by
Carin A. T. C. Lunenburg 1, Linda M. Henricks 2,3, André B. P. Van Kuilenburg 4, Ron H. J. Mathijssen 5, Jan H. M. Schellens 2,3, Hans Gelderblom 1 , Henk-Jan Guchelaar 6 and Jesse J. Swen 6,*
, Henk-Jan Guchelaar 6 and Jesse J. Swen 6,*
, Henk-Jan Guchelaar 6 and Jesse J. Swen 6,*
1
Department of Medical Oncology, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands
2
Department of Clinical Pharmacology, Division of Medical Oncology, The Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands
3
Division of Pharmacology, The Netherlands Cancer Institute, 1066 CX Amsterdam, The Netherlands
4
Department of Clinical Chemistry, Amsterdam University Medical Centre, 1105 AZ Amsterdam, The Netherlands
5
Department of Medical Oncology, Erasmus MC Cancer Institute, 3015 GD Rotterdam, The Netherlands
6
Department of Clinical Pharmacy & Toxicology, Leiden University Medical Center, 2333 ZA Leiden, The Netherlands
Genes 2018, 9(12), 585; https://doi.org/10.3390/genes9120585 - 28 Nov 2018
Cited by 12 | Viewed by 14478
Abstract
DPYD genotyping prior to fluoropyrimidine treatment is increasingly implemented in clinical care. Without phasing information (i.e., allelic location of variants), current genotype-based dosing guidelines cannot be applied to patients carrying multiple DPYD variants. The primary aim of this study is to examine diagnostic
[...] Read more.
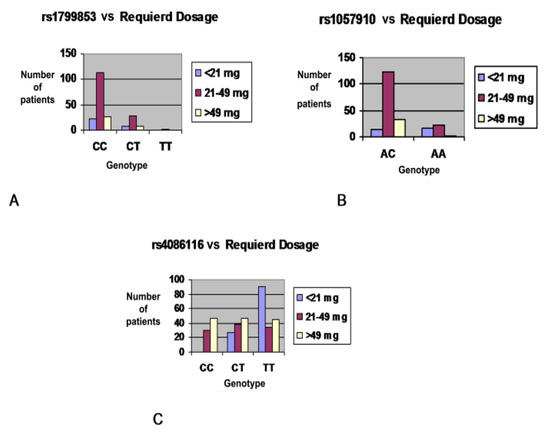
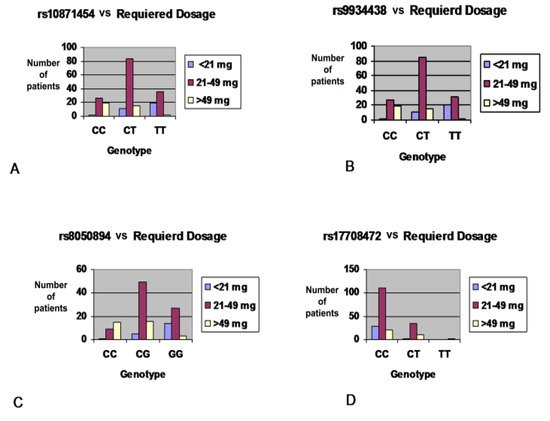
DPYD genotyping prior to fluoropyrimidine treatment is increasingly implemented in clinical care. Without phasing information (i.e., allelic location of variants), current genotype-based dosing guidelines cannot be applied to patients carrying multiple DPYD variants. The primary aim of this study is to examine diagnostic and therapeutic strategies for fluoropyrimidine treatment of patients carrying multiple DPYD variants. A case series of patients carrying multiple DPYD variants is presented. Different genotyping techniques were used to determine phasing information. Phenotyping was performed by dihydropyrimidine dehydrogenase (DPD) enzyme activity measurements. Publicly available databases were queried to explore the frequency and phasing of variants of patients carrying multiple DPYD variants. Four out of seven patients carrying multiple DPYD variants received a full dose of fluoropyrimidines and experienced severe toxicity. Phasing information could be retrieved for four patients. In three patients, variants were located on two different alleles, i.e., in trans. Recommended dose reductions based on the phased genotype differed from the phenotype-derived dose reductions in three out of four cases. Data from publicly available databases show that the frequency of patients carrying multiple DPYD variants is low (< 0.2%), but higher than the frequency of the commonly tested DPYD*13 variant (0.1%). Patients carrying multiple DPYD variants are at high risk of developing severe toxicity. Additional analyses are required to determine the correct dose of fluoropyrimidine treatment. In patients carrying multiple DPYD variants, we recommend that a DPD phenotyping assay be carried out to determine a safe starting dose.
Full article
(This article belongs to the Special Issue Pharmacogenomics and Personalized Medicine)
▼
Show Figures
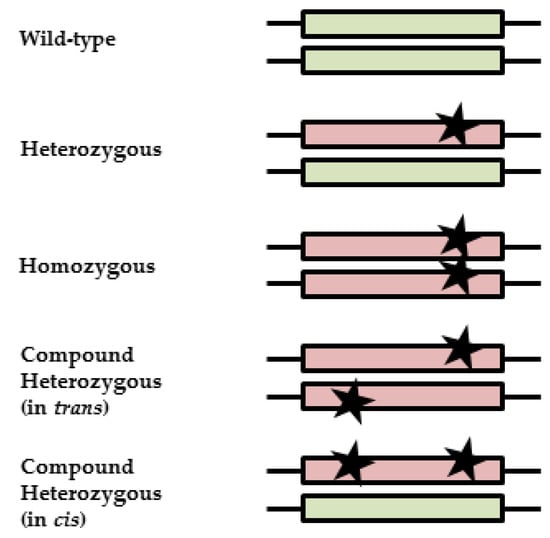
Figure 1



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}