Genes 2020, 11(7), 729; https://doi.org/10.3390/genes11070729 - 30 Jun 2020
Cited by 8 | Viewed by 5470
Abstract
The incidence of liver disease is increasing significantly worldwide and, as a result, there is a pressing need to develop new technologies and applications for end-stage liver diseases. For many of them, orthotopic liver transplantation is the only viable therapeutic option. Stem cells
[...] Read more.
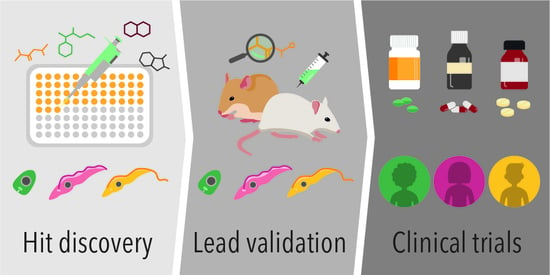
The incidence of liver disease is increasing significantly worldwide and, as a result, there is a pressing need to develop new technologies and applications for end-stage liver diseases. For many of them, orthotopic liver transplantation is the only viable therapeutic option. Stem cells that are capable of differentiating into all liver cell types and could closely mimic human liver disease are extremely valuable for disease modeling, tissue regeneration and repair, and for drug metabolism studies to develop novel therapeutic treatments. Despite the extensive research efforts, positive results from rodent models have not translated meaningfully into realistic preclinical models and therapies. The common marmoset Callithrix jacchus has emerged as a viable non-human primate model to study various human diseases because of its distinct features and close physiologic, genetic and metabolic similarities to humans. C. jacchus embryonic stem cells (cjESC) and recently generated cjESC-derived hepatocyte-like cells (cjESC-HLCs) could fill the gaps in disease modeling, liver regeneration and metabolic studies. They are extremely useful for cell therapy to regenerate and repair damaged liver tissues in vivo as they could efficiently engraft into the liver parenchyma. For in vitro studies, they would be advantageous for drug design and metabolism in developing novel drugs and cell-based therapies. Specifically, they express both phase I and II metabolic enzymes that share similar substrate specificities, inhibition and induction characteristics, and drug metabolism as their human counterparts. In addition, cjESCs and cjESC-HLCs are advantageous for investigations on emerging research areas, including blastocyst complementation to generate entire livers, and bioengineering of discarded livers to regenerate whole livers for transplantation.
Full article
(This article belongs to the Special Issue Genes at Ten)
►
Show Figures
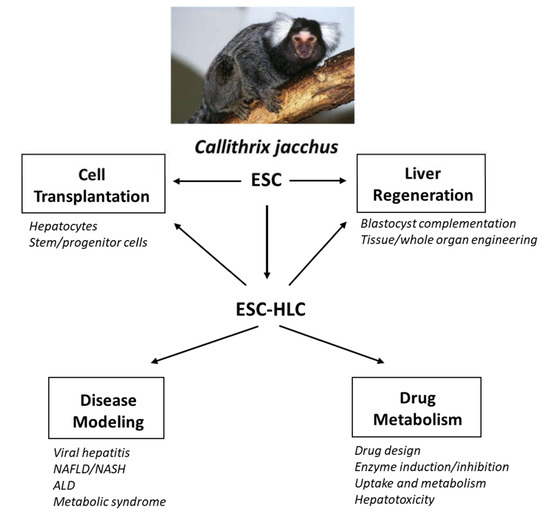
Figure 1








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}