Microorganisms 2020, 8(4), 529; https://doi.org/10.3390/microorganisms8040529 - 7 Apr 2020
Cited by 38 | Viewed by 4976
Abstract
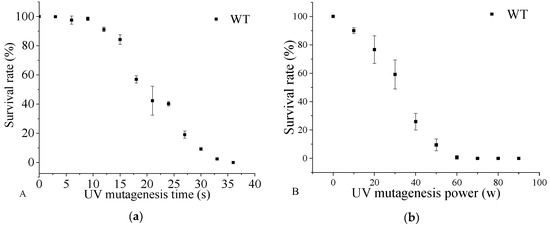
Docosahexaenoic acid (DHA), a n-3 long-chain polyunsaturated fatty acid, is critical for physiological activities of the human body. Marine eukaryote Aurantiochytrium sp. is considered a promising source for DHA production. Mutational studies have shown that ultraviolet (UV) irradiation (50 W, 30 s)
[...] Read more.
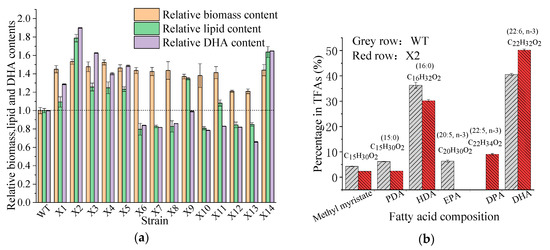
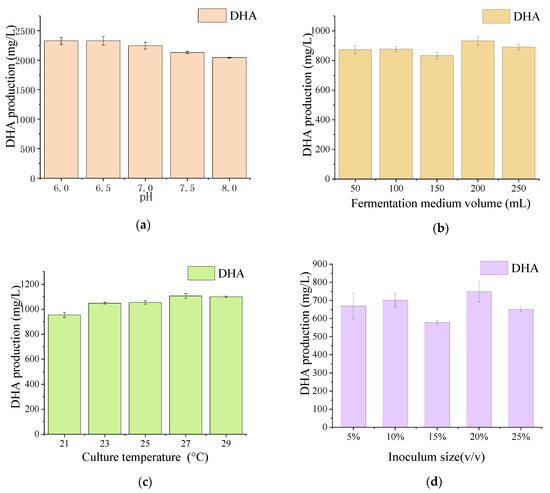
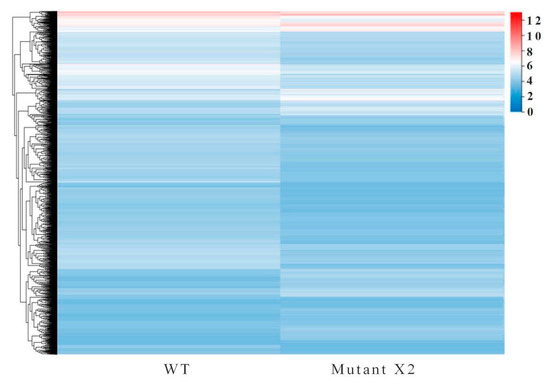
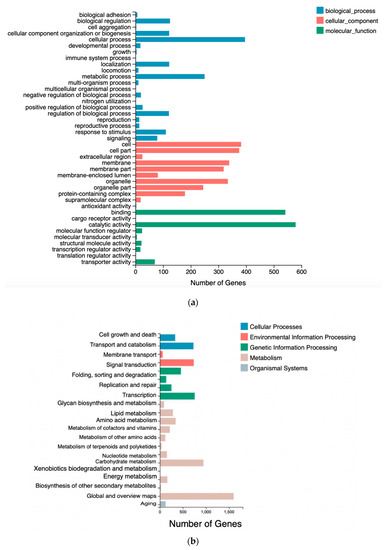
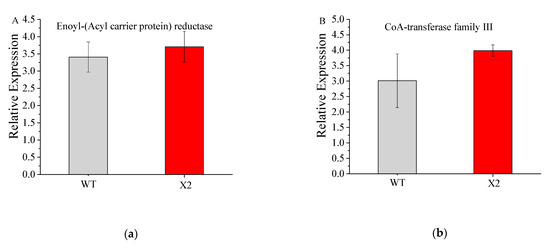
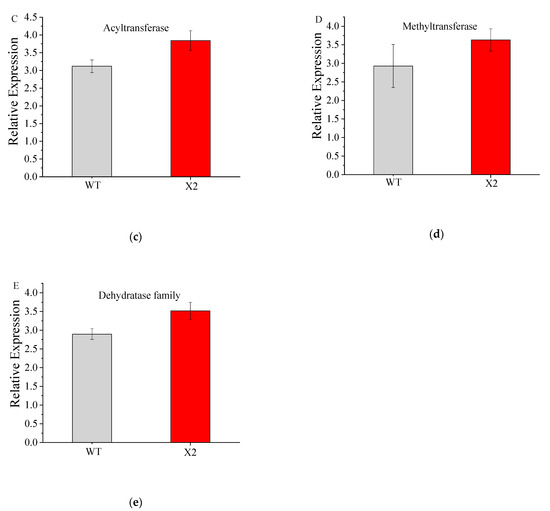
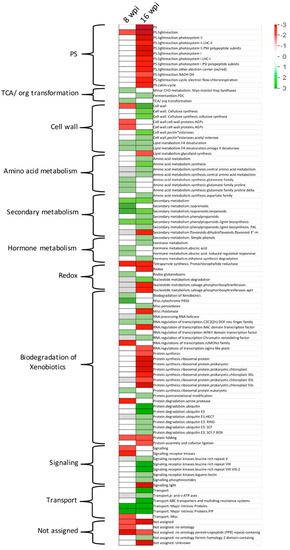
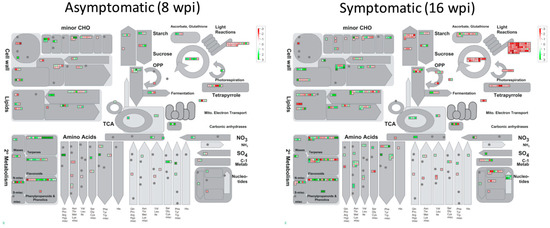
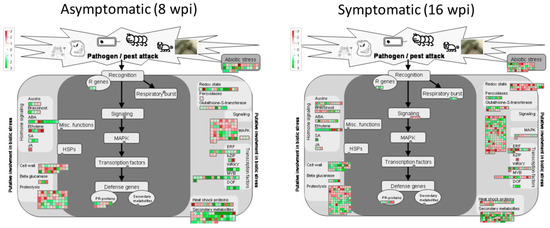
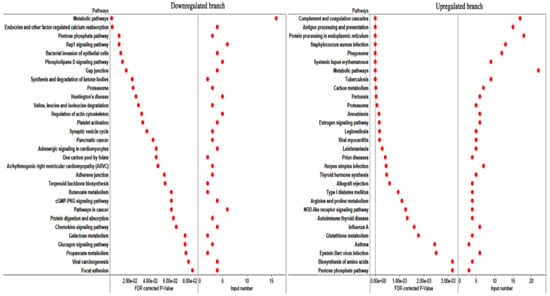
Docosahexaenoic acid (DHA), a n-3 long-chain polyunsaturated fatty acid, is critical for physiological activities of the human body. Marine eukaryote Aurantiochytrium sp. is considered a promising source for DHA production. Mutational studies have shown that ultraviolet (UV) irradiation (50 W, 30 s) could be utilized as a breeding strategy for obtaining high-yield DHA-producing Aurantiochytrium sp. After UV irradiation (50 W, 30 s), the mutant strain X2 which shows enhanced lipid (1.79-fold, 1417.37 mg/L) and DHA (1.90-fold, 624.93 mg/L) production, was selected from the wild Aurantiochytrium sp. Instead of eicosapentaenoic acid (EPA), 9.07% of docosapentaenoic acid (DPA) was observed in the mutant strain X2. The comparative transcriptomic analysis showed that in both wild type and mutant strain, the fatty acid synthesis (FAS) pathway was incomplete with key desaturases, but genes related to the polyketide synthase (PKS) pathway were observed. Results presented that mRNA expression levels of CoAT, AT, ER, DH, and MT down-regulated in wild type but up-regulated in mutant strain X2, corresponding to the increased intercellular DHA accumulation. These findings indicated that CoAT, AT, ER, DH, and MT can be exploited for high DHA yields in Aurantiochytrium.
Full article
(This article belongs to the Special Issue Microbial Secondary Metabolites and Biotechnology)
►
Show Figures
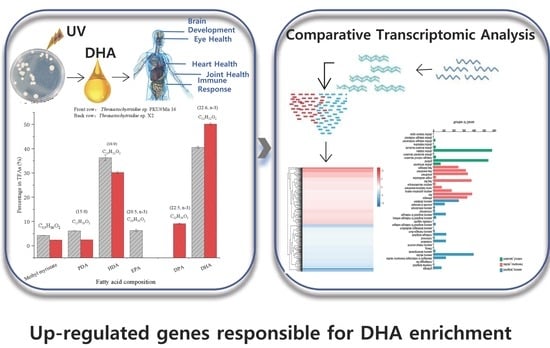
Graphical abstract







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}