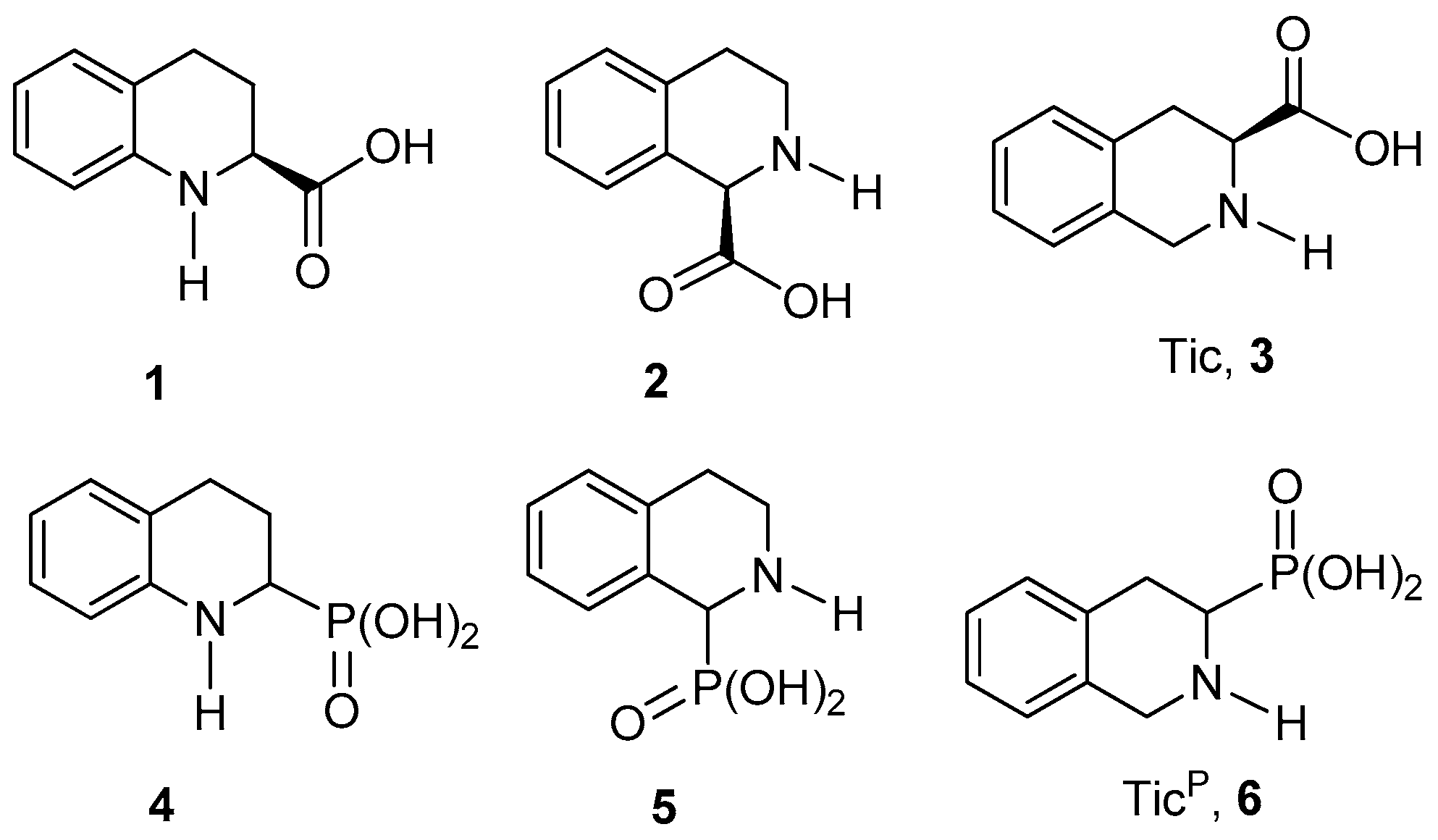
2. Results and Discussion
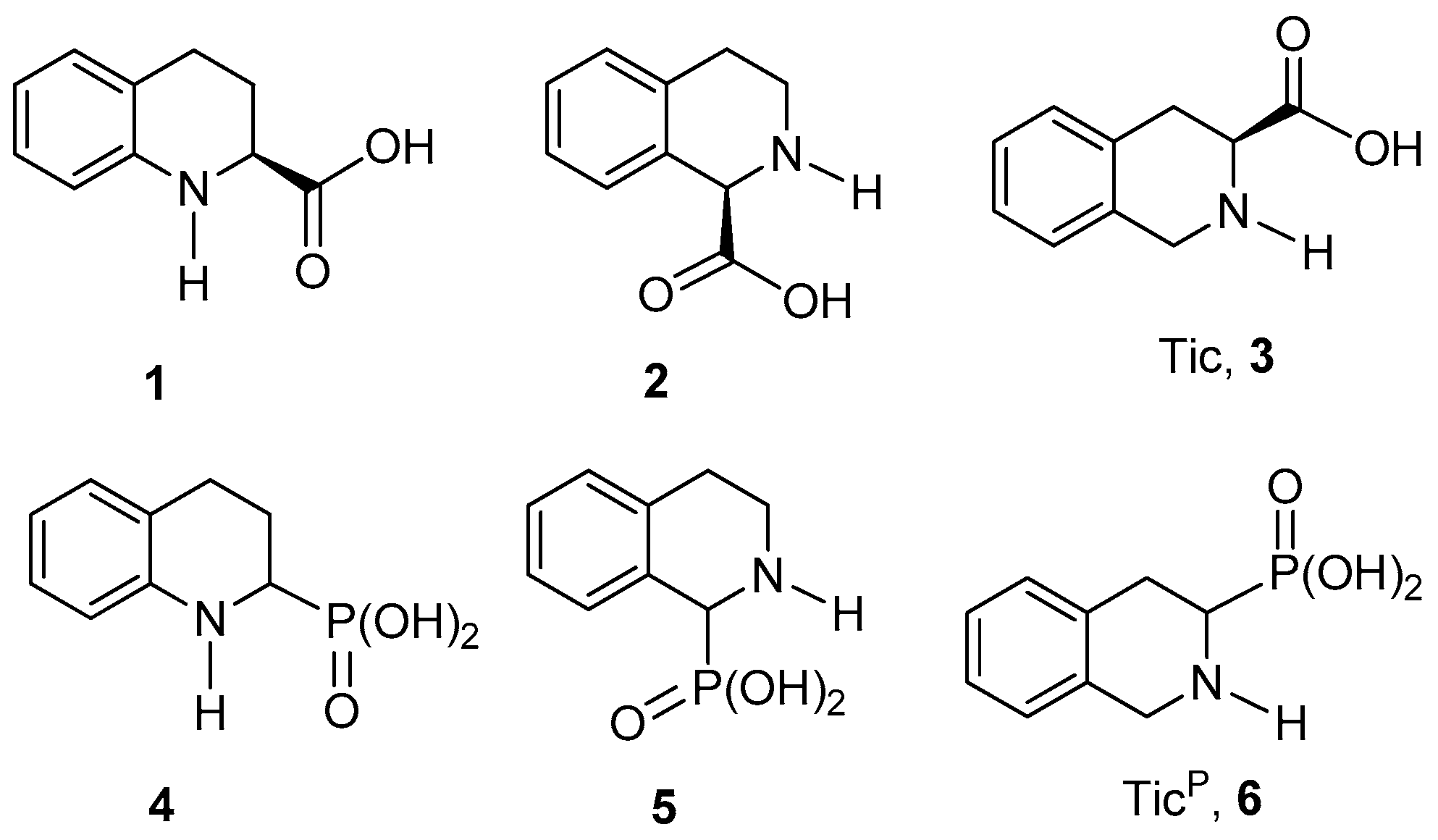
For the synthesis of 1,2,3,4-tetrahydroquinoline-2-phosphonic acid
4, we proposed the
N-acyliminium ions as suitable precursors, considering that the well-known reduction of the carbonyl group of lactams such as the 3,4-dihydro-2(1
H)-quinolinone
7, and their subsequent transformation into a
N-acyliminium ion, is one of the methods that allows the incorporation of the phosphonate functionality into the α position of the nitrogen atom such as we have previously described [
26,
27,
28]. These proposals prompted us to explore further application of the
N-acyliminium strategy for the synthesis of the target compound. In this context, the commercially available 3,4-dihydro-2(1
H)-quinolinone
7 was reacted with lithium bis(trimethylsilyl)amide (LiHMDS) as a base and benzyl chloroformate in THF at −78 °C, to obtain the
N-Cbz-3,4-dihydro-2(1
H)-quinolinone
8 in 91% yield. Reduction of the carbonyl group in
8 with diisobutylaluminium hydride (DIBAL-H) and subsequent reaction with methanol and catalytic amounts of pyridinium
p-toluenesulfonate (PPTS), gave the methoxyaminal
9, which was treated immediately with trimethyl phosphite in the presence of BF
3·OEt
2, obtaining the dimethyl
N-Cbz-1,2,3,4-tetrahydroquinoline-2-phosphonate
11 in 62% via the
N-acyliminium ion
10 in 62% yield from
8. Treatment of this
N-Cbz-protected phosphonate with a 33% solution of hydrogen bromide in acetic acid, afforded the 1,2,3,4-tetrahydroquinoline-2-phosphonic acid
4 as hydrobromide in quantitative yield (
Scheme 1).
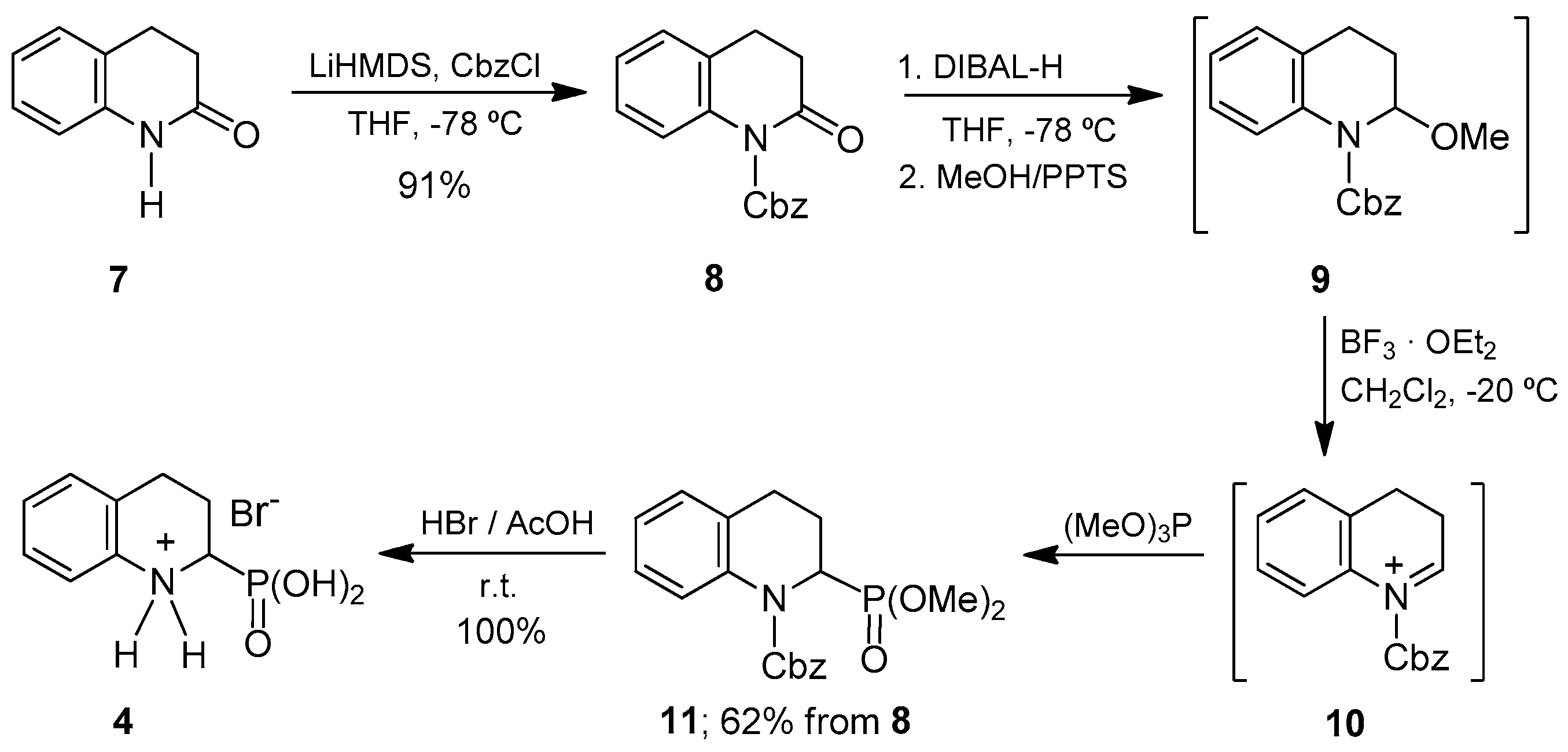
The literature has described the hydrophosphonylation of quinoline derivatives to obtain the 1,2-dihydroquinolin-2-ylphosphonate and 2,4-diphosphono-1,2,3,4-tetrahydroquinoline derivatives using activating agents [
44,
45,
46,
47,
48]. These reactions proceed via quinolinium ions that can be viewed as counterparts of the above-mentioned iminium species. Thus, the addition of an acyl chloride to quinoline can be considered as a way to generate the iminium ion necessary for subsequent regioselective incorporation of the phosphonate functionality α to the nitrogen atom.
Based on this precedent, we decided to compare the efficiency of the
N-acylquinolinium salts as intermediates in the synthesis of α-aminophosphonic acid
4. For this purpose, benzyl chloroformate was added to quinoline and the
N-Cbz-quinolinium chloride formed
12 was reacted with trimethyl phosphite in acetonitrile at 50 °C, obtaining the dimethyl
N-Cbz-1,2-dihydroquinoline-2-phosphonate derivative
13 in 74% yield from quinolone. The diphosphonylation products at the 2- and 4-positions of the quinoline ring were not detected. The regioselectivity observed in the addition of trimethyl phosphite to the
N-acylquinolinium ion
12 may be attributed to the electron-withdrawing character of the benzyloxycarbonyl group, which increases the electrophilic nature of the carbon adjacent to the nitrogen atom. Additionally, the benzyloxycarbonyl group was selected for its easy incorporation and removal under mild conditions. Thus, the catalytic hydrogenation resulted in simultaneous removal of the Cbz group and reduction of the double bond at positions 3,4 to afford the α-aminophosphonate
14, which, by treatment with hydrogen bromide in acetic acid, gave the 1,2,3,4-tetrahydroquinoline-2-phosphonic acid
4 as hydrobromide. The latter two steps proceeded quantitatively and obviated the need for purification (
Scheme 2).
The 1,2,3,4-tetrahydroquinoline-2-phosphonic acid
4 was obtained in 74% overall yield from quinoline following the synthetic strategy in
Scheme 2, which compares favorably with the 56% global yield achieved in
Scheme 1, when starting from quinolinone
1. Accordingly, the superior global yield, together with the much lower price of quinoline in comparison with 3,4-dihydro-2(1
H)-quinolinone
7, makes the methodology in
Scheme 2 more advantageous than the lactam-based one for the preparation of α-aminophosphonic acid
4. From an operational viewpoint, both routes required few purification steps—by column chromatography—of intermediate compounds.
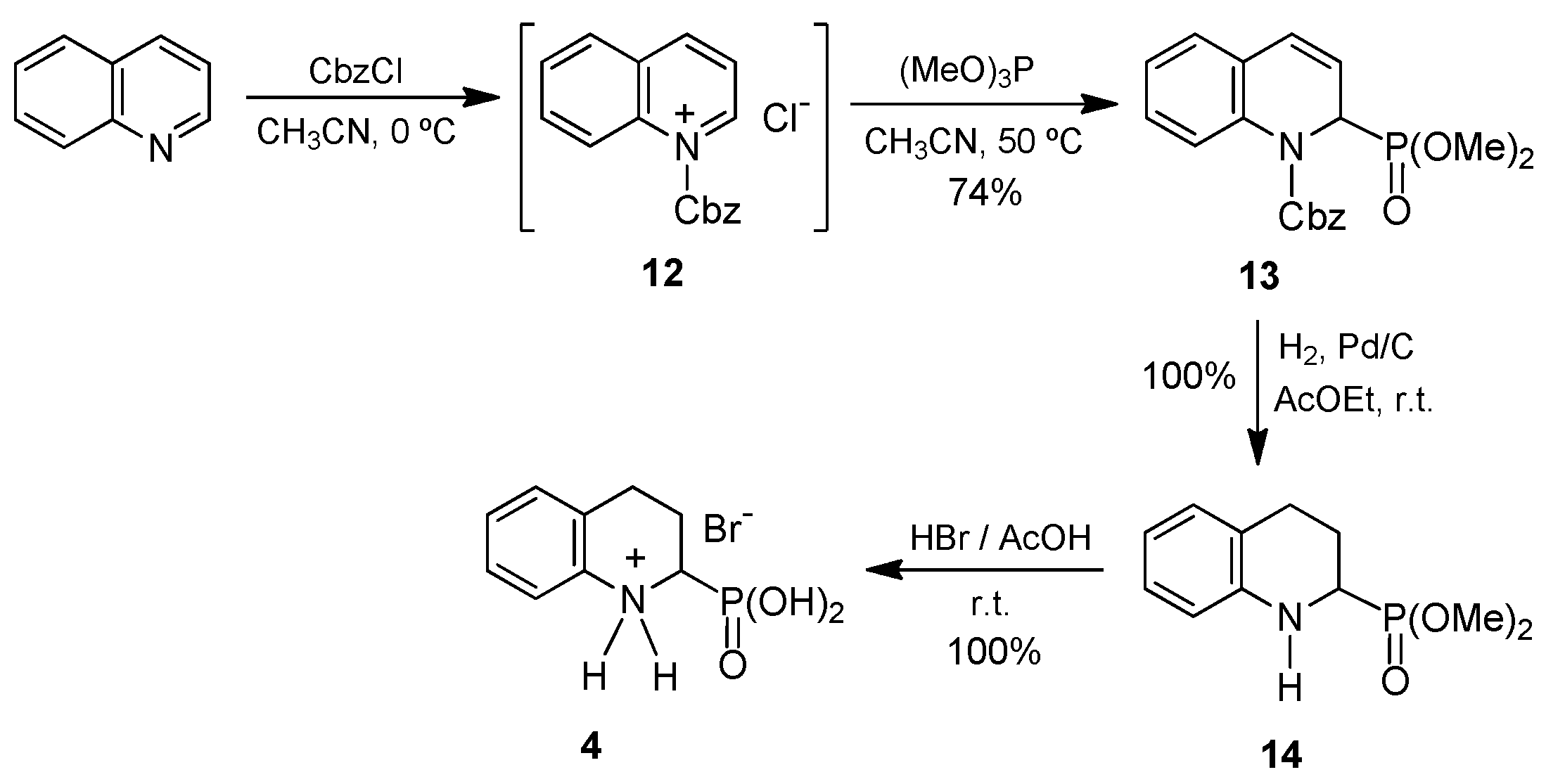
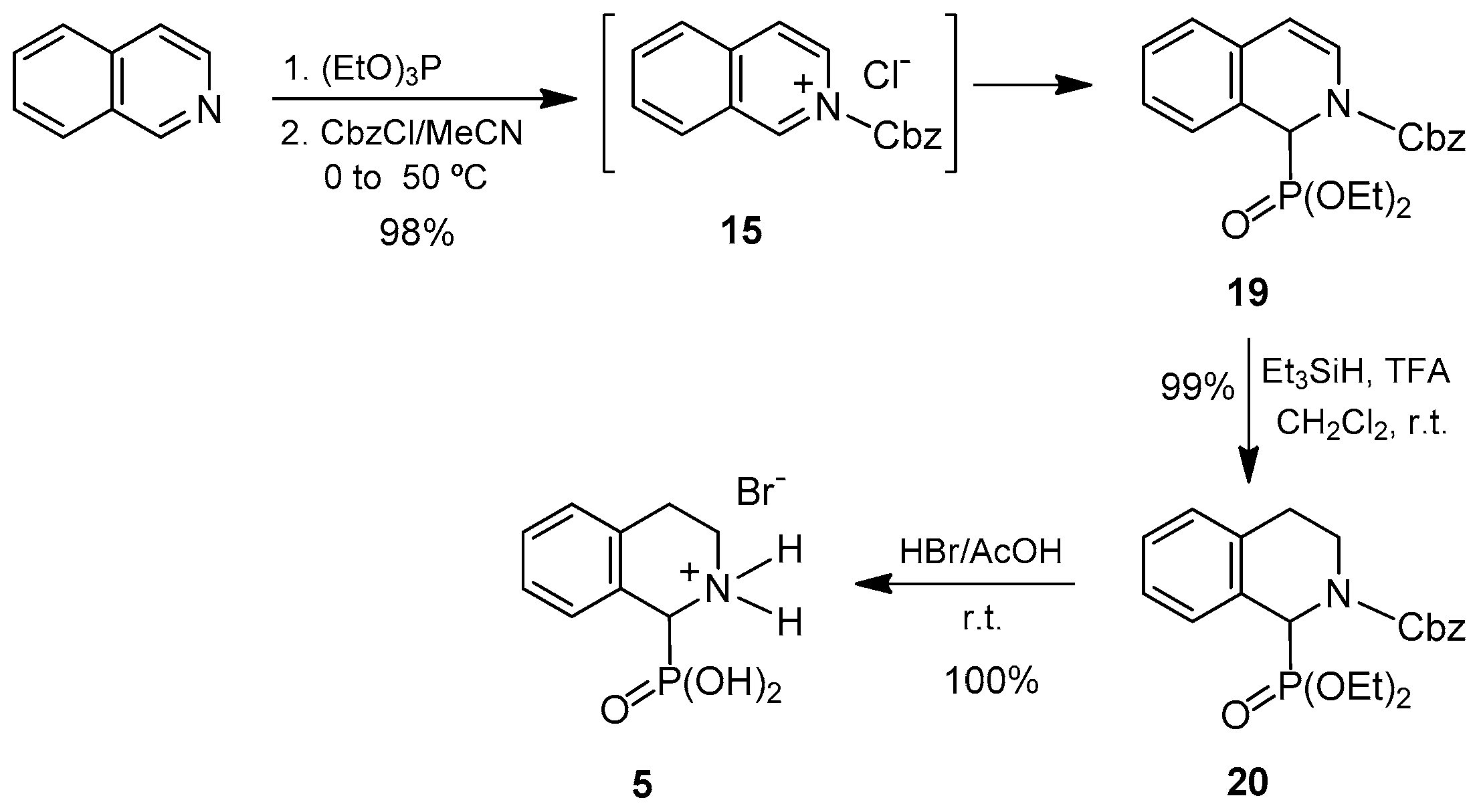
We next addressed the preparation of 1,2,3,4-tetrahydroisoquinoline-1-phosphonic acid
5. Similarly to that described above for the quinoline counterpart, the preparation of
N-substituted-1,2-dihydroisoquinoline-1-phosphonates through the isoquinolinium salts formed by reaction of isoquinoline with acyl chlorides and subsequent addition of trialkyl phosphites has been described [
49,
50]. Thus, the synthesis of dimethyl
N-Cbz-1,2-dihydroisoquinoline-1-phosphonate should be straightforward, as shown above for the preparation of dimethyl
N-Cbz-1,2-dihydroquinoline-2-phosphonate
13.
On the other hand, the lactam to be used as a starting product for the preparation of the target amino acid 5 is not easily available from commercial sources in this case. This fact together with the advantageous preparation of amino acid 4 via the formation of quinolinium salt prompted us to undertake the synthesis of 1,2,3,4-tetrahydroisoquinoline-1-phosphonic acid following an analogous synthetic route.
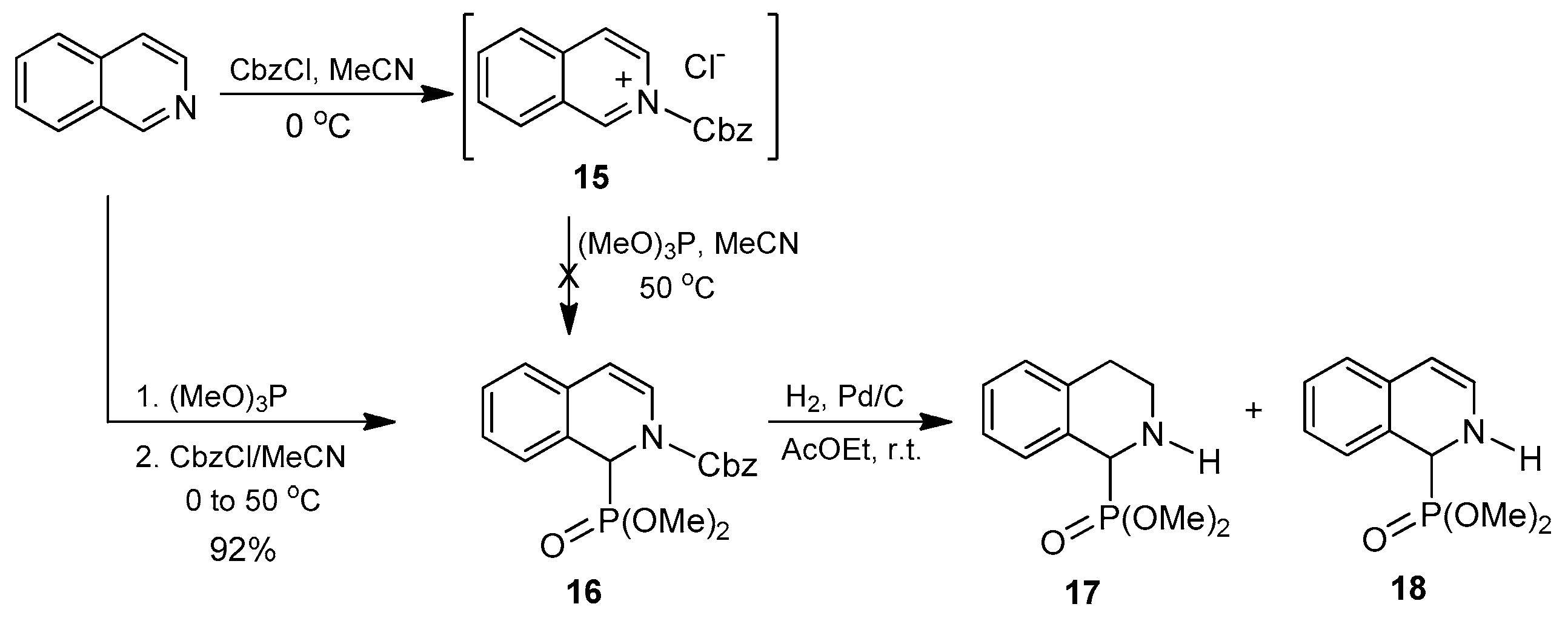
Thus, using the reaction conditions described above, the isoquinoline was reacted with benzyl chloroformate followed by the addition of trimethyl phosphite in acetonitrile at 50 °C. However, under these reaction conditions, the desired compound
16 was not obtained. It is noteworthy that during the addition of benzyl chloroformate to isoquinoline, a yellow solid was formed, which was later identified as the isoquinolinium salt
15. This compound remained unaltered upon addition of trimethyl phosphite so that the formation of the expected 1,2-dihydroisoquinoline-1-phosphonate
16 did not take place. We reasoned that if the trimethyl phosphite was present in the reaction medium before benzyl chloroformate was added, the isoquinolinium ion formed
16 would immediately react with it, thus preventing precipitation. To our delight, when the order of addition of these reagents was exchanged (that is, trimethyl phosphite prior to benzyl chloroformate), the dimethyl
N-Cbz-1,2-dihydroisoquinoline-1-phosphonate
16 was obtained at 92% yield. In the next step, we carried out the catalytic hydrogenation of the double bond in
16 under an atmospheric pressure of hydrogen gas and using Pd/C as a catalyst, to generate the tetrahydroisoquinoline moiety. However, the Cbz group in
16 was readily eliminated at room temperature, whereas the double bond at positions 3,4 was only partially hydrogenated even after long reaction times. As a consequence, mixtures of the desired tetrahydroisoquinoline derivative
17 and the analogous dihydroisoquinoline
18 were obtained. Attempts to improve this result by changing the solvent as well as by increasing the reaction temperature or hydrogen gas pressure proved unsuccessful and a mixture of
17 and
18 was always obtained (
Scheme 3).
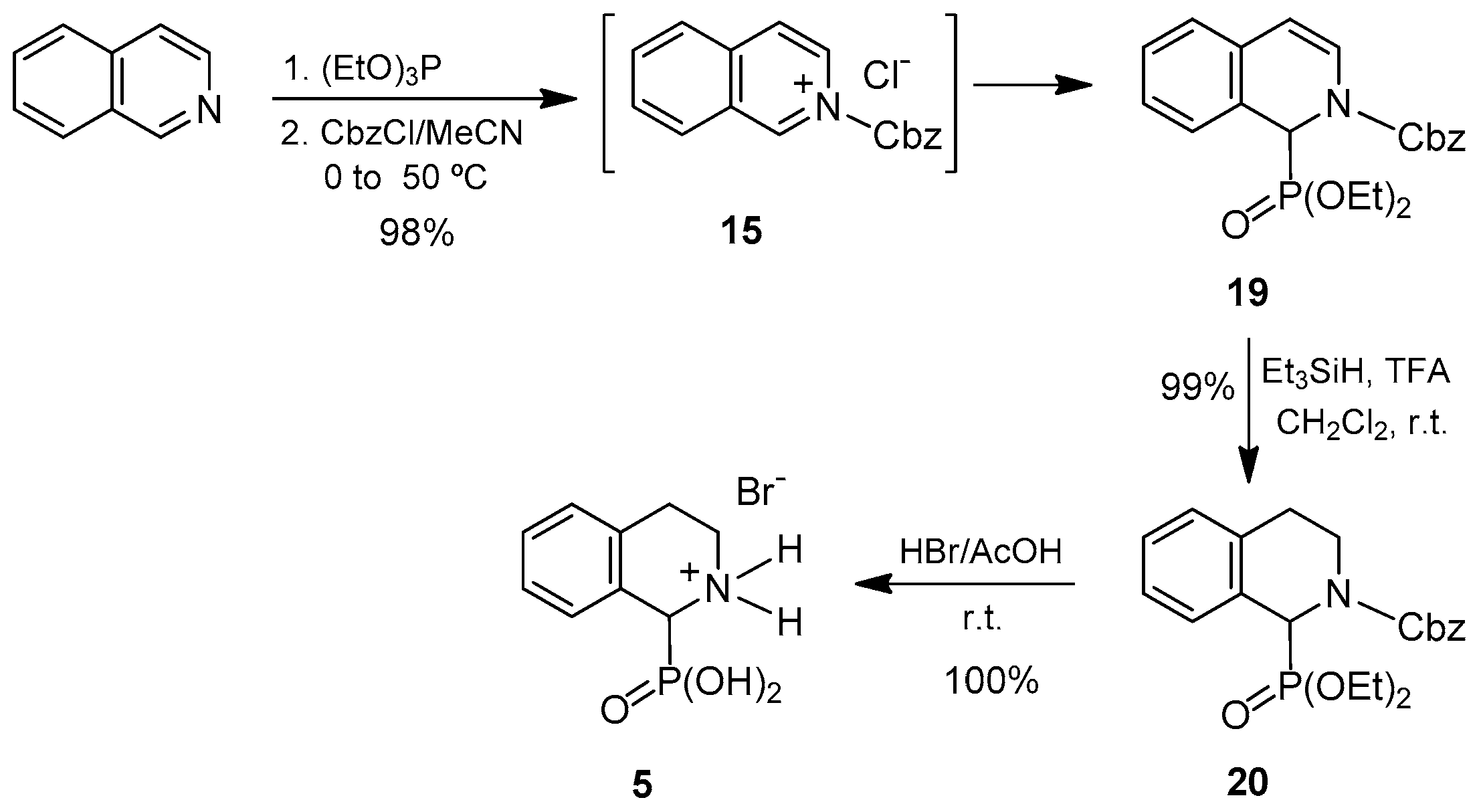
To circumvent this problem, we synthesized the compound analogous to
16 that bears the less acid-sensitive diethyl phosphonate group
19. A procedure identical to that established for the dimethyl derivative but using triethyl phosphite as the phosphorus source provided the desired diethyl
N-Cbz-1,2-dihydroisoquinoline-1-phosphonate
19 in 98% yield from isoquinoline. Taking into account the fact that compound
19 can be considered not only as an
N-acyl-α-aminophosphonate, but also as an enamide, we decided to perform the reduction of the double bond using trifluoroacetic acid and triethylsilane as the reducing agent, following the conditions described by Jacobsen et al. [
51] for cyclic enamides. In this case, the diethyl
N-Cbz-1,2,3,4-tetrahydroisoquinoline-1-phosphonate
20 was readily obtained in quantitative yield by reaction with triethylsilane and trifluoroacetic acid. Subsequent cleavage of the protecting groups in
20 by treatment with hydrogen bromide in acetic acid, afforded the 1,2,3,4-tetrahydroisoquinoline-1-phosphonic acid
5 as hydrobromide in quantitative yield. This compound was isolated in 97% overall yield from isoquinoline (
Scheme 4).
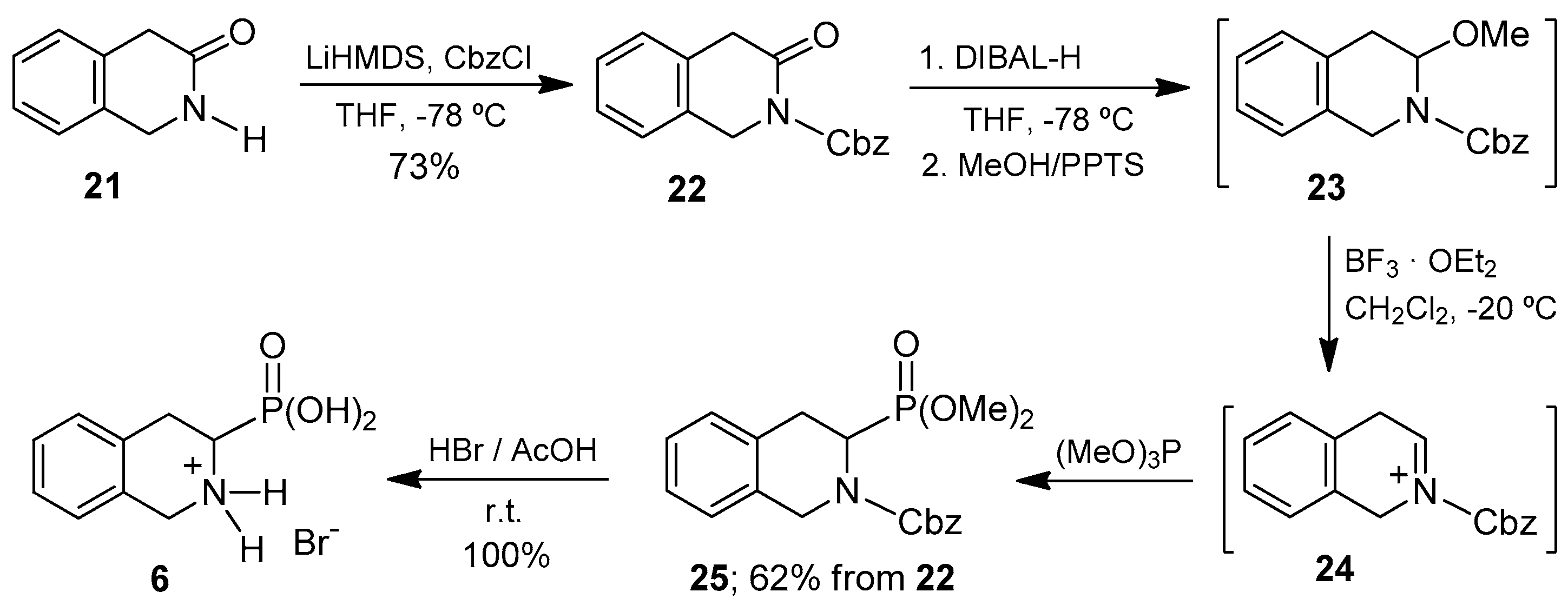
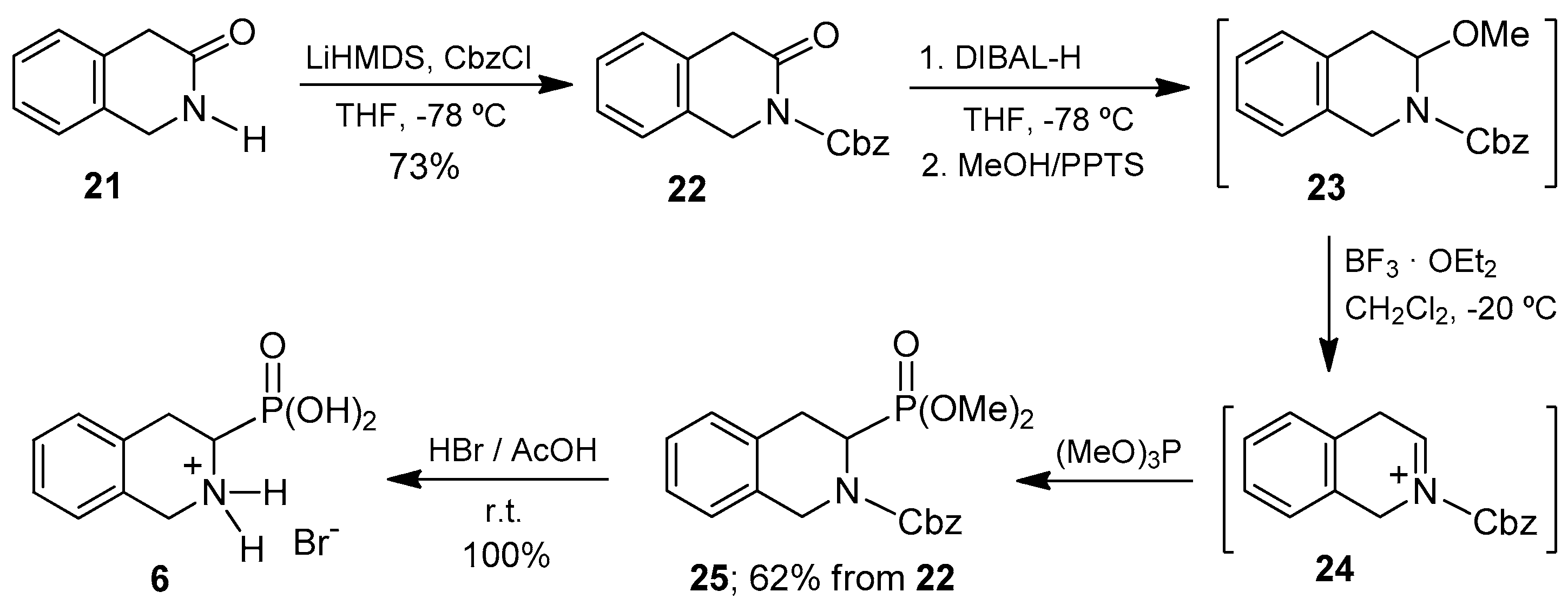
Finally, we focused on the preparation of 1,2,3,4-tetrahydroisoquinoline-3-phosphonic acid
6, that is, the phosphonic analogue of Tic (Tic
P). This compound exhibits a tetrahydroisoquinoline core, as does the aminophosphonic acid
5. However, in the case of Tic
P, the phosphonate group is at the 3-position. However, the isoquinoline selectively provides the phosphonate group at the 1-position of the ring. Therefore, we believe that the generation of a suitable iminium ion for the introduction of the phosphonate moiety at the desired 3-position, the lactam precursor was the only route to consider in this case. It should be noted that, following this strategy, the position of the carbonyl group in the starting lactam determines with complete regiocontrol the introduction of the phosphonate substituent, which is a distinctive advantage of this methodology. The adequate lactam
21 used in the synthesis of the target amino acid was easily prepared following reported procedures that involved reaction of 2-phenylacetyl chloride with aqueous ammonia to give 2-phenylacetamide [
52] and subsequent condensation with formaldehyde [
53]. Lactam
21 was readily transformed into 1,2,3,4-tetrahydroisoquinoline-3-phosphonic acid (Tic
P,
6) following the synthetic route strictly similar to that reported above for the preparation of the α-aminophosphonic acid
4 from lactam
7 (
Scheme 1). The 1,2,3,4-tetrahydroisoquinoline-3-phosphonic acid
6 was thus obtained as hydrobromide in 52% global yield from the starting substrate
21, with isolation and purification of only two synthetic intermediates
22,
25 (
Scheme 5).
The results obtained show the successful use of the lactam 21 to generate the key intermediate N-acyliminium ion, which by phosphonylation, afforded the desired diethyl N-Cbz-1,2,3,4-tetrahydroisoquinoline-3-phosphonate 25, demonstrating thus the versatility of this methodology in the preparation of α-aminophosphonic acids structurally related to pipecolic acid.
3. Materials and Methods
3.1. General
All reagents were used as received from commercial suppliers (Sigma-Aldrich Chemie GmbH, Buchs, Switzerland) without further purification. Thin-layer chromatography (TLC) was performed on Macherey-Nagel Polygram
® SIL G/UV
254 (Macherey-Nagel, Duren, Germany) precoated silica gel polyester plates. The products were visualized by exposure to UV light (254 nm), iodine vapour or ethanolic solution of phosphomolybdic acid. Column chromatography was performed using 60 M (0.04–0.063 mm) silica gel from Macherey-Nagel. Melting points were determined on a Gallenkamp apparatus (Weiss Gallenkamp, Loughborough, UK). IR spectra were registered on a Nicolet Avatar 360 FTIR spectrophotometer (Thermo Electron Corporation, Madison, WI, USA); ν
max is given for the main absorption bands.
1H-,
13C- and
31P-NMR spectra were recorded on Bruker AV-400 or AV-300 instruments (Bruker BioSpin GmbH, Rheinstetten, Germany) at room temperature, unless otherwise indicated, using the residual solvent signal as the internal standard; chemical shifts (δ) are expressed in ppm and coupling constants (
J) in Hertz. High-resolution mass spectra were obtained on a Bruker Microtof-Q spectrometer. Compound
22 was prepared by reaction of 2-phenylacetamide [
52] with formaldehyde [
53].
1H-,
13C- and
31P- NMR spectra of all final compounds are showed in the supplementary material (
Figures S1–S34).
3.2. Synthesis of N-Benzyloxycarbonyl-3,4-dihydro-2-quinolinone 8
A 1 M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (2.80 mL, 2.80 mmol) was slowly added to a solution of 3,4-dihydro-2(1H)-quinolinone 7 (400 mg, 2.72 mmol) in anhydrous tetrahydrofuran (10 mL) kept at −78 °C under argon. After 30 min, benzyl chloroformate (0.39 mL, 464 mg, 2.72 mmol) was added dropwise and stirring was continued for additional 3 h. The reaction mixture was then treated with saturated aqueous ammonium chloride (10 mL) and allowed to warm to room temperature. The two layers were separated and the aqueous phase was extracted with dichloromethane (2 × 20 mL). The combined organic extracts were dried, filtered, and concentrated. Purification by column chromatography (eluent:hexanes/ethyl acetate 4:1) afforded 8 as a colourless oil (694 mg, 2.47 mmol, 91% yield). IR (neat) νmax 1773, 1699 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 7.48–7.33 (m, 5H, Ar), 7.20–7.13 (m, 2H, Ar), 7.10–7.04 (m, 1H, Ar), 6.92–6.88 (m, 1H, Ar), 5.41 (s, 2H, CH2Ph), 2.98–2.92 (m, 2H, H-4), 2.72–2.67 (m, 2H, H-3) ppm. 13C-NMR (100 MHz, CDCl3): δ = 169.90 (CO), 153.46 (COO), 136.94 (Ar), 134.52 (Ar), 128.85 (Ar), 128.76 (Ar), 128.75 (Ar), 127.95 (Ar), 127.39 (Ar), 126.95 (Ar), 124.78 (Ar), 118.65 (Ar), 70.05 (CH2Ph), 33.05 (C-3), 25.55 (C-4) ppm. HRMS (ESI): calcd. for C17H15NNaO3 [M + Na]+ 304.0944; found 304.0947.
3.3. Synthesis of Dimethyl N-benzyloxycarbonyl-1,2,3,4-tetrahydroquinoline-2-phosphonate 11
A 1 M solution of diisobutylaluminium hydride in hexanes (2.40 mL, 2.40 mmol) was slowly added to a solution of 8 (448 mg, 1.59 mmol) in anhydrous tetrahydrofuran (8 mL) kept at −78 °C under argon. After stirring at this temperature for 2 h, the reaction was treated with saturated aqueous sodium acetate (5 mL) and warmed to room temperature. A 3:1 mixture of diethyl ether and saturated aqueous ammonium chloride (16 mL) was then added and the resulting mixture was stirred at room temperature until a suspension was formed. The solid was filtered off under reduced pressure and washed with diethyl ether (2 × 10 mL). The organic layer was separated and the aqueous phase was extracted with diethyl ether (2 × 20 mL). The combined organic extracts were washed with brine (20 mL), dried, filtered, and evaporated to provide the hemiaminal as an oil. It was dissolved in methanol (6 mL) and treated with pyridinium p-toluenesulfonate (40 mg, 0.16 mmol). After stirring at room temperature for 2 h, triethylamine (0.10 mL, 74 mg, 0.73 mmol) was added. The solvent was evaporated and the crude methoxyaminal 9 thus obtained was dissolved in anhydrous dichloromethane (7 mL) and kept under argon. Trimethyl phosphite (0.19 mL, 198 mg, 1.59 mmol) was added and the resulting solution was cooled to −20 °C. Boron trifluoride-diethyl ether (0.20 mL, 226 mg, 1.59 mmol) was added dropwise and the reaction mixture was slowly warmed to room temperature and stirred for 12 h. After quenching with water (2 mL), the two layers were separated and the aqueous phase was extracted with dichloromethane (2 × 10 mL). The combined organic extracts were dried, filtered, and concentrated. Purification by column chromatography (eluent:ethyl acetate/hexanes 4:1) afforded 11 as a colourless oil (372 mg, 0.99 mmol, 62% yield). IR (neat) νmax 1702, 1279, 1029 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 7.53–7.46 (m, 1H, Ar), 7.37–7.28 (m, 5H, Ar), 7.22–7.16 (m, 1H, Ar), 7.14–7.06 (m, 2H, Ar), 5.31 (d, J = 12.5 Hz, 1H, CH2Ph), 5.16 (d, J = 12.5 Hz, 1H, CH2Ph), 5.04 (ddd, J = 13.4, 8.7, 7.3 Hz, 1H, H-2), 3.63 (d, J = 10.6 Hz, 3H, OMe), 3.51 (d, J = 10.6 Hz, 3H, OMe), 2.85–2.75 (m, 1H, H-4), 2.67–2.57 (m, 1H, H-4′), 2.54–2.40 (m, 1H, H-3), 2.17–2.03 (m, 1H, H-3′) ppm. 13C-NMR (100 MHz, CDCl3): δ = 154.68 (CO), 136.84 (Ar), 136.08 (Ar), 132.78 (Ar), 128.54 (Ar), 128.16 (Ar), 127.88 (Ar), 127.56 (Ar), 126.36 (Ar), 125.97 (Ar), 125.22 (Ar), 68.11 (CH2Ph), 53.07 (d, J = 6.2 Hz, OMe), 52.82 (d, J = 7.2 Hz, OMe), 49.33 (d, J = 158.2 Hz, C-2), 25.75 (C-3), 25.66 (C-4) ppm. 31P-NMR (162 MHz, CDCl3): δ = 26.39 ppm. HRMS (ESI): calcd. for C19H22NNaO5P [M + Na]+ 398.1128; found 398.1145.
3.4. Synthesis of Dimethyl N-Benzyloxycarbonyl-1,2-dihydroquinoline-2-phosphonate 13
Benzyl chloroformate (0.61 mL, 726 mg, 4.26 mmol) was added dropwise to a solution of quinoline (0.46 mL, 500 mg, 3.87 mmol) in anhydrous acetonitrile (6 mL) kept at 0 °C under argon. After stirring at this temperature for 10 min, trimethyl phosphite (0.50 mL, 528 mg, 4.26 mmol) was slowly added followed by sodium iodide (853 mg, 5.69 mmol). The mixture was heated at 50 °C for 10 min. The solvent was evaporated and the crude product was partitioned between dichloromethane (15 mL) and saturated aqueous sodium bicarbonate (15 mL). The organic phase was separated and the aqueous layer was further extracted with dichloromethane (3 × 15 mL). The combined organic extracts were washed with brine (10 mL), dried, filtered, and concentrated. Purification by column chromatography (eluent:ethyl acetate/hexanes 4:1) afforded 13 as a colourless oil (1.07 g, 2.87 mmol, 74% yield). IR (neat) νmax 1706, 1653, 1603, 1263, 1125, 1026 cm−1. 1H-NMR (400 MHz, DMSO-d6, 80 °C): δ = 7.58–7.52 (m, 1H, Ar), 7.44–7.30 (m, 5H, Ar), 7.25–7.07 (m, 3H, Ar), 6.71–6.65 (m, 1H, H-4), 6.07 (ddd, J = 9.5, 6.3, 4.5 Hz, 1H, H-3), 5.61 (ddd, J = 21.4, 6.3, 1.2 Hz, 1H, H-2), 5.27 (s, 2H, CH2Ph), 3.47 (d, J = 10.6 Hz, 6H, OMe) ppm. 13C-NMR (100 MHz, DMSO-d6, 80 °C): δ = 152.90 (d, J = 4.7 Hz, CO), 135.68 (Ar), 134.66 (Ar), 128.05 (Ar), 127.70 (Ar), 127.40 (Ar), 127.35 (Ar), 127.01 (d, J = 9.7 Hz, C-4), 126.77 (d, J = 4.0 Hz, Ar), 126.06 (d, J = 1.5 Hz, Ar), 124.35 (Ar), 123.66 (Ar), 122.55 (d, J = 3.0 Hz, C-3), 67.44 (CH2Ph), 52.70 (d, J = 7.0 Hz, OMe), 52.39 (d, J = 6.6 Hz, OMe), 50.84 (d, J = 151.5 Hz, C-2) ppm. 31P-NMR (162 MHz, DMSO-d6, 80 °C): δ = 20.41 ppm. HRMS (ESI): calcd. for C19H20NNaO5P [M + Na]+ 396.0971; found 396.0991.
3.5. Synthesis of Dimethyl 1,2,3,4-Tetrahydroquinoline-2-phosphonate 14
A mixture of 13 (200 mg, 0.54 mmol) and 10% Pd/C (20 mg) in ethyl acetate (10 mL) was stirred at room temperature under an atmospheric pressure of hydrogen gas for 12 h. Filtration of the catalyst and evaporation of the solvent provided pure 14 as a colourless oil (130 mg, 0.54 mmol, 100% yield). IR (neat) νmax 3316, 1231, 1057, 1029 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 7.02–6.93 (m, 2H, Ar), 6.70–6.63 (m, 1H, Ar), 6.58–6.53 (m, 1H, Ar), 4.16 (br s, 1H, NH), 3.82 (d, J = 10.3 Hz, 3H, OMe), 3.81 (d, J = 10.5 Hz, 3H, OMe), 3.71 (ddd, J = 10.1, 6.7, 3.4 Hz, 1H, H-2), 2.88–2.75 (m, 2H, H-4), 2.29–2.19 (m, 1H, H-3), 2.12–1.98 (m, 1H, H-3′) ppm. 13C-NMR (100 MHz, CDCl3): δ = 143.08 (d, J = 10.9 Hz, Ar), 129.40 (Ar), 127.11 (Ar), 121.05 (Ar), 118.26 (Ar), 114.97 (Ar), 53.83 (d, J = 6.7 Hz, OMe), 53.08 (d, J = 7.3 Hz, OMe), 49.05 (d, J = 161.5 Hz, C-2), 26.09 (d, J = 13.7 Hz, C-4), 22.39 (d, J = 5.2 Hz, C-3) ppm. 31P-NMR (162 MHz, CDCl3): δ = 27.39 ppm. HRMS (ESI): calcd. for C11H16NNaO3P [M + Na]+ 264.0760; found 264.0752.
3.6. Synthesis of 1,2,3,4-Tetrahydroquinoline-2-phosphonic Acid Hydrobromide 4
From 11: A 33% solution of hydrogen bromide in acetic acid (2 mL) was added to compound 11 (110 mg, 0.29 mmol) and the reaction mixture was stirred at room temperature for 3 h. The solvent was evaporated and the residue was taken up in water and lyophilised to afford 4 as a white solid (86 mg, 0.29 mmol, 100% yield). M.p. 94–96 °C (dec.). IR (nujol) νmax 3381, 1171, 1077, 1057 cm−1. 1H-NMR (400 MHz, CD3OD): δ = 7.40–7.28 (m, 4H, Ar), 3.78 (ddd, J = 13.1, 12.1, 2.5 Hz, 1H, H-2), 3.08–3.01 (m, 2H, H-4), 2.52–2.43 (m, 1H, H-3), 2.19–2.06 (m, 1H, H-3′) ppm. 13C-NMR (100 MHz, CD3OD): δ = 132.21 (d, J = 0.7 Hz, Ar), 131.87 (Ar), 131.78 (Ar), 130.25 (Ar), 128.64 (Ar), 124.49 (Ar), 52.52 (d, J = 153.2 Hz, C-2), 25.94 (d, J = 12.4 Hz, C-4), 22.37 (d, J = 2.2 Hz, C-3) ppm. 31P-NMR (162 MHz, CD3OD): δ = 14.61 ppm. HRMS (ESI): calcd. for C9H13NO3P [M − Br]+ 214.0628; found 214.0632.
From 14: A 33% solution of hydrogen bromide in acetic acid (2 mL) was added to compound 14 (130 mg, 0.54 mmol) and the reaction mixture was stirred at room temperature for 3 h. The solvent was evaporated and the residue was taken up in water and lyophilised to afford 4 as a white solid (158 mg, 0.54 mmol, 100% yield). Spectroscopic data were identical to those described above.
3.7. Synthesis of Dimethyl N-Benzyloxycarbonyl-1,2-dihydroisoquinoline-1-phosphonate 16
Benzyl chloroformate (0.61 mL, 726 mg, 4.26 mmol) was added dropwise to a solution of isoquinoline (500 mg, 3.87 mmol) and trimethyl phosphite (0.50 mL, 528 mg, 4.26 mmol) in anhydrous acetonitrile (6 mL) kept at 0 °C under argon. Sodium iodide (853 mg, 5.69 mmol) was slowly added and the mixture was heated at 50 °C for 10 min. The solvent was evaporated and the crude product was partitioned between dichloromethane (15 mL) and saturated aqueous sodium bicarbonate (15 mL). The organic phase was separated and the aqueous layer was further extracted with dichloromethane (3 × 15 mL). The combined organic extracts were washed with brine (10 mL), dried, filtered, and concentrated. Purification by column chromatography (eluent: ethyl acetate/hexanes 4:1) afforded 16 as a colourless oil (1.33 g, 3.56 mmol, 92% yield). IR (neat) νmax 1715, 1342, 1295, 1238, 1121, 1028 cm−1. 1H-NMR (300 MHz, DMSO-d6, 50 °C): δ = 7.51–7.11 (m, 9H, Ar), 6.91 (d, J = 7.7 Hz, 1H, H-3), 6.08–5.94 (m, 1H, H-4), 5.83 (d, J = 15.8 Hz, 1H, H-1), 5.27 (s, 2H, CH2Ph), 3.57–3.38 (m, 6H, OMe) ppm. 13C-NMR (100 MHz, DMSO-d6): δ = (duplicate signals are observed for some carbons; asterisks indicate those corresponding to the minor rotamer) 152.37* (CO), 151.88 (CO), 136.01 (Ar), 135.91* (Ar), 131.03* (d, J = 3.6 Hz, Ar), 130.84 (d, J = 3.6 Hz, Ar), 128.75* (d J = 3.0 Hz, Ar), 128.68 (d, J = 3.1 Hz, Ar), 128.54 (Ar), 128.47* (Ar), 128.26 (Ar), 128.11* (Ar), 127.92 (Ar), 127.55 (d, J = 5.2 Hz, Ar), 127.30 (d, J = 2.3 Hz, Ar), 127.18 (d, J = 2.0 Hz, Ar), 125.43 (Ar), 125.35 (Ar), 124.86 (d, J = 2.9 Hz, C-3), 124.79* (C-3), 109.86 (C-4), 67.86* (CH2Ph), 67.77 (CH2Ph), 53.40* (d, J = 148.4 Hz, C-1), 53.16* (d, J = 6.1 Hz, OMe), 53.12 (d, J = 7.0 Hz, OMe), 52.64 (d, J = 149.6 Hz, C-1) ppm. 31P-NMR (122 MHz, DMSO-d6, 50 °C): δ = 21.34 ppm. HRMS (ESI): calcd. for C19H20NNaO5P [M + Na]+ 396.0971; found 396.0982.
3.8. Synthesis of Diethyl N-Benzyloxycarbonyl-1,2-dihydroisoquinoline-1-phosphonate 19
Benzyl chloroformate (0.61 mL, 726 mg, 4.26 mmol) was added dropwise to a solution of isoquinoline (500 mg, 3.87 mmol) and triethyl phosphite (0.73 mL, 708 mg, 4.26 mmol) in anhydrous acetonitrile (6 mL) kept at 0 °C under argon. Sodium iodide (853 mg, 5.69 mmol) was slowly added and the mixture was heated at 50 °C for 10 min. The solvent was evaporated and the crude product was partitioned between dichloromethane (15 mL) and saturated aqueous sodium bicarbonate (15 mL). The organic phase was separated and the aqueous layer was further extracted with dichloromethane (3 × 15 mL). The combined organic extracts were washed with brine (10 mL), dried, filtered, and concentrated. Purification by column chromatography (eluent:ethyl acetate/hexanes 3:2) afforded 19 as a colourless oil (1.52 g, 3.79 mmol, 98% yield). IR (neat) νmax 1715, 1294, 1253, 1120, 1023 cm−1. 1H-NMR (300 MHz, DMSO-d6, 70 °C): δ = 7.50–7.10 (m, 9H, Ar), 6.91 (d, J = 7.8 Hz, 1H, H-3), 5.98 (d, J = 7.8 Hz, 1H, H-4), 5.77 (d, J = 15.9 Hz, 1H, H-1), 5.26 (s, 2H, CH2Ph), 3.95–3.69 (m, 4H, OCH2), 1.08 (t, J = 7.0 Hz, 3H, Me), 1.07 (t, J = 7.0 Hz, 3H, Me) ppm. 13C-NMR (100 MHz, DMSO-d6): δ = (duplicate signals are observed for some carbons; asterisks indicate those corresponding to the minor rotamer) 152.48* (CO), 151.91 (CO), 136.05 (Ar), 135.88* (Ar), 131.15* (d, J = 3.6 Hz, Ar), 130.98 (d, J = 3.7 Hz, Ar), 128.69* (d, J = 3.3 Hz, Ar), 128.61 (d, J = 3.3 Hz, Ar), 128.56 (Ar), 128.47* (Ar), 128.28 (Ar), 128.25* (Ar), 128.13* (Ar), 127.97 (Ar), 127.53 (d, J = 5.2 Hz, Ar), 127.22 (d, J = 2.6 Hz, Ar), 127.11* (d, J = 2.7 Hz, Ar), 125.59 (d, J = 1.8 Hz, Ar), 125.56* (d, J = 2.4 Hz, Ar), 125.42 (Ar), 124.84* (C-3), 124.81 (C-3), 109.95 (C-4), 67.84* (CH2Ph), 67.71 (CH2Ph), 62.54 (d, J = 7.1 Hz, OCH2), 62.42* (d, J = 6.6 Hz, OCH2), 62.38* (d, J = 7.1 Hz, OCH2), 53.97* (d, J = 148.9 Hz, C-1), 53.15 (d, J = 150.2 Hz, C-1), 16.17 (d, J = 5.3 Hz, Me), 16.15* (d, J = 5.7 Hz, Me) ppm. 31P-NMR (122 MHz, DMSO-d6, 70 °C): δ = 18.77 ppm. HRMS (ESI): calcd. for C21H25NO5P [M + H]+ 402.1465; found 402.1466.
3.9. Synthesis of Diethyl N-Benzyloxycarbonyl-1,2,3,4-tetrahydroisoquinoline-1-phosphonate 20
Triethylsilane (1.0 mL, 730 mg, 6.28 mmol) and trifluoroacetic acid (0.48 mL, 716 mg, 6.28 mmol) were added to a solution of 19 (300 mg, 0.75 mmol) in anhydrous dichloromethane (15 mL) kept at 0 °C under argon. The solution was allowed to warm to room temperature and stirred for 18 h. Evaporation of the solvent followed by column chromatography (eluent:hexanes/ethyl acetate 1:1) afforded 20 as a colourless oil (298 mg, 0.74 mmol, 99% yield). IR (neat) νmax 1701, 1294, 1249, 1230, 1051, 1022 cm−1. 1H-NMR (300 MHz, DMSO-d6, 70 °C): δ = 7.44–7.16 (m, 9H, Ar), 5.53 (d, J = 20.4 Hz, 1H, H-1), 5.17 (s, 2H, CH2Ph), 4.14–3.77 (m, 5H, H-3, OCH2), 3.72–3.52 (m, 1H, H-3′), 2.98–2.78 (m, 2H, H-4), 1.17 (t, J = 7.0 Hz, 3H, Me), 1.08 (t, J = 6.9 Hz, 3H, Me) ppm. 13C-NMR (100 MHz, DMSO-d6): δ = (duplicate signals are observed for some carbons; asterisks indicate those corresponding to the minor rotamer) 154.60 (d, J = 3.9 Hz, CO), 154.18* (d, J = 2.4 Hz, CO), 136.66 (Ar), 136.46* (Ar), 134.73 (d, J = 5.6 Hz, Ar), 134.63* (d, J = 5.6 Hz, Ar), 129.22 (Ar), 129.13* (d, J = 2.2 Hz, Ar), 128.95 (d, J = 2.3 Hz, Ar), 128.85 (Ar), 128.38 (Ar), 128.33* (Ar), 127.94* (Ar), 127.91 (Ar), 127.86* (Ar), 127.67 (d, J = 4.0 Hz, Ar), 127.58 (Ar), 127.38 (Ar), 125.88 (d, J = 2.8 Hz, Ar), 125.83* (d, J = 2.8 Hz, Ar), 66.92* (CH2Ph), 66.82 (CH2Ph), 62.58 (d, J = 7.2 Hz, OCH2), 62.19 (d, J = 7.1 Hz, OCH2), 52.76* (d, J = 149.9 Hz, C-1), 52.34 (d, J = 152.0 Hz, C-1), 39.05* (C-3), 38.84 (C-3), 27.43 (C-4), 27.15* (C-4), 16.08* (d, J = 5.6 Hz, Me), 16.05 (d, J = 5.3 Hz, Me). 31P-NMR (122 MHz, DMSO-d6, 70 °C): δ = 20.75. HRMS (ESI): calcd. for C21H27NO5P [M + H]+ 404.1621; found 404.1611.
3.10. Synthesis of 1,2,3,4-Tetrahydroisoquinoline-1-phosphonic Acid Hydrobromide 5
A 33% solution of hydrogen bromide in acetic acid (2 mL) was added to 20 (160 mg, 0.40 mmol) and the reaction mixture was stirred at room temperature for 3 h. The solvent was evaporated and the residue was taken up in water and lyophilised to afford 5 as a white solid (117 mg, 0.40 mmol, 100% yield). M.p. 85–87 °C (dec.). IR (nujol) νmax 3421, 1212, 1118, 1019 cm−1. 1H-NMR (400 MHz, D2O): δ = 7.45–7.41 (m, 1H, Ar), 7.37–7.27 (m, 3H, Ar), 4.70 (d, J = 17.6 Hz, 1H, H-1), 3.78 (ddd, J = 12.7, 9.2, 6.5 Hz, 1H, H-3), 3.58–3.51 (m, 1H, H-3′), 3.18–3.12 (m, 2H, H-4) ppm. 13C-NMR (100 MHz, CD3OD): δ = 132.88 (d, J = 5.4 Hz, Ar), 130.40 (d, J = 2.2 Hz, Ar), 129.47 (d, J = 2.7 Hz, Ar), 128.94 (d, J = 3.6 Hz, Ar), 127.92 (d, J = 2.6 Hz, Ar), 127.40 (d, J = 5.1 Hz, Ar), 54.00 (d, J = 147.3 Hz, C-1), 41.15 (d, J = 2.1 Hz, C-3), 25.88 (C-4) ppm. 31P-NMR (162 MHz, D2O): δ = 10.02 ppm. HRMS (ESI): calcd. for C9H13NO3P [M − Br]+ 214.0628; found 214.0633.
3.11. Synthesis of N-Benzyloxycarbonyl-1,4-dihydro-3-isoquinolinone 22
A 1 M solution of lithium bis(trimethylsilyl)amide in tetrahydrofuran (0.83 mL, 0.83 mmol) was slowly added to a solution of 1,4-dihydro-3(2H)-isoquinolinone 21 (122 mg, 0.83 mmol) in anhydrous tetrahydrofuran (5 mL) kept at −78 °C under argon. After 30 min, benzyl chloroformate (0.12 mL, 142 mg, 0.83 mmol) was added dropwise and stirring was continued for additional 3 h. The reaction was then treated with saturated aqueous ammonium chloride (10 mL) and allowed to warm to room temperature. The two layers were separated and the aqueous phase was extracted with dichloromethane (2 × 10 mL). The combined organic extracts were dried, filtered, and concentrated. Purification by column chromatography (eluent:hexanes/ethyl acetate 7:3) afforded 22 as a white solid (172 mg, 0.61 mmol, 73% yield). M.p. 63–65 °C. IR (nujol) νmax 1692 cm−1. 1H-NMR (400 MHz, CDCl3): δ = 7.49–7.44 (m, 2H, Ar), 7.40–7.25 (m, 6H, Ar), 7.23–7.19 (m, 1H, Ar), 5.34 (s, 2H, CH2Ph), 4.92 (s, 2H, H-1), 3.73 (s, 2H, H-4) ppm. 13C-NMR (100 MHz, CDCl3): δ = 169.43 (CO), 153.62 (COO), 135.38 (Ar), 132.25 (Ar), 132.13 (Ar), 128.74 (Ar), 128.50 (Ar), 128.46 (Ar), 128.22 (Ar), 127.35 (Ar), 126.99 (Ar), 125.81 (Ar), 68.97 (CH2Ph), 48.75 (C-1), 41.53 (C-4) ppm. HRMS (ESI): calcd. for C17H15NNaO3 [M + Na]+ 304.0944; found 304.0943.
3.12. Synthesis of Dimethyl N-Benzyloxycarbonyl-1,2,3,4-tetrahydroisoquinoline-3-phosphonate 25
A 1 M solution of diisobutylaluminium hydride in hexanes (0.78 mL, 0.78 mmol) was slowly added to a solution of 22 (146 mg, 0.52 mmol) in anhydrous tetrahydrofuran (5 mL) kept at −78 °C under argon. After stirring at this temperature for 2 h, the reaction was treated with saturated aqueous sodium acetate (10 mL) and allowed to warm to room temperature. A 3:1 mixture of diethyl ether and saturated aqueous ammonium chloride (16 mL) was then added and the resulting mixture was stirred at room temperature until a suspension was formed. The solid was filtered off under reduced pressure and washed with diethyl ether (2 × 10 mL). The organic layer was separated and the aqueous phase was extracted with diethyl ether (2 × 10 mL). The combined organic extracts were washed with brine (10 mL), dried, filtered, and evaporated to provide the hemiaminal as an oil. It was dissolved in methanol (5 mL) and treated with pyridinium p-toluenesulfonate (13 mg, 0.05 mmol). After stirring at room temperature for 2 h, triethylamine (31 µL, 22 mg, 0.22 mmol) was added. The solvent was evaporated and the crude methoxyaminal obtained 23 was dissolved in anhydrous dichloromethane (5 mL) and kept under argon. Trimethyl phosphite (61 µL, 65 mg, 0.52 mmol) was added and the resulting solution was cooled to −20 °C. Boron trifluoride-diethyl ether (65 µL, 74 mg, 0.52 mmol) was added and the reaction mixture was slowly warmed to room temperature and stirred for 12 h. After quenching with water (10 mL), the two layers were separated and the aqueous phase was extracted with dichloromethane (2 × 10 mL). The combined organic extracts were dried, filtered, and concentrated. Purification by column chromatography (eluent:ethyl acetate/hexanes 9:1) afforded 25 as a colourless oil (138 mg, 0.37 mmol, 71% yield). IR (neat) νmax 1703, 1410, 1246, 1055, 1031 cm−1. 1H-NMR (400 MHz, CDCl3): δ = (duplicate signals are observed for some protons; asterisks indicate those corresponding to the minor rotamer) 7.42–7.30 (m, 5H, Ar), 7.23–7.04 (m, 4H, Ar), 5.31–5.12 (m, 2H, CH2Ph), 5.11–5.03 (m, 1H, H-3), 5.00–4.86 (m, 1H, H-1) overlapped with 4.97*–4.89* (m, 1H, H-3), 4.52 (d, J = 16.6 Hz, 1H, H-1′), 4.45* (d, J = 16.6 Hz, 1H, H-1′), 3.65 (d, J = 10.7 Hz, 3H, OMe), 3.51* (d, J = 10.7 Hz, 3H, OMe), 3.35 (d, J = 10.7 Hz, 3H, OMe), 3.33–3.16 (m, 2H, H-4) overlapped with 3.24* (d, J = 10.7 Hz, 3H, OMe) ppm. 13C-NMR (100 MHz, CDCl3): δ = (duplicate signals are observed for some carbons; asterisks indicate those corresponding to the minor rotamer) 155.72 (CO), 155.05* (CO), 136.37 (Ar), 136.18* (Ar), 132.61* (Ar), 132.46 (Ar), 131.41 (Ar), 131.00* (Ar), 128.64 (Ar), 128.53 (Ar), 128.45* (Ar), 128.24 (Ar), 128.17* (Ar), 127.95 (Ar), 126.70 (Ar), 126.64* (Ar), 126.60 (Ar), 126.09* (Ar), 125.86 (Ar), 67.79 (CH2Ph), 52.74 (d, J = 6.4 Hz, OMe), 52.58 (d, J = 6.6 Hz, OMe), 52.34* (d, J = 6.9 Hz, OMe), 47.07* (d, J = 154.2 Hz, C-3), 46.29 (d, J = 154.1 Hz, C-3), 44.37 (C-1), 28.56* (C-4), 28.32 (C-4) ppm. 31P-NMR (162 MHz, CDCl3): δ = (a duplicate signal is observed; an asterisk indicates that corresponding to the minor rotamer) 27.67, 27.07* ppm. HRMS (ESI): calcd. for C19H22NNaO5P [M + Na]+ 398.1128; found 398.1144.
3.13. Synthesis of 1,2,3,4-Tetrahydroisoquinoline-3-phosphonic Acid Hydrobromide 6
A 33% solution of hydrogen bromide in acetic acid (2 mL) was added to 25 (100 mg, 0.27 mmol) and the reaction mixture was stirred at room temperature for 3 h. The solvent was evaporated and the residue was taken up in water and lyophilised to afford 6 as a white solid (79 mg, 0.27 mmol, 100% yield). M.p. 102–103 °C (dec.). IR (nujol) νmax 3404, 1232, 1010 cm−1. 1H-NMR (400 MHz, CD3OD): δ = 7.35–7.22 (m, 4H, Ar), 4.52 (d, J = 15.7 Hz, 1H, H-1), 4.42 (dd, J = 15.7, 3.1 Hz, 1H, H-1′), 3.94–3.83 (m, 1H, H-3), 3.34–3.25 (m, 2H, H-4) ppm. 13C-NMR (100 MHz, CD3OD): δ = 131.71 (d, J = 12.2 Hz, Ar), 129.97 (Ar), 129.27 (Ar), 128.69 (Ar), 128.32 (Ar), 127.77 (Ar), 51.34 (d, J = 153.1 Hz, C-3), 47.01 (d, J = 7.7 Hz, C-1), 27.44 (C-4) ppm. 31P-NMR (162 MHz, CD3OD): δ = 14.06 ppm. HRMS (ESI): calcd. for C9H13NO3P [M − Br]+ 214.0628; found 214.0631.


 ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}