Integrated Amplification Microarrays for Infectious Disease Diagnostics

Abstract
:1. Introduction
2. Solid Phase Amplification
3. Integrated Biochemistry for Single-Step, Closed-Amplicon Microarrays
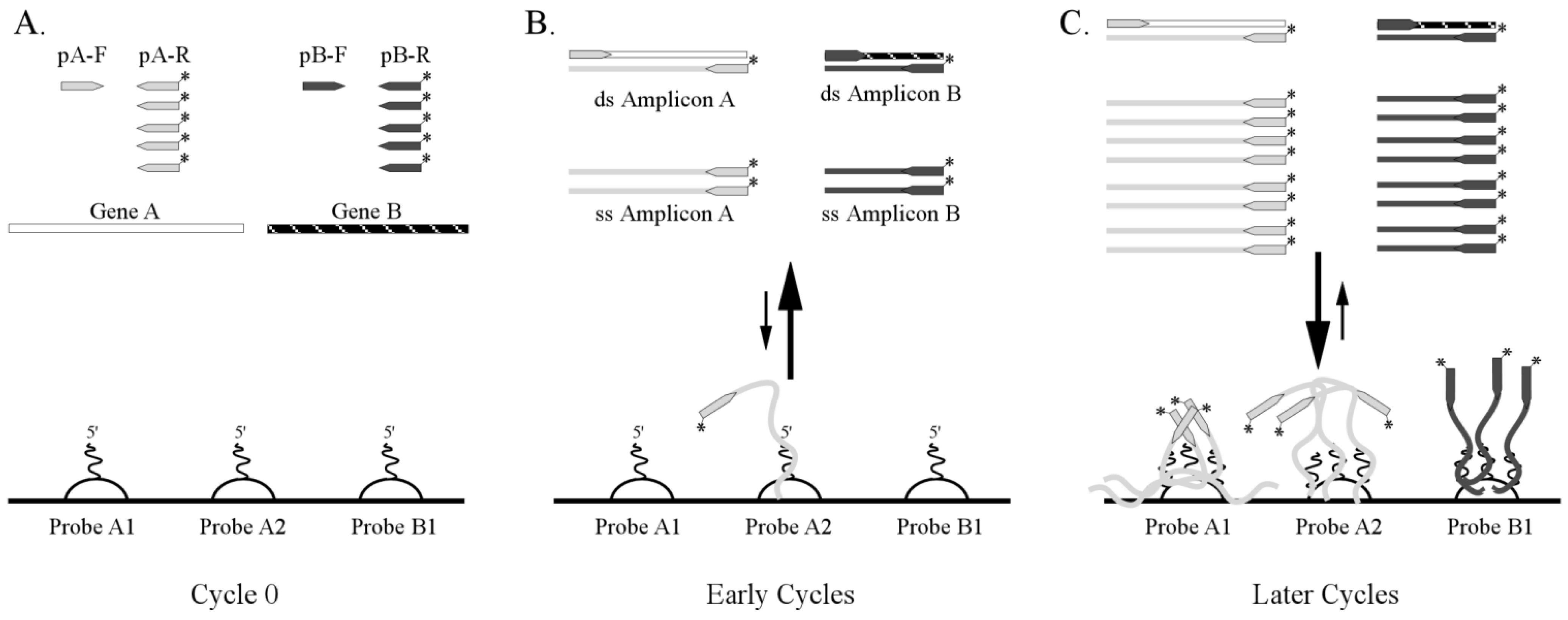
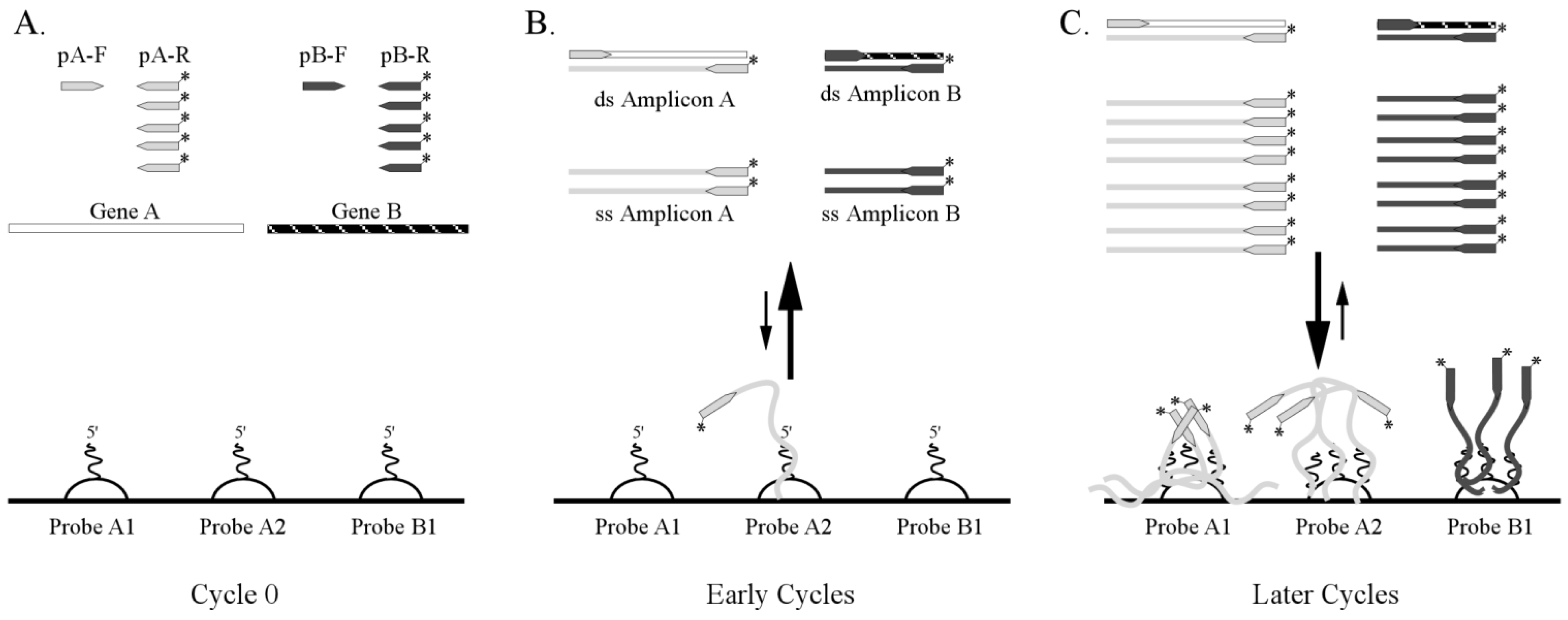
3.1. One-Step RT-PCR Amplification Microarray
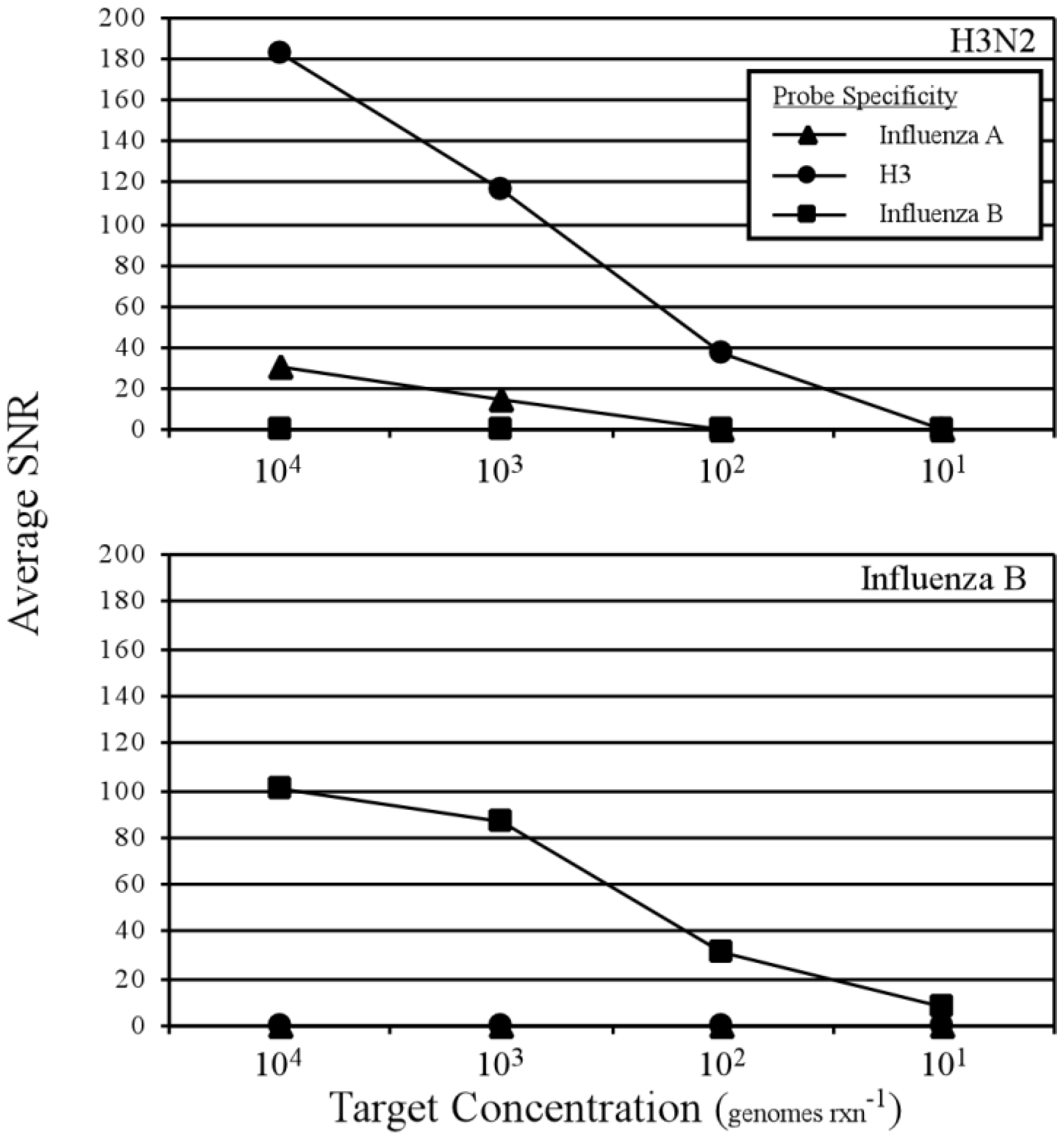
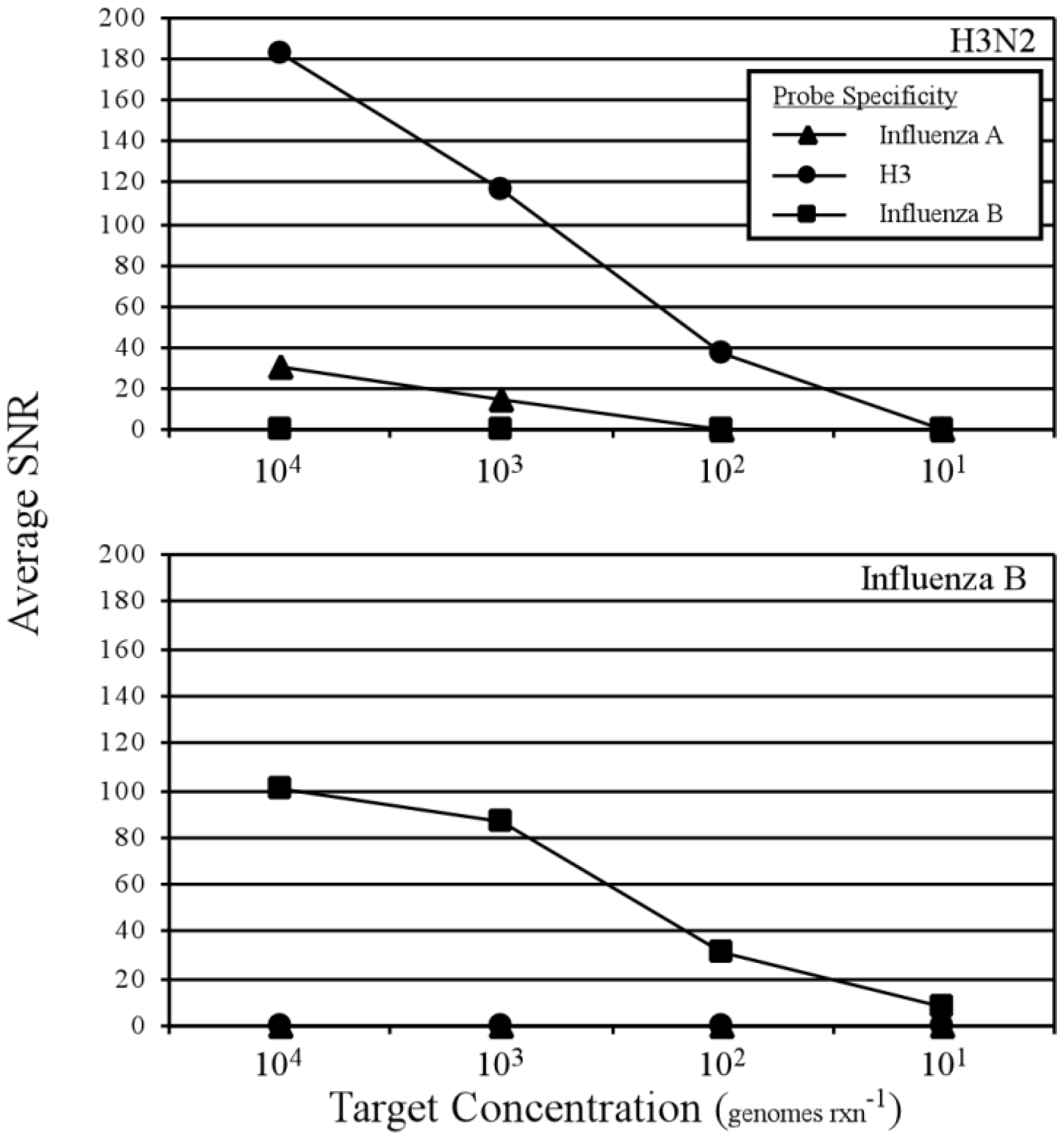
3.2. Two-Step RT-PCR Amplification Microarray for DNA and RNA Genomes

{kind=link}
{kind=link}
{kind=link}
| Target (gene copies per reaction) | ||||||||
|---|---|---|---|---|---|---|---|---|
| Probe Specificity | HSV1 (5,000) | HSV2 (500) | VZV (500) | GFP (5,500) | EVU (500) | CMV (500) | HHV6 (500) | WNV (500) |
| HSV1 | 34.56 | 0.72 | 0.67 | 0.44 | 0.68 | 0.55 | 0.54 | 0.17 |
| HSV2 | 1.70 | 66.94 | 0.14 | 0.21 | −0.08 | 0.16 | 0.64 | −0.01 |
| VZV | 0.86 | 0.64 | 24.82 | 0.76 | 0.44 | −0.21 | 0.90 | 0.05 |
| GFP | 0.76 | 1.10 | 0.43 | 244.26 | 0.52 | 0.53 | 0.04 | 0.21 |
| EVU | 1.03 | 0.57 | 0.63 | 0.27 | 198.73 | −0.03 | 1.21 | 0.14 |
| CMV | 0.81 | 0.92 | 0.57 | 0.62 | 4.31 | 346.28 | 0.36 | 0.20 |
| HHV6 | 1.65 | 0.67 | 1.16 | 0.22 | 0.72 | 0.70 | 132.13 | −0.31 |
| WNV | 0.74 | 0.64 | 0.75 | 0.34 | 0.65 | 0.52 | 1.72 | 472.55 |
| Target (gene copies per reaction) | ||||||
|---|---|---|---|---|---|---|
| Probe Specificity | Enterovirus RNA | West Nile virus RNA | ||||
| 104 | 103 | 102 | 104 | 103 | 102 | |
| HSV1 | 0.21 | 0.05 | 0.09 | 0.18 | 0.39 | 0.12 |
| HSV2 | −0.25 | 0.08 | −0.08 | −0.46 | −0.20 | −0.30 |
| VZV | 0.76 | 0.38 | 0.39 | 0.20 | 0.07 | −0.01 |
| GFP | −0.25 | 0.25 | 0.01 | 0.13 | −0.27 | 0.07 |
| EVU | 405.00 | 198.73 | 29.57 | −0.16 | 0.08 | −0.44 |
| CMV | 3.49 | 1.38 | 0.74 | 0.13 | 0.15 | 0.11 |
| HHV6 | 0.03 | 0.57 | 0.11 | 0.27 | 0.22 | −0.35 |
| WNV | −0.26 | 0.03 | 0.01 | 1,093.61 | 824.32 | 1,263.02 |
3.3. Integrated Waste Chamber and Entirely Closed-Amplicon Consumable

| DST Phenotype and DNA amounts | MDR 250 pg | MDR 15 pg | WT 250 pg | WT 15 pg | |
|---|---|---|---|---|---|
| Gene | Mutation | Wildtype to Mutant Ratios | |||
| rpoB | D516V | 5.8 | 9.1 | 5.4 | 8.4 |
| H526D | 3.5 | 7.9 | 4.4 | 8.5 | |
| H526Y1 | 3.9 | 5.1 | 2.7 | 3.4 | |
| S531L | 0.1 | 0.1 | 2.2 | 3.1 | |
| L533P | 9.1 | 7.9 | 7.2 | 13.4 | |
| katG | S315T | 0.2 | 0.3 | 12.0 | 11.5 |
| inhA | T8A | 9.1 | 4.9 | 8.2 | 4.5 |
| T8C | 5.1 | 2.8 | 4.7 | 2.5 | |
| C15T | 7.0 | 7.7 | 7.4 | 7.8 | |
| G17T | 8.0 | 3.1 | 7.1 | 2.9 | |
4. Discussion and Future Prospects
Acknowledgments
References
- Lyon, E.; Wittwer, C.T. LightCycler technology in molecular diagnostics. J. Mol. Diagn. 2009, 11, 93–101. [Google Scholar] [CrossRef]
- Liu, Y.T. A technological update of molecular diagnostics for infectious diseases. Infect. Disord. Drug Targets 2008, 8, 183–188. [Google Scholar]
- Dong, J.; Olano, J.P.; McBride, J.W.; Walker, D.H. Emerging pathogens: Challenges and successes of molecular diagnostics. J. Mol. Diagn. 2008, 10, 185–197. [Google Scholar] [CrossRef]
- Yang, S.; Rothman, R.E. PCR-based diagnostics for infectious diseases: Uses, limitations, and future applications in acute-care setting. Lancet Infect Dis. 2004, 4, 337–348. [Google Scholar] [CrossRef]
- Dumler, J.S.; Valsamakis, A. Molecular diagnostics for existing and emerging infections. Complementary tools for a new era of clinical microbiology. Am. J. Clin. Pathol. 1999, 112, S33–S39. [Google Scholar]
- Millar, B.C.; Xu, J.; Moore, J.E. Molecular diagnostics of medically important bacterial infections. Curr. Issues Mol. Biol. 2007, 9, 21–39. [Google Scholar]
- Robertson, B.H.; Nicholson, J.K. New microbiology tools for public health and their implications. Annu. Rev. Public Health 2005, 26, 281–302. [Google Scholar] [CrossRef]
- O’Connor, L.; Glynn, B. Recent advances in the development of nucleic acid diagnostics. Expert Rev. Med. Devices 2010, 7, 529–539. [Google Scholar] [CrossRef]
- Kaltenboeck, B.; Wang, C. Advances in real-time PCR: Application to clinical laboratory diagnostics. Adv. Clin. Chem. 2005, 40, 219–259. [Google Scholar] [CrossRef]
- Mackay, I.M. Real-time PCR in the microbiology laboratory. Clin. Microbiol. Infect. 2005, 10, 190–212. [Google Scholar] [CrossRef]
- Procop, G.W. Molecular diagnostics for the detection and characterization of microbial pathogens. Clin. Infect. Dis. 2007, 45, S99–S111. [Google Scholar] [CrossRef]
- Easley, C.J.; Karlinsey, J.M.; Bienvenue, J.M.; Legendre, L.A.; Roper, M.G.; Feldman, S.H.; Hughes, M.A.; Hewlett, E.L.; Merkel, T.J.; Ferrance, J.P.; Landers, J.P. A fully integrated microfluidic genetic analysis system with sample-in-answer-out capability. Proc. Natl. Acad. Sci. USA 2006, 103, 19272–19277. [Google Scholar]
- Mahalanabis, M.; Do, J.; Al Muayad, H.; Zhang, J.Y.; Klapperich, C.M. An integrated disposable device for DNA extraction and helicase dependent amplification. Biomed. Microdevices 2010, 12, 353–359. [Google Scholar] [CrossRef]
- Chen, D.; Mauk, M.; Qiu, X.; Liu, C.; Kim, J.; Ramprasad, S.; Ongagna, S.; Abrams, W.R.; Malamud, D.; Corstjens, P.L.; Bau, H.H. An integrated, self-contained microfluidic cassette for isolation, amplification, and detection of nucleic acid. Biomed. Microdevices 2010, 12, 705–719. [Google Scholar] [CrossRef]
- Njoroge, S.K.; Chen, H.W.; Witek, M.A.; Soper, S.A. Integrated microfluidic systems for DNA analysis. Top. Curr. Chem. 2011, 304, 203–260. [Google Scholar] [CrossRef]
- Bissonnette, L.; Bergeron, M.G. Diagnosing infections—Current and anticipated technologies for point-of-care diagnostics and home-based testing. Clin. Microbiol. Infect. 2010, 16, 1044–1053. [Google Scholar] [CrossRef]
- Park, S.; Zhang, Y.; Lin, S.; Wang, T.H.; Yang, S. Advances in microfluidic PCR for point-of-care infectious disease diagnostics. Biotechnol. Adv. 2011, 29, 830–839. [Google Scholar] [CrossRef]
- Niemz, A.; Ferguson, T.M.; Boyle, D.S. Point-of-care nucleic acid testing for infectious diseases. Trends Biotechnol. 2011, 29, 240–250. [Google Scholar] [CrossRef]
- Peeling, R.W.; Mabey, D. Point-of-care tests for diagnosing infections in the developing world. Clin. Microbiol. Infect. 2010, 16, 1062–1069. [Google Scholar] [CrossRef]
- Taylor, T.B.; Winn-Deen, E.S.; Picozza, E.; Woudenberg, T.M.; Albin, M. Optimization of the performance of the polymerase chain reaction in silicon-based microstructures. Nucl. Acids Res. 1997, 25, 3164–3168. [Google Scholar] [CrossRef]
- Irwin, P.L.; Nguyen, L.H.; Chen, C.Y. The relationship between purely stochastic sampling error and the number of technical replicates used to estimate concentration at an extreme dilution. Anal. Bioanal. Chem. 2010, 398, 895–903. [Google Scholar] [CrossRef]
- Walsh, P.S.; Erlich, H.A.; Higuchi, R. Preferential PCR amplification of alleles: Mechanisms and solutions. PCR Methods Appl. 1992, 1, 241–250. [Google Scholar] [CrossRef]
- Sykes, P.J.; Neoh, S.H.; Brisco, M.J.; Hughes, E.; Condon, J.; Morley, A.A. Quantitation of targets for PCR by use of limiting dilution. Biotechniques 1992, 13, 444–449. [Google Scholar]
- Vogelstein, B.; Kinzler, K.W. Digital PCR. Proc. Natl. Acad. Sci. USA 1999, 96, 9236–9241. [Google Scholar] [CrossRef]
- Zhong, Q.; Bhattacharya, S.; Kotsopoulos, S.; Olson, J.; Taly, V.; Griffiths, A.D.; Link, D.R.; Larson, J.W. Multiplex digital PCR: Breaking the one target per color barrier of quantitative PCR. Lab Chip 2011, 11, 2167–2174. [Google Scholar] [CrossRef]
- Shen, F.; Davydova, E.K.; Du, W.; Kreutz, J.E.; Piepenburg, O.; SIsmagilov, R.F. Digital isothermal quantification of nucleic acids via simultaneous chemical initiation of recombinase polymerase amplification reactions on SlipChip. Anal. Chem. 2011, 83, 3533–3540. [Google Scholar]
- Weile, J.; Knabbe, C. Current applications and future trends of molecular diagnostics in clinical bacteriology. Anal. Bioanal. Chem. 2009, 394, 731–742. [Google Scholar] [CrossRef]
- Shen, Y.; Wu, B.L. Microarray-based genomic DNA profiling technologies in clinical molecular diagnostics. Clin. Chem. 2009, 55, 659–669. [Google Scholar] [CrossRef]
- Miller, M.B.; Tang, Y.-W. Basic concepts of microarrays and potential applications in clinical microbiology. Clin. Microbiol. Rev. 2009, 22, 611–633. [Google Scholar] [CrossRef]
- Wu, L.; Williams, P.M.; Kock, W. Clinical applications of microarray-based diagnostic tests. Biotechniques 2005, 39, S577–S582. [Google Scholar]
- Clerc, O.; Greub, G. Routine use of point-of-care tests: Usefulness and application in clinical microbiology. Clin. Microbiol. Infect. 2010, 16, 1054–1061. [Google Scholar] [CrossRef]
- Metzgar, D.; Myers, C.A.; Russell, K.L.; Faix, D.; Blair, P.J.; Brown, J.; Vo, S.; Swayne, D.E.; Thomas, C.; Stenger, D.A.; et al. Single assay for simultaneous detection and differential identification of human and avian influenza virus types, subtypes, and emergent variants. PLoS One 2010, 5, e8995. [Google Scholar]
- Chen, E.C.; Miller, S.A.; DeRisi, J.L.; Chiu, C.Y. Using a pan-viral microarray assay (Virochip) to screen clinical samples for viral pathogens. J. Vis. Exp. 2011, 50. [Google Scholar] [CrossRef]
- Mahony, J.; Chong, S.; Merante, F.; Yaghoubian, S.; Sinha, T.; Lisle, C.; Janeczko, R. Development of a respiratory virus panel test for detection of twenty human respiratory viruses by use of multiplex PCR and a fluid microbead-based assay. J. Clin. Microbiol. 2007, 45, 2965–2970. [Google Scholar] [CrossRef]
- Kessler, N.; Ferraris, O.; Palmer, K.; Marsh, W.; Steel, A. Use of the DNA flow-thru chip, a three-dimensional biochip, for typing and subtyping of influenza viruses. J. Clin. Microbiol. 2004, 42, 2173–2185. [Google Scholar] [CrossRef]
- Palacios, G.; Quan, P.-L.; Jabado, O.M.; Conlan, S.; Hirschberg, D.L.; Liu, Y.; Zhai, J.; Renwick, N.; Hui, J.; Hegyi, H.; et al. Panmicrobial oligonucleotide array for diagnosis of infectious diseases. Emerg. Infect. Dis. 2007, 13, 73–81. [Google Scholar] [CrossRef]
- Lodes, M.J.; Suciu, D.; Wilmoth, J.L.; Ross, M.; Munro, S.; Dix, K.; Bernards, K.; Stover, A.G.; Quintana, M.; Iihoshi, N.; Lyon, W.J.; Danley, D.L.; McShea, A. Identification of upper respiratory tract pathogens using electrochemical detection on an oligonucleotide array. PLoS ONE 2007, 2, e924. [Google Scholar]
- Caoili, J.C.; Mayorova, A.; Sikes, D.; Hickman, L.; Plikaytis, B.B.; Shinnick, T.M. Evaluation of the TB-biochip oligonucleotide microarray system for rapid detection of rifampin resistance in Mycobacterium tuberculosis. J. Clin. Microbiol. 2006, 44, 2378–2381. [Google Scholar] [CrossRef]
- Liu, R.H.; Yang, J.; Lenigk, R.; Bonanno, J.; Grodzinski, P. Self-contained, fully integrated biochip for sample preparation, polymerase chain reaction amplification, and DNA microarray detection. Anal. Chem. 2004, 76, 1824–1831. [Google Scholar] [CrossRef]
- Liu, R.H.; Lodes, M.J.; Nguyen, T.; Siuda, T.; Slota, M.; Fuji, H.S.; McShea, A. Validation of a fully integrated microfluidic array device for influenza A subtype identification and sequencing. Anal. Chem. 2006, 78, 4184–4193. [Google Scholar]
- Summerer, D.; Hevroni, D.; Jain, A.; Oldenburger, O.; Parker, J.; Caruso, A.; Stähler, C.F.; Stähler, P.F.; Beier, M. A flexible and fully integrated system for amplification, detection and genotyping of genomic DNA targets based on microfluidic oligonucleotide arrays. New Biotechnol. 2010, 27, 149–155. [Google Scholar]
- Trau, D.; Lee, T.M.H.; Lao, A.I.K.; Lenigk, R.; Hsing, I.-M.; Ip, N.Y.; Carles, M.C.; Sucher, N.J. Genotyping on a complementary metal oxide semiconductor silicon polymerase chain reaction chip with integrated DNA microarray. Anal. Chem. 2002, 74, 3168–3173. [Google Scholar] [CrossRef]
- Regan, J.; Létant, S.; Adams, K.; Nguyen, N.; Derlet, R.; Cohen, S.; Vitalis, E.; Tammero, L.; Ortiz, J.; McBride, M.; Birch, J. A sample-in-answer-out instrument for the detection of multiple respiratory pathogens in unprepared nasopharyngeal swab samples. Analyst 2010, 135, 2316–2322. [Google Scholar] [CrossRef]
- Teo, J.; Pietro, P.D.; Biagio, F.S.; Capozzoli, M.; Deng, Y.M.; Barr, I.; Caldwell, N.; Ong, K.L.; Sato, M.; Tan, R.; Lin, R. VereFlu™: An integrated multiplex RT-PCR and microarray assay for rapid detection and identification of human influenza A and B viruses using lab-on-chip technology. Arch. Virol. 2011, 156, 1371–1378. [Google Scholar] [CrossRef]
- Yeung, S.-W.; Lee, T.M.-H.; Cai, H.; Hsing, I.M. A DNA biochip for on-the-spot multiplexed pathogen identification. Nucl. Acids Res. 2006, 34. [Google Scholar] [CrossRef]
- Dorris, D.R.; Ramakrishnan, R.; Trakas, D.; Dudzik, F.; Belval, R.; Zhao, C.; Nguyen, A.; Domanus, M.; Mazumder, A. A highly reproducible, linear, and automated sample preparation method for DNA microarrays. Genome Res. 2002, 12, 976–984. [Google Scholar] [CrossRef]
- Liu, R.H.; Dill, K.; Fuji, H.S.; McShea, A. Integrated microfluidic biochips for DNA microarray analysis. Expert Rev. Mol. Diagn. 2006, 6, 253–261. [Google Scholar] [CrossRef]
- Raymond, F.; Carbonneau, J.; Boucher, N.; Robitaille, L.; Boisvert, S.; Wu, W.-K.; De Serres, G.; Boivin, G.; Corbeil, J. Comparison of automated microarray detection with real-time PCR assays for detection of respiratory viruses in specimens obtained from children. J. Clin. Microbiol. 2009, 47, 743–750. [Google Scholar] [CrossRef]
- Kumar, S.; Wang, L.; Fan, J.; Kraft, A.; Bose, M.E.; Tiwari, S.; Van Dyke, M.; Haigis, R.; Luo, T.; Ghosh, M.; et al. Detection of 11 common viral and bacterial pathogens causing community-acquired pneumonia or sepsis in asymptomatic patients by using a multiplex reverse transcription-PCR assay with manual (enzyme hybridization) or automated (electronic microarray) detection. J. Clin. Microbiol. 2008, 46, 3063–3072. [Google Scholar] [CrossRef]
- Foglieni, B.; Brisci, A.; San Biagio, F.; Di Pietro, P.; Petralia, S.; Conoci, S.; Ferrari, M.; Cremonesi, L. Integrated PCR amplification and detection processes on a Lab-on-Chip platform: A new advanced solution for molecular diagnostics. Clin. Chem. Lab. Med. 2010, 48, 329–336. [Google Scholar]
- Stamm, S.; Brosius, J. Sanchored PCR: PCR with cDNA coupled to a solid phase. Nucl. Acid. Res. 1991, 19, 1350. [Google Scholar] [CrossRef]
- Erdogan, F.; Kirchner, R.; Mann, W.; Ropers, H.-H.; Nuber, U.A. Detection of mitochondrial single nucleotide polymorphisms using a primer elongation reaction on oligonucleotide microarrays. Nucl. Acid. Res. 2001, 29. [Google Scholar] [CrossRef]
- Shapero, M.H.; Leuther, K.K.; Nguyen, A.; Scott, M.; Jones, K.W. SNP genotyping by multiplexed solid-phase amplification and fluorescent minisequencing. Genome Res. 2001, 11, 1926–1934. [Google Scholar]
- Lockley, A.K.; Jones, C.G.; Bruce, J.S.; Franklin, S.J.; Bardsley, R.G. Colorimetric detection of immobilised PCR products generated on a solid support. Nucl. Acid. Res. 1997, 25, 1313–1314. [Google Scholar] [CrossRef]
- Adessi, C.; Matton, G.; Ayala, G.; Turcatti, G.; Mermod, J.-J.; Mayer, P.; Kawashima, E. Solid phase DNA amplification: characterisation of primer attachment and amplification mechanisms. Nucl. Acid. Res. 2000, 28, e87. [Google Scholar] [CrossRef]
- Adams, C.P.; Kron, S.J. Method for Performing Amplification of Nucleic Acid with Two Primers Bound to a Single Solid Support. U.S. Patent 5,641,658, 24 June 1997. [Google Scholar]
- Onodera, K.; d’Offay, J.; Melcher, U. Nylon membrane-immobilized PCR for detection of bovine viruses. Biotechniques 2002, 32, 74–80. [Google Scholar]
- Westin, L.; Xu, X.; Miller, C.; Wang, L.; Edman, C.F.; Nerenberg, M. Anchored multiplex amplification on a microelectronic chip array. Nat. Biotechnol. 2000, 18, 199–204. [Google Scholar] [CrossRef]
- Strizhkov, B.N.; Drobyshev, A.L.; Mikhailovich, V.M.; Mirzabekov, A.D. PCR amplification on a microarray of gel-immobilized oligonucleotides: detection of bacterial toxin- and drug-resistant genes and their mutations. Biotechniques 2000, 29, 844–857. [Google Scholar]
- Turner, M.S.; Penning, S.; Sharp, A.; Hyland, V.J.; Harris, R.; Morris, C.P.; van Daal, A. Solid-phase amplification for detection of C282y and H63D hemochromatosis (HFE) gene mutations. Clin. Chem. 2001, 47, 1384–1389. [Google Scholar]
- Huber, M.; Losert, D.; Hiller, R.; Harwanegg, C.; Mueller, M.W.; Schmidt, W.M. Detection of single base alterations in genomic DNA by solid phase polymerase chain reaction on oligonucleotide microarrays. Anal. Biochem. 2001, 299, 24–30. [Google Scholar]
- Tillib, S.V.; Strizhkov, B.N.; Mirzabekov, A.D. Integration of multiple PCR amplifications and DNA mutation analyses by using oligonucleotide microchip. Anal. Biochem. 2001, 292, 155–160. [Google Scholar]
- Mitterer, G.; Huber, M.; Leidinger, E.; Kirisits, C.; Lubitz, W.; Mueller, M.W.; Schmidt, W.M. Microarray-based identification of bacteria in clinical samples by solid-phase PCR amplification of 23S ribosomal DNA sequences. J. Clin. Microbiol. 2004, 42, 1048–1057. [Google Scholar] [CrossRef]
- Mitterer, G.; Schmidt, W.M. Microarray-based detection of bacteria by on-chip PCR. Methods Mol. Biol. 2006, 345, 37–51. [Google Scholar]
- Pemov, A.; Modi, H.; Chandler, D.P.; Bavykin, S. DNA analysis with multiplex microarray-enhanced PCR. Nucl. Acid. Res. 2005, 33, e11. [Google Scholar] [CrossRef]
- Li, Y.; Guo, S.J.; Shao, N.; Tu, S.; Xu, M.; Ren, Z.R.; Ling, X.; Wang, G.Q.; Lin, Z.X.; Tao, S.C. A universal multiplex PCR strategy for 100-plex amplification using a hydrophobically patterned microarray. Lab Chip 2011, 11, 3609–3618. [Google Scholar] [CrossRef]
- Sun, Y.; Dhumpa, R.; Bang, D.D.; Handberg, K.; Wolff, A. DNA microarray-based solid-phase RT-PCR for rapid detection and identification of influenza virus type A and subtypes H5 and H7. Diagn. Microbiol. Infect. Dis. 2011, 69, 432–439. [Google Scholar] [CrossRef]
- von Nickisch-Rosenegk, M.; Marschan, X.; Andresen, D.; Bier, F.F. Reverse transcription-polymerase chain reaction on a microarray: The integrating concept of “active arrays”. Anal. Bioanal. Chem. 2008, 391, 1671–1678. [Google Scholar] [CrossRef]
- Andresen, D.; Von Nickisch-Rosenegk, M.; Bier, F.F. Helicase-dependent amplification: Use in onchip amplification and potential for point-of-care diagnostics. Expert Rev. Mol. Diagn. 2009, 9, 645–650. [Google Scholar] [CrossRef]
- Vora, G.J.; Meador, C.E.; Stenger, D.A.; Andreadis, J.D. Nucleic acid amplification strategies for DNA microarray-based pathogen detection. Appl. Environ. Microbiol. 2004, 70, 3047–3054. [Google Scholar] [CrossRef]
- Fredriksson, S.; Banér, J.; Dahl, F.; Chu, A.; Ji, H.; Welch, K.; Davis, R.W. Multiplex amplification of all coding sequences within 10 cancer genes by Gene-Collector. Nucl. Acid. Res. 2007, 35. [Google Scholar] [CrossRef]
- Borel, N.; Kempf, E.; Hotzel, H.; Schubert, E.; Torgerson, P.; Slickers, P.; Ehricht, R.; Tasara, T.; Pospischil, A.; Sachse, K. Direct identification of chlamydiae from clinical samples using a DNA microarray assay: A validation study. Mol. Cell. Probe 2008, 22, 55–64. [Google Scholar] [CrossRef]
- Khodakov, D.A.; Zakharova, N.V.; Gryadunov, D.A.; Filatov, F.P.; Zasedatelev, A.S.; Mikhailovich, V.M. An oligonucleotide microarray for multiplex real-time PCR identification of HIV-1, HBV, and HCV. Biotechniques 2008, 44, 241–248. [Google Scholar]
- Pierik, A.; Boamfa, M.; van Zelst, M.; Clout, D.; Stapert, H.; Dijksman, F.; Broer, D.; Wimberger-Friedl, R. Real time quantitative amplification detection on a microarray: Towards high multiplex quantitative PCR. Lab Chip 2012, 12, 1897–1902. [Google Scholar] [CrossRef]
- Yershov, G.; Barsky, V.; Belgovskiy, A.; Kirillov, E.; Kreindlin, E.; Ivanov, I.; Parinov, S.; Guschin, D.; Drobishev, A.; Dubiley, S.; Mirzabekov, A. DNA analysis and diagnostics on oligonucleotide microchips. Proc. Natl. Acad. Sci. USA 1996, 93, 4913–4918. [Google Scholar]
- Cooney, C.G.; Sipes, D.; Thakore, N.; Holmberg, R.; Belgrader, P. A plastic, disposable microfluidic flow cell for coupled on-chip PCR and microarray detection of infectious agents. Biomed. Microdevices 2012, 14, 45–53. [Google Scholar] [CrossRef]
- Brammer, T.L.; Murray, E.L.; Fukuda, K.; Hall, H.E.; Klimov, A.; Cox, N.J. Surveillance for influenza—United States, 1997–98, 1998–99, and 1999–00 seasons. MMWR Surveillance Summaries 2002, 51, 1–10. [Google Scholar]
- Murphy, B.R.; Webster, R.G. Orthomyxoviruses. In Virology, 3rd; Fields, B.N., Knipe, D.M., Howley, P.M., Eds.; Lippincott-Raven Press: Philadelphia, PA, USA, 1996; pp. 1337–1420. [Google Scholar]
- Webster, R.G.; Bean, W.J.; Gorman, O.T.; Chambers, T.M.; Kawaoka, Y. Evolution and ecology of influenza A viruses. Microbiol. Rev. 1992, 56, 152–179. [Google Scholar]
- Golova, J.B.; Chernov, B.K.; Perov, A.N.; Reynolds, J.; Linger, Y.L.; Kukhtin, A.; Chandler, D.P. Non-volatile copolymer compositions for fabricating gel element microarrays. Anal. Biochem. 2012, 421, 526–533. [Google Scholar]
- Sandgren, A.; Strong, M.; Muthukrishnan, P.; Weiner, B.K.; Church, G.M.; Murray, M.B. Tuberculosis drug resistance mutation database. PLoS Med. 2009, 6, e1000002. [Google Scholar]
- WHO Global Tuberculosis Control 2011; Publication No. WHO/HTM/TB/2011.16, World Health Organization, WHO Press: Geneva, Switzerland, 2011; 248.
- Sanchez, J.A.; Pierce, K.E.; Rice, J.E.; Wangh, L.J. Linear-After-The-Exponential (LATE)–PCR: An advanced method of asymmetric PCR and its uses in quantitative real-time analysis. Proc. Natl. Acad. Sci. USA 2004, 101, 1933–1938. [Google Scholar]
- Edman, C.F.; Mehta, P.; Press, R.; Spargo, C.A.; Walker, G.T.; Nerenberg, M. Pathogen analysis and genetic predisposition testing using microelectronic arrays and isothermal amplification. Amer. J. Inv. Med. 2000, 48, 93–101. [Google Scholar]
- Detter, J.C.; Jett, J.M.; Lucas, S.M.; Dalin, E.; Arellano, A.R.; Wang, M.; Nelson, J.R.; Chapman, J.; Lou, Y.; Rokhsar, D.; Hawkins, T.L.; Richardson, P.M. Isothermal strand-displacement amplification applications for high-throughput genomics. Genomics 2002, 80, 691–698. [Google Scholar]
- Hataoka, Y.; Zhang, L.; Mori, Y.; Tomita, N.; Notomi, T.; Baba, Y. Analysis of specific gene by integration of isothermal amplification and electrophoresis on poly(methyl methacrylate) microchips. Anal. Chem. 2004, 76, 3689–3693. [Google Scholar] [CrossRef]
- Parida, M.; Horioke, K.; Ishida, H.; Dash, P.K.; Saxena, P.; Jana, A.M.; Islam, M.A.; Inoue, S.; Hosaka, N.; Morita, K. Rapid detection and differentiation of dengue virus serotypes by a real-time reverse transcription-loop-mediated isothermal amplification assay. J. Clin. Microbiol. 2005, 43, 2895–2903. [Google Scholar] [CrossRef]
- Takakura, S.; Tsuchiya, S.; Fujihara, N.; Kudo, T.; Iinuma, Y.; Mitarai, S.; Ichiyama, S.; Yasukawa, K.; Ishiguro, T. Isothermal RNA sequence amplification method for rapid antituberculosis drug susceptibility testing of Mycobacterium tuberculosis. J. Clin. Microbiol. 2005, 43, 2489–2491. [Google Scholar]
- Demidov, V.V. Rolling-circle amplification in DNA diagnostics: the power of simplicity. Expert Rev. Mol. Diagn. 2002, 2, 89–95. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Chandler, D.P.; Bryant, L.; Griesemer, S.B.; Gu, R.; Knickerbocker, C.; Kukhtin, A.; Parker, J.; Zimmerman, C.; George, K.S.; Cooney, C.G. Integrated Amplification Microarrays for Infectious Disease Diagnostics. Microarrays 2012, 1, 107-124. https://doi.org/10.3390/microarrays1030107
Chandler DP, Bryant L, Griesemer SB, Gu R, Knickerbocker C, Kukhtin A, Parker J, Zimmerman C, George KS, Cooney CG. Integrated Amplification Microarrays for Infectious Disease Diagnostics. Microarrays. 2012; 1(3):107-124. https://doi.org/10.3390/microarrays1030107
Chicago/Turabian StyleChandler, Darrell P., Lexi Bryant, Sara B. Griesemer, Rui Gu, Christopher Knickerbocker, Alexander Kukhtin, Jennifer Parker, Cynthia Zimmerman, Kirsten St. George, and Christopher G. Cooney. 2012. "Integrated Amplification Microarrays for Infectious Disease Diagnostics" Microarrays 1, no. 3: 107-124. https://doi.org/10.3390/microarrays1030107
APA StyleChandler, D. P., Bryant, L., Griesemer, S. B., Gu, R., Knickerbocker, C., Kukhtin, A., Parker, J., Zimmerman, C., George, K. S., & Cooney, C. G. (2012). Integrated Amplification Microarrays for Infectious Disease Diagnostics. Microarrays, 1(3), 107-124. https://doi.org/10.3390/microarrays1030107