac fibrous matrix with decreased coronary vessel penetration; neonatal lethality | Ctsk | [9,87,88] |
| Snai1 | b,cPhenotypically normal and viable | E-cadherin, Mmp15 | [92,93,96,97] |
| Snai2 | Phenotypically normal and viable | None identified | [40] |
| Sox9 | Hypoplastic endocardial cushions. Embryonic lethality at E11.5-E12 due to congestive heart failure. | None identified | [102,104,108] |
| Scleraxis | Thickened valves; viable | Col1a2 | [106,107] |
| C/EBP | dImproved cardiac function after ischemia/reperfusion injury | Raldh2, Wt1 | [32] |
| Hand2 | eEpicardial blistering; abnormal coronary vessel development; loss of cardiac fibroblasts; persistent truncus arteriosus. Embryonic lethality by E14.5. | Pdgfra | [101] |
| Twist1 | Abnormal outflow tract endocardial cushion mesenchyme. Embryonic lethality by E11.5. | Tbx20, Snai2 | [30,76,100] |
3.2. Tcf21

3.3. Tbx18
3.4. Nfatc1
3.5. Snai1 and Snal2
3.6. Twist1 and Hand2
3.7. Scleraxis and Sox9
3.8. C/EBP
4. Transcriptional Regulation of EPDC Lineages in Adult Cardiac Regeneration, Injury, and Fibrosis
5. Conclusions and Future Perspectives
Acknowledgments
Conflict of Interest
References
- Gittenberger-de Groot, A.C.; Vrancken Peeters, M.P.; Mentink, M.M.; Gourdie, R.G.; Poelmann, R.E. Epicardium-derived cells contribute a novel population to the myocardial wall and the atrioventricular cushions. Circ. Res. 1998, 82, 1043–1052. [Google Scholar] [CrossRef]
- Mikawa, T.; Gourdie, R.G. Pericardial mesoderm generates a population of coronary smooth muscle cells migrating into the heart along with ingrowth of the epicardial organ. Dev. Biol. 1996, 174, 221–232. [Google Scholar] [CrossRef]
- Cai, C.L.; Martin, J.C.; Sun, Y.; Cui, L.; Wang, L.; Ouyang, K.; Yang, L.; Bu, L.; Liang, X.; Zhang, X.; et al. A myocardial lineage derives from Tbx18 epicardial cells. Nature 2008, 454, 104–108. [Google Scholar] [CrossRef]
- Gittenberger-de Groot, A.C.; Winter, E.M.; Poelmann, R.E. Epicardium-derived cells (EPDCs) in development, cardiac disease and repair of ischemia. J. Cell. Mol. Med. 2010, 14, 1056–1060. [Google Scholar]
- Red-Horse, K.; Ueno, H.; Weissman, I.L.; Krasnow, M.A. Coronary arteries form by developmental reprogramming of venous cells. Nature 2010, 464, 549–553. [Google Scholar] [CrossRef]
- Katz, T.C.; Singh, M.K.; Degenhardt, K.; Rivera-Feliciano, J.; Johnson, R.L.; Epstein, J.A.; Tabin, C.J. Distinct compartments of the proepicardial organ give rise to coronary vascular endothelial cells. Dev. Cell 2012, 22, 639–650. [Google Scholar] [CrossRef]
- Zhou, B.; Ma, Q.; Rajagopal, S.; Wu, S.M.; Domian, I.; Rivera-Feliciano, J.; Jiang, D.; von Gise, A.; Ikeda, S.; Chien, K.R.; et al. Epicardial progenitors contribute to the cardiomyocyte lineage in the developing heart. Nature 2008, 454, 109–113. [Google Scholar] [CrossRef]
- Braitsch, C.M.; Combs, M.D.; Quaggin, S.E.; Yutzey, K.E. Pod1/Tcf21 is regulated by retinoic acid signaling and inhibits differentiation of epicardium-derived cells into smooth muscle in the developing heart. Dev. Biol. 2012, 368, 345–357. [Google Scholar] [CrossRef]
- Combs, M.D.; Braitsch, C.M.; Lange, A.W.; James, J.F.; Yutzey, K.E. Nfatc1 promotes epicardium-derived cell invasion into myocardium. Development 2011, 138, 1747–1757. [Google Scholar] [CrossRef]
- Greulich, F.; Farin, H.F.; Schuster-Gossler, K.; Kispert, A. Tbx18 function in epicardial development. Cardiovasc. Res. 2012, 96, 476–483. [Google Scholar] [CrossRef]
- Kirschner, K.M.; Wagner, N.; Wagner, K.D.; Wellmann, S.; Scholz, H. The Wilms tumor suppressor Wt1 promotes cell adhesion through transcriptional activation of the alpha4integrin gene. J. Biol. Chem. 2006, 281, 31930–31939. [Google Scholar] [CrossRef]
- von Gise, A.; Zhou, B.; Honor, L.B.; Ma, Q.; Petryk, A.; Pu, W.T. Wt1 regulates epicardial epithelial to mesenchymal transition through beta-catenin and retinoic acid signaling pathways. Dev. Biol. 2011, 356, 421–431. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Huang, G.; Eskiocak, B.; Goetsch, S.; Sung, C.Y.; Banfi, S.; Sauer, M.F.; Olsen, G.S.; Duffield, J.S.; et al. The bHLH transcription factor Tcf21 is required for lineage-specific EMT of cardiac fibroblast progenitors. Development 2012, 139, 2139–2149. [Google Scholar] [CrossRef]
- Kayalar, N.; Burkhart, H.M.; Dearani, J.A.; Cetta, F.; Schaff, H.V. Congenital coronary anomalies and surgical treatment. Congenit. Heart Dis. 2009, 4, 239–251. [Google Scholar] [CrossRef]
- Smart, N.; Bollini, S.; Dube, K.N.; Vieira, J.M.; Zhou, B.; Davidson, S.; Yellon, D.; Riegler, J.; Price, A.N.; Lythgoe, M.F.; et al. De novo cardiomyocytes from within the activated adult heart after injury. Nature 2011, 474, 640–644. [Google Scholar] [CrossRef]
- Limana, F.; Bertolami, C.; Mangoni, A.; Di Carlo, A.; Avitabile, D.; Mocini, D.; Iannelli, P.; De- Mori, R.; Marchetti, C.; Pozzoli, O.; et al. Myocardial infarction induces embryonic reprogramming of epicardial c-kit(+) cells: Role of the pericardial fluid. J. Mol. Cell. Cardiol. 2010, 48, 609–618. [Google Scholar] [CrossRef]
- Smart, N.; Riley, P.R. The epicardium as a candidate for heart regeneration. Future Cardiol. 2012, 8, 53–69. [Google Scholar] [CrossRef]
- Zhou, B.; Honor, L.B.; He, H.; Ma, Q.; Oh, J.H.; Butterfield, C.; Lin, R.Z.; Melero-Martin, J.M.; Dolmatova, E.; Duffy, H.S.; et al. Adult mouse epicardium modulates myocardial injury by secreting paracrine factors. J. Clin. Invest. 2011, 121, 1894–1904. [Google Scholar] [CrossRef]
- Munoz-Chapuli, R.; Macias, D.; Gonzalez-Iriarte, M.; Carmona, R.; Atencia, G.; Perez-Pomares, J.M. [the epicardium and epicardial-derived cells: Multiple functions in cardiac development]. Rev. Esp. Cardiol. 2002, 55, 1070–1082. [Google Scholar] [CrossRef]
- Lie-Venema, H.; van den Akker, N.M.; Bax, N.A.; Winter, E.M.; Maas, S.; Kekarainen, T.; Hoeben, R.C.; de Ruiter, M.C.; Poelmann, R.E.; Gittenberger-de Groot, A.C. Origin, fate, and function of epicardium-derived cells (EPDCs) in normal and abnormal cardiac development. ScientificWorldJournal 2007, 7, 1777–1798. [Google Scholar] [CrossRef]
- Reese, D.E.; Mikawa, T.; Bader, D.M. Development of the coronary vessel system. Circ. Res. 2002, 91, 761–768. [Google Scholar] [CrossRef]
- Lavine, K.J.; Ornitz, D.M. Fibroblast growth factors and hedgehogs: At the heart of the epicardial signaling center. Trends Genet. 2008, 24, 33–40. [Google Scholar] [CrossRef]
- Winter, E.M.; Gittenberger-de Groot, A.C. Epicardium-derived cells in cardiogenesis and cardiac regeneration. Cell. Mol. Life Sci. 2007, 64, 692–703. [Google Scholar] [CrossRef]
- Wessels, A.; van den Hoff, M.J.; Adamo, R.F.; Phelps, A.L.; Lockhart, M.M.; Sauls, K.; Briggs, L.E.; Norris, R.A.; van Wijk, B.; Perez-Pomares, J.M.; et al. Epicardially derived fibroblasts preferentially contribute to the parietal leaflets of the atrioventricular valves in the murine heart. Dev. Biol. 2012, 366, 111–124. [Google Scholar] [CrossRef]
- Kispert, A. No muscle for a damaged heart: Thymosin beta 4 treatment after myocardial infarction does not induce myocardial differentiation of epicardial cells. J. Mol. Cell. Cardiol. 2012, 52, 10–12. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; Carmona, R.; Gonzalez-Iriarte, M.; Atencia, G.; Wessels, A.; Munoz-Chapuli, R. Origin of coronary endothelial cells from epicardial mesothelium in avian embryos. Int. J. Dev. Biol. 2002, 46, 1005–1013. [Google Scholar]
- Wu, B.; Zhang, Z.; Lui, W.; Chen, X.; Wang, Y.; Chamberlain, A.A.; Moreno-Rodriguez, R.A.; Markwald, R.R.; O'Rourke, B.P.; Sharp, D.J.; et al. Endocardial cells form the coronary arteries by angiogenesis through myocardial-endocardial VEGF signaling. Cell 2012, 151, 1083–1096. [Google Scholar] [CrossRef]
- Mu, H.; Ohashi, R.; Lin, P.; Yao, Q.; Chen, C. Cellular and molecular mechanisms of coronary vessel development. Vasc. Med. 2005, 10, 37–44. [Google Scholar] [CrossRef]
- Lie-Venema, H.; Eralp, I.; Markwald, R.R.; van den Akker, N.M.; Wijffels, M.C.; Kolditz, D.P.; van der Laarse, A.; Schalij, M.J.; Poelmann, R.E.; Bogers, A.J.; et al. Periostin expression by epicardium-derived cells is involved in the development of the atrioventricular valves and fibrous heart skeleton. Differentiation 2008, 76, 809–819. [Google Scholar] [CrossRef]
- Zhou, B.; von Gise, A.; Ma, Q.; Hu, Y.W.; Pu, W.T. Genetic fate mapping demonstrates contribution of epicardium-derived cells to the annulus fibrosis of the mammalian heart. Dev. Biol. 2010, 338, 251–261. [Google Scholar] [CrossRef]
- van Wijk, B.; Gunst, Q.D.; Moorman, A.F.; van den Hoff, M.J. Cardiac regeneration from activated epicardium. PLoS One 2012, 7, e44692. [Google Scholar]
- Huang, G.N.; Thatcher, J.E.; McAnally, J.; Kong, Y.; Qi, X.; Tan, W.; DiMaio, J.M.; Amatruda, J.F.; Gerard, R.D.; Hill, J.A.; et al. C/EBP transcription factors mediate epicardial activation during heart development and injury. Science 2012, 338, 1599–1603. [Google Scholar] [CrossRef]
- Huff, V.; Miwa, H.; Haber, D.A.; Call, K.M.; Housman, D.; Strong, L.C.; Saunders, G.F. Evidence for WT1 as a Wilms tumor (WT) gene: Intragenic germinal deletion in bilateral WT. Am. J. Hum. Genet. 1991, 48, 997–1003. [Google Scholar]
- Armstrong, J.F.; Pritchard-Jones, K.; Bickmore, W.A.; Hastie, N.D.; Bard, J.B. The expression of the Wilms' tumour gene, WT1, in the developing mammalian embryo. Mech. Dev. 1993, 40, 85–97. [Google Scholar] [CrossRef]
- Carmona, R.; Gonzalez-Iriarte, M.; Perez-Pomares, J.M.; Munoz-Chapuli, R. Localization of the Wilm's tumour protein WT1 in avian embryos. Cell Tissue Res. 2001, 303, 173–186. [Google Scholar] [CrossRef]
- Perez-Pomares, J.M.; Phelps, A.; Sedmerova, M.; Carmona, R.; Gonzalez-Iriarte, M.; Munoz-Chapuli, R.; Wessels, A. Experimental studies on the spatiotemporal expression of WT1 and RALDH2 in the embryonic avian heart: A model for the regulation of myocardial and valvuloseptal development by epicardially derived cells (EPDCs). Dev. Biol. 2002, 247, 307–326. [Google Scholar] [CrossRef]
- Guadix, J.A.; Ruiz-Villalba, A.; Lettice, L.; Velecela, V.; Munoz-Chapuli, R.; Hastie, N.D.; Perez-Pomares, J.M.; Martinez-Estrada, O.M. Wt1 controls retinoic acid signalling in embryonic epicardium through transcriptional activation of raldh2. Development 2011, 138, 1093–1097. [Google Scholar] [CrossRef]
- Moore, A.W.; McInnes, L.; Kreidberg, J.; Hastie, N.D.; Schedl, A. YAC complementation shows a requirement for Wt1 in the development of epicardium, adrenal gland and throughout nephrogenesis. Development 1999, 126, 1845–1857. [Google Scholar]
- Martinez-Estrada, O.M.; Lettice, L.A.; Essafi, A.; Guadix, J.A.; Slight, J.; Velecela, V.; Hall, E.; Reichmann, J.; Devenney, P.S.; Hohenstein, P.; et al. Wt1 is required for cardiovascular progenitor cell formation through transcriptional control of Snail and E-cadherin. Nat. Genet. 2010, 42, 89–93. [Google Scholar]
- Takeichi, M.; Nimura, K.; Mori, M.; Nakagami, H.; Kaneda, Y. The transcription factors Tbx18 and Wt1 control the epicardial epithelial-mesenchymal transition through bi-directional regulation of Slug in murine primary epicardial cells. PLoS One 2013, 8, e57829. [Google Scholar]
- Hsu, W.H.; Yu, Y.R.; Hsu, S.H.; Yu, W.C.; Chu, Y.H.; Chen, Y.J.; Chen, C.M.; You, L.R. The Wilms' tumor suppressor Wt1 regulates Coronin 1b expression in the epicardium. Exp. Cell Res. 2013, 319, 1365–1381. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D.; Scholz, H.; Kirschner, K.M.; Schedl, A. Intermediate filament protein nestin is expressed in developing kidney and heart and might be regulated by the Wilms' tumor suppressor Wt1. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2006, 291, R779–R787. [Google Scholar] [CrossRef]
- Wagner, N.; Wagner, K.D.; Theres, H.; Englert, C.; Schedl, A.; Scholz, H. Coronary vessel development requires activation of the TrkB neurotrophin receptor by the Wilms' tumor transcription factor Wt1. Genes Dev. 2005, 19, 2631–2642. [Google Scholar] [CrossRef]
- Niederreither, K.; Vermot, J.; Messaddeq, N.; Schuhbaur, B.; Chambon, P.; Dolle, P. Embryonic retinoic acid synthesis is essential for heart morphogenesis in the mouse. Development 2001, 128, 1019–1031. [Google Scholar]
- Chen, T.H.; Chang, T.C.; Kang, J.O.; Choudhary, B.; Makita, T.; Tran, C.M.; Burch, J.B.; Eid, H.; Sucov, H.M. Epicardial induction of fetal cardiomyocyte proliferation via a retinoic acid-inducible trophic factor. Dev. Biol. 2002, 250, 198–207. [Google Scholar]
- Dyson, E.; Sucov, H.M.; Kubalak, S.W.; Schmid-Schonbein, G.W.; DeLano, F.A.; Evans, R.M.; Ross, J., Jr.; Chien, K.R. Atrial-like phenotype is associated with embryonic ventricular failure in retinoid x receptor alpha -/- mice. Proc. Natl. Acad. Sci. USA 1995, 92, 7386–7390. [Google Scholar] [CrossRef]
- Jenkins, S.J.; Hutson, D.R.; Kubalak, S.W. Analysis of the proepicardium-epicardium transition during the malformation of the RXRalpha-/- epicardium. Dev. Dyn. 2005, 233, 1091–1101. [Google Scholar] [CrossRef]
- Zamora, M.; Manner, J.; Ruiz-Lozano, P. Epicardium-derived progenitor cells require beta-catenin for coronary artery formation. Proc. Natl. Acad. Sci. USA 2007, 104, 18109–18114. [Google Scholar] [CrossRef]
- Wu, M.; Smith, C.L.; Hall, J.A.; Lee, I.; Luby-Phelps, K.; Tallquist, M.D. Epicardial spindle orientation controls cell entry into the myocardium. Dev. Cell 2010, 19, 114–125. [Google Scholar] [CrossRef]
- Duan, J.; Gherghe, C.; Liu, D.; Hamlett, E.; Srikantha, L.; Rodgers, L.; Regan, J.N.; Rojas, M.; Willis, M.; Leask, A.; et al. Wnt1/betacatenin injury response activates the epicardium and cardiac fibroblasts to promote cardiac repair. EMBO J. 2012, 31, 429–442. [Google Scholar]
- Rudat, C.; Kispert, A. Wt1 and epicardial fate mapping. Circ. Res. 2012, 111, 165–169. [Google Scholar] [CrossRef]
- Zhou, B.; Pu, W.T. Genetic Cre-loxP assessment of epicardial cell fate using Wt1-driven Cre alleles. Circ. Res. 2012, 111, e276–e280. [Google Scholar] [CrossRef]
- Hidai, H.; Bardales, R.; Goodwin, R.; Quertermous, T.; Quertermous, E.E. Cloning of capsulin, a basic helix-loop-helix factor expressed in progenitor cells of the pericardium and the coronary arteries. Mech. Dev. 1998, 73, 33–43. [Google Scholar] [CrossRef]
- Lu, J.; Richardson, J.A.; Olson, E.N. Capsulin: A novel bHLH transcription factor expressed in epicardial progenitors and mesenchyme of visceral organs. Mech. Dev. 1998, 73, 23–32. [Google Scholar] [CrossRef]
- Quaggin, S.E.; Vanden Heuvel, G.B.; Igarashi, P. Pod-1, a mesoderm-specific basic-helix-loop-helix protein expressed in mesenchymal and glomerular epithelial cells in the developing kidney. Mech. Dev. 1998, 71, 37–48. [Google Scholar] [CrossRef]
- Lu, J.; Chang, P.; Richardson, J.A.; Gan, L.; Weiler, H.; Olson, E.N. The basic helix-loop-helix transcription factor capsulin controls spleen organogenesis. Proc. Natl. Acad. Sci. USA 2000, 97, 9525–9530. [Google Scholar] [CrossRef]
- Quaggin, S.E.; Schwartz, L.; Cui, S.; Igarashi, P.; Deimling, J.; Post, M.; Rossant, J. The basic-helix-loop-helix protein pod1 is critically important for kidney and lung organogenesis. Development 1999, 126, 5771–5783. [Google Scholar]
- Azambuja, A.P.; Portillo-Sanchez, V.; Rodrigues, M.V.; Omae, S.V.; Schechtman, D.; Strauss, B.E.; Costanzi-Strauss, E.; Krieger, J.E.; Perez-Pomares, J.M.; Xavier-Neto, J. Retinoic acid and VEGF delay smooth muscle relative to endothelial differentiation to coordinate inner and outer coronary vessel wall morphogenesis. Circ. Res. 2010, 107, 204–216. [Google Scholar] [CrossRef]
- Acharya, A.; Baek, S.T.; Banfi, S.; Eskiocak, B.; Tallquist, M.D. Efficient inducible Cre-mediated recombination in Tcf21cell lineages in the heart and kidney. Genesis 2011, 49, 870–877. [Google Scholar] [CrossRef]
- Funato, N.; Ohyama, K.; Kuroda, T.; Nakamura, M. Basic helix-loop-helix transcription factor epicardin/capsulin/pod-1 suppresses differentiation by negative regulation of transcription. J. Biol. Chem. 2003, 278, 7486–7493. [Google Scholar]
- Watada, H.; Kajimoto, Y.; Umayahara, Y.; Matsuoka, T.; Morishima, T.; Yamasaki, Y.; Kawamori, R.; Kamada, T. Ubiquitous, but variable, expression of two alternatively spliced mrnas encoding mouse homologues of transcription factors E47 and E12. Gene 1995, 153, 255–259. [Google Scholar] [CrossRef]
- Barnes, R.M.; Firulli, A.B. A twist of insight - the role of Twist-family bHLH factors in development. Int. J. Dev. Biol. 2009, 53, 909–924. [Google Scholar] [CrossRef]
- Tandon, P.; Miteva, Y.V.; Kuchenbrod, L.M.; Cristea, I.M.; Conlon, F.L. Tcf21 regulates the specification and maturation of proepicardial cells. Development 2013, 140, 2409–2421. [Google Scholar] [CrossRef]
- Plotkin, M.; Mudunuri, V. Pod1 induces myofibroblast differentiation in mesenchymal progenitor cells from mouse kidney. J. Cell. Biochem. 2008, 103, 675–690. [Google Scholar] [CrossRef]
- Murre, C.; McCaw, P.S.; Baltimore, D. A new DNA binding and dimerization motif in immunoglobulin enhancer binding, daughterless, MyoD, and myc proteins. Cell 1989, 56, 777–783. [Google Scholar] [CrossRef]
- Miyagishi, M.; Hatta, M.; Ohshima, T.; Ishida, J.; Fujii, R.; Nakajima, T.; Fukamizu, A. Cell type-dependent transactivation or repression of mesoderm-restricted basic helix-loop-helix protein, POD-1/capsulin. Mol. Cell. Biochem. 2000, 205, 141–147. [Google Scholar] [CrossRef]
- Miyagishi, M.; Nakajima, T.; Fukamizu, A. Molecular characterization of mesoderm-restricted basic helix-loop-helix protein, POD-1/capsulin. Int. J. Mol. Med. 2000, 5, 27–31. [Google Scholar]
- Kikuchi, K.; Gupta, V.; Wang, J.; Holdway, J.E.; Wills, A.A.; Fang, Y.; Poss, K.D. Tcf21+ epicardial cells adopt non-myocardial fates during zebrafish heart development and regeneration. Development 2011, 138, 2895–2902. [Google Scholar] [CrossRef]
- Lu, X.; Wang, L.; Chen, S.; He, L.; Yang, X.; Shi, Y.; Cheng, J.; Zhang, L.; Gu, C.C.; Huang, J.; et al. Genome-wide association study in Han Chinese identifies four new susceptibility loci for coronary artery disease. Nat. Genet. 2012, 44, 890–894. [Google Scholar] [CrossRef]
- Schunkert, H.; Konig, I.R.; Kathiresan, S.; Reilly, M.P.; Assimes, T.L.; Holm, H.; Preuss, M.; Stewart, A.F.; Barbalic, M.; Gieger, C.; et al. Large-scale association analysis identifies 13 new susceptibility loci for coronary artery disease. Nat. Genet. 2011, 43, 333–338. [Google Scholar] [CrossRef]
- Di Meglio, F.; Castaldo, C.; Nurzynska, D.; Romano, V.; Miraglia, R.; Bancone, C.; Langella, G.; Vosa, C.; Montagnani, S. Epithelial-mesenchymal transition of epicardial mesothelium is a source of cardiac CD117-positive stem cells in adult human heart. J. Mol. Cell. Cardiol. 2010, 49, 719–727. [Google Scholar] [CrossRef]
- Kraus, F.; Haenig, B.; Kispert, A. Cloning and expression analysis of the mouse T-box gene Tbx18. Mech. Dev. 2001, 100, 83–86. [Google Scholar] [CrossRef]
- Plageman, T.F., Jr.; Yutzey, K.E. T-box genes and heart development: Putting the "t" in heart. Dev. Dyn. 2005, 232, 11–20. [Google Scholar] [CrossRef]
- Airik, R.; Bussen, M.; Singh, M.K.; Petry, M.; Kispert, A. Tbx18 regulates the development of the ureteral mesenchyme. J. Clin. Invest. 2006, 116, 663–674. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Mommersteeg, M.T.; Trowe, M.O.; Prall, O.W.; de Gier-de Vries, C.; Soufan, A.T.; Bussen, M.; Schuster-Gossler, K.; Harvey, R.P.; Moorman, A.F.; et al. Formation of the venous pole of the heart from an Nkx2–5-negative precursor population requires Tbx18. Circ. Res. 2006, 98, 1555–1563. [Google Scholar] [CrossRef]
- Shelton, E.L.; Yutzey, K.E. Twist1 function in endocardial cushion cell proliferation, migration, and differentiation during heart valve development. Dev. Biol. 2008, 317, 282–295. [Google Scholar] [CrossRef]
- Christoffels, V.M.; Grieskamp, T.; Norden, J.; Mommersteeg, M.T.; Rudat, C.; Kispert, A. Tbx18 and the fate of epicardial progenitors. Nature 2009, 458, E8–E9; Discussion E9–E10. [Google Scholar]
- Grieskamp, T.; Rudat, C.; Ludtke, T.H.; Norden, J.; Kispert, A. Notch signaling regulates smooth muscle differentiation of epicardium-derived cells. Circ. Res. 2011, 108, 813–823. [Google Scholar] [CrossRef]
- Farin, H.F.; Bussen, M.; Schmidt, M.K.; Singh, M.K.; Schuster-Gossler, K.; Kispert, A. Transcriptional repression by the T-box proteins Tbx18 and Tbx15 depends on Groucho corepressors. J. Biol. Chem. 2007, 282, 25748–25759. [Google Scholar]
- Hogan, P.G.; Chen, L.; Nardone, J.; Rao, A. Transcriptional regulation by calcium, calcineurin, and NFAT. Genes Dev. 2003, 17, 2205–2232. [Google Scholar] [CrossRef]
- de la Pompa, J.L.; Timmerman, L.A.; Takimoto, H.; Yoshida, H.; Elia, A.J.; Samper, E.; Potter, J.; Wakeham, A.; Marengere, L.; Langille, B.L.; et al. Role of the NF-ATc transcription factor in morphogenesis of cardiac valves and septum. Nature 1998, 392, 182–186. [Google Scholar] [CrossRef]
- Ranger, A.M.; Grusby, M.J.; Hodge, M.R.; Gravallese, E.M.; de la Brousse, F.C.; Hoey, T.; Mickanin, C.; Baldwin, H.S.; Glimcher, L.H. The transcription factor NF-ATc is essential for cardiac valve formation. Nature 1998, 392, 186–190. [Google Scholar] [CrossRef]
- Combs, M.D.; Yutzey, K.E. VEGF and RANKL regulation of NFATc1 in heart valve development. Circ. Res. 2009, 105, 565–574. [Google Scholar] [CrossRef]
- Zhou, B.; Wu, B.; Tompkins, K.L.; Boyer, K.L.; Grindley, J.C.; Baldwin, H.S. Characterization of Nfatc1 regulation identifies an enhancer required for gene expression that is specific to pro-valve endocardial cells in the developing heart. Development 2005, 132, 1137–1146. [Google Scholar] [CrossRef]
- Combs, M.D.; Yutzey, K.E. Heart valve development: Regulatory networks in development and disease. Circ. Res. 2009, 105, 408–421. [Google Scholar] [CrossRef]
- Takayanagi, H.; Kim, S.; Koga, T.; Nishina, H.; Isshiki, M.; Yoshida, H.; Saiura, A.; Isobe, M.; Yokochi, T.; Inoue, J.; et al. Induction and activation of the transcription factor NFATc1 (NFAT2) integrate RANKL signaling in terminal differentiation of osteoclasts. Dev. Cell 2002, 3, 889–901. [Google Scholar] [CrossRef]
- Rapa, I.; Volante, M.; Cappia, S.; Rosas, R.; Scagliotti, G.V.; Papotti, M. Cathepsin K is selectively expressed in the stroma of lung adenocarcinoma but not in bronchioloalveolar carcinoma. A useful marker of invasive growth. Am. J. Clin. Pathol. 2006, 125, 847–854. [Google Scholar] [CrossRef]
- Matsumoto, M.; Kogawa, M.; Wada, S.; Takayanagi, H.; Tsujimoto, M.; Katayama, S.; Hisatake, K.; Nogi, Y. Essential role of p38 mitogen-activated protein kinase in cathepsin K gene expression during osteoclastogenesis through association of NFATc1 and PU.1. J. Biol. Chem. 2004, 279, 45969–45979. [Google Scholar] [CrossRef]
- Zeini, M.; Hang, C.T.; Lehrer-Graiwer, J.; Dao, T.; Zhou, B.; Chang, C.P. Spatial and temporal regulation of coronary vessel formation by calcineurin-NFAT signaling. Development 2009, 136, 3335–3345. [Google Scholar] [CrossRef]
- Casanova, J.C.; Travisano, S.; de la Pompa, J.L. Epithelial-to-mesenchymal transition in epicardium is independent of Snail1. Genesis 2013, 51, 32–40. [Google Scholar] [CrossRef]
- Carmona, R.; Gonzalez-Iriarte, M.; Macias, D.; Perez-Pomares, J.M.; Garcia-Garrido, L.; Munoz-Chapuli, R. Immunolocalization of the transcription factor Slug in the developing avian heart. Anat. Embryol. 2000, 201, 103–109. [Google Scholar] [CrossRef]
- Cano, A.; Perez-Moreno, M.A.; Rodrigo, I.; Locascio, A.; Blanco, M.J.; del Barrio, M.G.; Portillo, F.; Nieto, M.A. The transcription factor snail controls epithelial-mesenchymal transitions by repressing E-cadherin expression. Nat. Cell Biol. 2000, 2, 76–83. [Google Scholar] [CrossRef]
- Tao, G.; Levay, A.K.; Gridley, T.; Lincoln, J. Mmp15 is a direct target of Snai1 during endothelial to mesenchymal transformation and endocardial cushion development. Dev. Biol. 2011, 359, 209–221. [Google Scholar]
- Tao, G.; Miller, L.J.; Lincoln, J. Snai1 is important for avian epicardial cell transformation and motility. Dev. Dyn. 2013, 242, 699–708. [Google Scholar] [CrossRef]
- Merki, E.; Zamora, M.; Raya, A.; Kawakami, Y.; Wang, J.; Zhang, X.; Burch, J.; Kubalak, S.W.; Kaliman, P.; Belmonte, J.C.; et al. Epicardial retinoid X receptor alpha is required for myocardial growth and coronary artery formation. Proc. Natl. Acad. Sci. USA 2005, 102, 18455–18460. [Google Scholar] [CrossRef]
- Mahtab, E.A.; Wijffels, M.C.; van den Akker, N.M.; Hahurij, N.D.; Lie-Venema, H.; Wisse, L.J.; Deruiter, M.C.; Uhrin, P.; Zaujec, J.; Binder, B.R.; et al. Cardiac malformations and myocardial abnormalities in Podoplanin knockout mouse embryos: Correlation with abnormal epicardial development. Dev. Dyn. 2008, 237, 847–857. [Google Scholar] [CrossRef]
- Batlle, E.; Sancho, E.; Franci, C.; Dominguez, D.; Monfar, M.; Baulida, J.; Garcia de Herreros, A. The transcription factor snail is a repressor of E-cadherin gene expression in epithelial tumour cells. Nat. Cell Biol. 2000, 2, 84–89. [Google Scholar] [CrossRef]
- del Monte, G.; Casanova, J.C.; Guadix, J.A.; MacGrogan, D.; Burch, J.B.; Perez-Pomares, J.M.; de la Pompa, J.L. Differential Notch signaling in the epicardium is required for cardiac inflow development and coronary vessel morphogenesis. Circ. Res. 2011, 108, 824–836. [Google Scholar] [CrossRef]
- Timmerman, L.A.; Grego-Bessa, J.; Raya, A.; Bertran, E.; Perez-Pomares, J.M.; Diez, J.; Aranda, S.; Palomo, S.; McCormick, F.; Izpisua-Belmonte, J.C.; et al. Notch promotes epithelial-mesenchymal transition during cardiac development and oncogenic transformation. Genes Dev. 2004, 18, 99–115. [Google Scholar] [CrossRef]
- Lee, M.P.; Yutzey, K.E. Twist1 directly regulates genes that promote cell proliferation and migration in developing heart valves. PLoS One 2011, 6, e29758. [Google Scholar]
- Barnes, R.M.; Firulli, B.A.; VanDusen, N.J.; Morikawa, Y.; Conway, S.J.; Cserjesi, P.; Vincentz, J.W.; Firulli, A.B. Hand2 loss-of-function in Hand1-expressing cells reveals distinct roles in epicardial and coronary vessel development. Circ. Res. 2011, 108, 940–949. [Google Scholar] [CrossRef]
- Smith, C.L.; Baek, S.T.; Sung, C.Y.; Tallquist, M.D. Epicardial-derived cell epithelial-to-mesenchymal transition and fate specification require PDGF receptor signaling. Circ. Res. 2011, 108, e15–e26. [Google Scholar] [CrossRef]
- Cserjesi, P.; Brown, D.; Ligon, K.L.; Lyons, G.E.; Copeland, N.G.; Gilbert, D.J.; Jenkins, N.A.; Olson, E.N. Scleraxis: A basic helix-loop-helix protein that prefigures skeletal formation during mouse embryogenesis. Development 1995, 121, 1099–1110. [Google Scholar]
- Lincoln, J.; Alfieri, C.M.; Yutzey, K.E. BMP and FGF regulatory pathways control cell lineage diversification of heart valve precursor cells. Dev. Biol. 2006, 292, 292–302. [Google Scholar] [CrossRef]
- Schweitzer, R.; Chyung, J.H.; Murtaugh, L.C.; Brent, A.E.; Rosen, V.; Olson, E.N.; Lassar, A.; Tabin, C.J. Analysis of the tendon cell fate using Scleraxis, a specific marker for tendons and ligaments. Development 2001, 128, 3855–3866. [Google Scholar]
- Levay, A.K.; Peacock, J.D.; Lu, Y.; Koch, M.; Hinton, R.B., Jr.; Kadler, K.E.; Lincoln, J. Scleraxis is required for cell lineage differentiation and extracellular matrix remodeling during murine heart valve formation in vivo. Circ. Res. 2008, 103, 948–956. [Google Scholar] [CrossRef]
- Espira, L.; Lamoureux, L.; Jones, S.C.; Gerard, R.D.; Dixon, I.M.; Czubryt, M.P. The basic helix-loop-helix transcription factor Scleraxis regulates fibroblast collagen synthesis. J. Mol. Cell. Cardiol. 2009, 47, 188–195. [Google Scholar] [CrossRef]
- Akiyama, H.; Chaboissier, M.C.; Behringer, R.R.; Rowitch, D.H.; Schedl, A.; Epstein, J.A.; de Crombrugghe, B. Essential role of Sox9 in the pathway that controls formation of cardiac valves and septa. Proc. Natl. Acad. Sci. USA 2004, 101, 6502–6507. [Google Scholar] [CrossRef]
- Kikuchi, K.; Poss, K.D. Cardiac regenerative capacity and mechanisms. Ann. Rev. Cell Dev. Biol. 2012, 28, 719–741. [Google Scholar] [CrossRef]
- Poss, K.D.; Wilson, L.G.; Keating, M.T. Heart regeneration in zebrafish. Science 2002, 298, 2188–2190. [Google Scholar] [CrossRef]
- Kikuchi, K.; Holdway, J.E.; Major, R.J.; Blum, N.; Dahn, R.D.; Begemann, G.; Poss, K.D. Retinoic acid production by endocardium and epicardium is an injury response essential for zebrafish heart regeneration. Dev. Cell 2011, 20, 397–404. [Google Scholar] [CrossRef]
- Lepilina, A.; Coon, A.N.; Kikuchi, K.; Holdway, J.E.; Roberts, R.W.; Burns, C.G.; Poss, K.D. A dynamic epicardial injury response supports progenitor cell activity during zebrafish heart regeneration. Cell 2006, 127, 607–619. [Google Scholar] [CrossRef]
- Jopling, C.; Sleep, E.; Raya, M.; Marti, M.; Raya, A.; Izpisua Belmonte, J.C. Zebrafish heart regeneration occurs by cardiomyocyte dedifferentiation and proliferation. Nature 2010, 464, 606–609. [Google Scholar] [CrossRef]
- Kikuchi, K.; Holdway, J.E.; Werdich, A.A.; Anderson, R.M.; Fang, Y.; Egnaczyk, G.F.; Evans, T.; Macrae, C.A.; Stainier, D.Y.; Poss, K.D. Primary contribution to zebrafish heart regeneration by gata4(+) cardiomyocytes. Nature 2010, 464, 601–605. [Google Scholar] [CrossRef]
- Porrello, E.R.; Mahmoud, A.I.; Simpson, E.; Hill, J.A.; Richardson, J.A.; Olson, E.N.; Sadek, H.A. Transient regenerative potential of the neonatal mouse heart. Science 2011, 331, 1078–1080. [Google Scholar] [CrossRef]
- Bock-Marquette, I.; Shrivastava, S.; Pipes, G.C.; Thatcher, J.E.; Blystone, A.; Shelton, J.M.; Galindo, C.L.; Melegh, B.; Srivastava, D.; Olson, E.N.; et al. Thymosin beta4 mediated PKC activation is essential to initiate the embryonic coronary developmental program and epicardial progenitor cell activation in adult mice in vivo. J. Mol. Cell. Cardiol. 2009, 46, 728–738. [Google Scholar] [CrossRef]
- Russell, J.L.; Goetsch, S.C.; Gaiano, N.R.; Hill, J.A.; Olson, E.N.; Schneider, J.W. A dynamic Notch injury response activates epicardium and contributes to fibrosis repair. Circ. Res. 2011, 108, 51–59. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Braitsch, C.M.; Yutzey, K.E. Transcriptional Control of Cell Lineage Development in Epicardium-Derived Cells. J. Dev. Biol. 2013, 1, 92-111. https://doi.org/10.3390/jdb1020092
Braitsch CM, Yutzey KE. Transcriptional Control of Cell Lineage Development in Epicardium-Derived Cells. Journal of Developmental Biology. 2013; 1(2):92-111. https://doi.org/10.3390/jdb1020092
Chicago/Turabian StyleBraitsch, Caitlin M., and Katherine E. Yutzey. 2013. "Transcriptional Control of Cell Lineage Development in Epicardium-Derived Cells" Journal of Developmental Biology 1, no. 2: 92-111. https://doi.org/10.3390/jdb1020092
APA StyleBraitsch, C. M., & Yutzey, K. E. (2013). Transcriptional Control of Cell Lineage Development in Epicardium-Derived Cells. Journal of Developmental Biology, 1(2), 92-111. https://doi.org/10.3390/jdb1020092