Pathogens 2019, 8(4), 196; https://doi.org/10.3390/pathogens8040196 - 18 Oct 2019
Cited by 9 | Viewed by 4714
Abstract
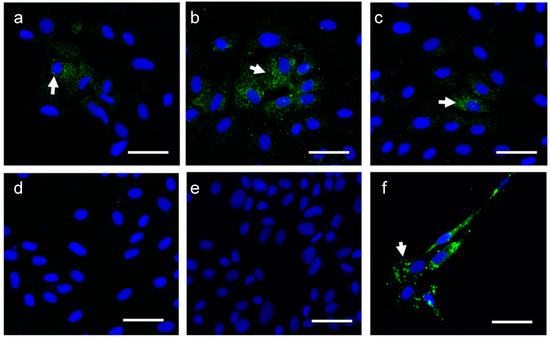
Tissue resident memory (TRM) CD8 T cells comprise a memory population that forms in peripheral, non-lymphoid tissues after an infection that does not recirculate into the bloodstream or other tissues. TRM cells often recognize conserved peptide epitopes shared among different
[...] Read more.
Tissue resident memory (TRM) CD8 T cells comprise a memory population that forms in peripheral, non-lymphoid tissues after an infection that does not recirculate into the bloodstream or other tissues. TRM cells often recognize conserved peptide epitopes shared among different strains of a pathogen and so offer a protective role upon secondary encounter with the same or related pathogens. Several recent studies have begun to shed light on the intrinsic and extrinsic factors regulating TRM. In addition, work is being done to understand how canonical “markers” of TRM actually affect the function of these cells. Many of these markers regulate the generation or persistence of these TRM cells, an important point of study due to the differences in persistence of TRM between tissues, which may impact future vaccine development to cater towards these important differences. In this review, we will discuss recent advances in TRM biology that may lead to strategies designed to promote this important protective immune subset.
Full article
(This article belongs to the Special Issue Influenza Virus and Vaccination)
►
Show Figures
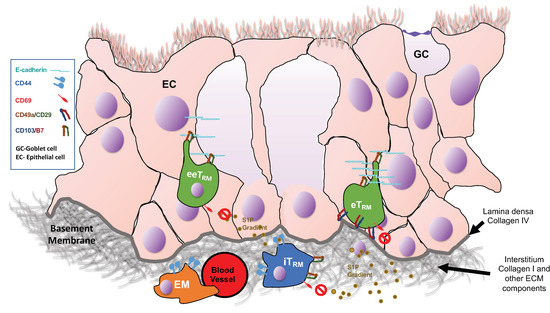
Figure 1






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}