Molecular Motions as a Drug Target: Mechanistic Simulations of Anthrax Toxin Edema Factor Function Led to the Discovery of Novel Allosteric Inhibitors

Abstract
:1. Introduction: Edema Factor, a Target of Choice to Fight Anthrax?
1.1. Virulence Factors
1.2. Bioterrorism Threat
1.3. Prevention and Treatments
1.4. Targets
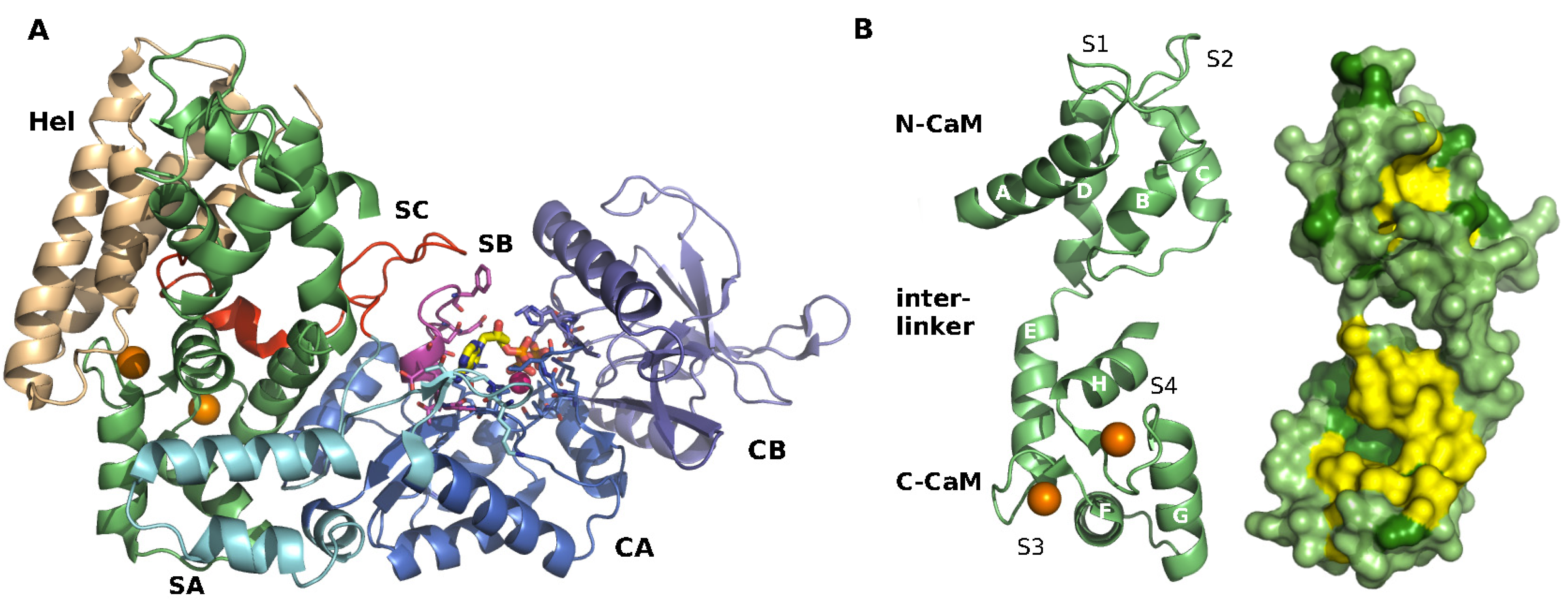
1.5. Structural Data

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| PDB id | Reference | Description |
|---|---|---|
| 1K8T | [23] | EF in free inactive form |
| 1K90 | [23] | EF in complex with CaM and 3'-deoxy-ATP |
| 1K93 | [23] | EF in complex with CaM |
| 1LVC | [25] | EF in complex with CaM and 2'deoxy 3' anthraniloyl ATP |
| 1PK0 | [26] | EF in complex with CaM and PMEApp a |
| 1S26 | [27] | EF in complex with CaM and 5' met-ATP |
| 1SK6 | [28] | EF in complex with CaM and cAMP, PPi b |
| 1XFU | [29] | EFΔ64 c in complex with CaM |
| 1XFV | [29] | EF in complex with CaM and 3'-deoxy-ATP |
| 1XFW | [29] | EF in complex with CaM and cAMP |
| 1XFX | [29] | EF in complex with CaM, 10 mM calcium |
| 1XFY | [29] | EF in complex with CaM |
| 1Y0V | [29] | EF in complex with CaM and PPi |
1.6. EF Inhibitors

1.6.1. Inhibitors Targeting EF Active Site
1.6.2. Inhibitors Targeting the EF-CaM Interaction
1.7. Objectives
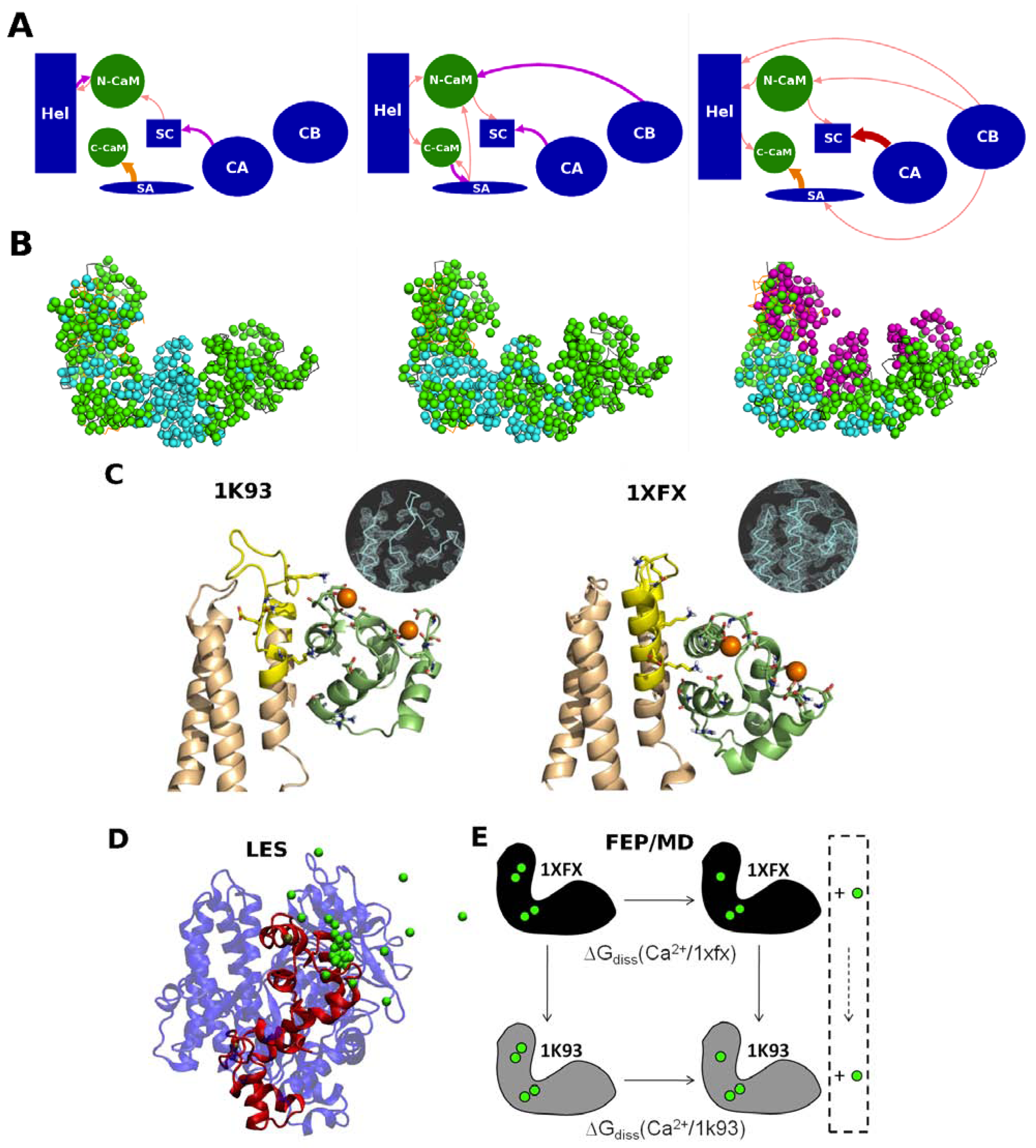
2. Interplay between EF, Calmodulin and Calcium Ions
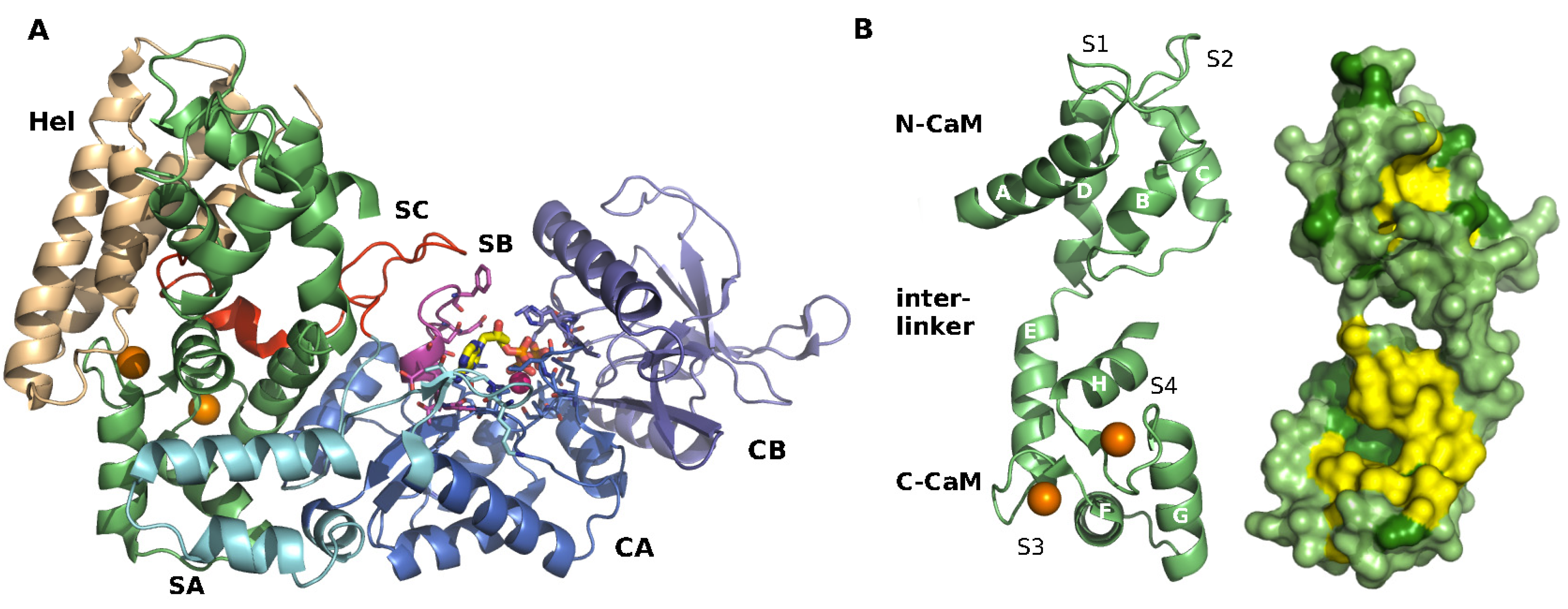
2.1. Structure of EF-CaM Complex
2.2. Calcium Probes the Modulation of EF Activity by CaM
2.3. Calcium Signal Propagation throughout EF-CaM Residue Network
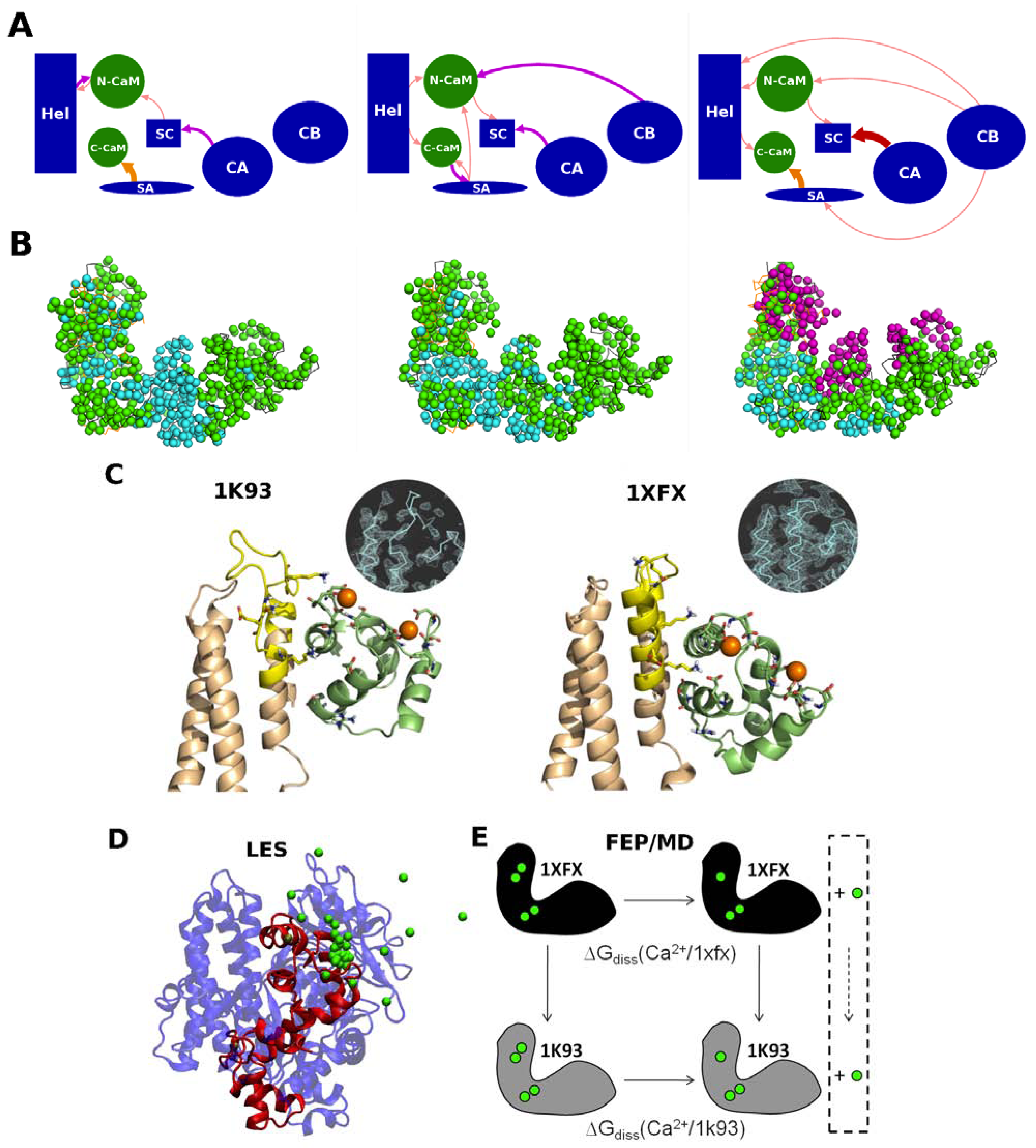
2.4. Interplay between EF/CaM and Ca/CaM
3. Catalytic Properties
3.1. EF Catalytic Site Structures

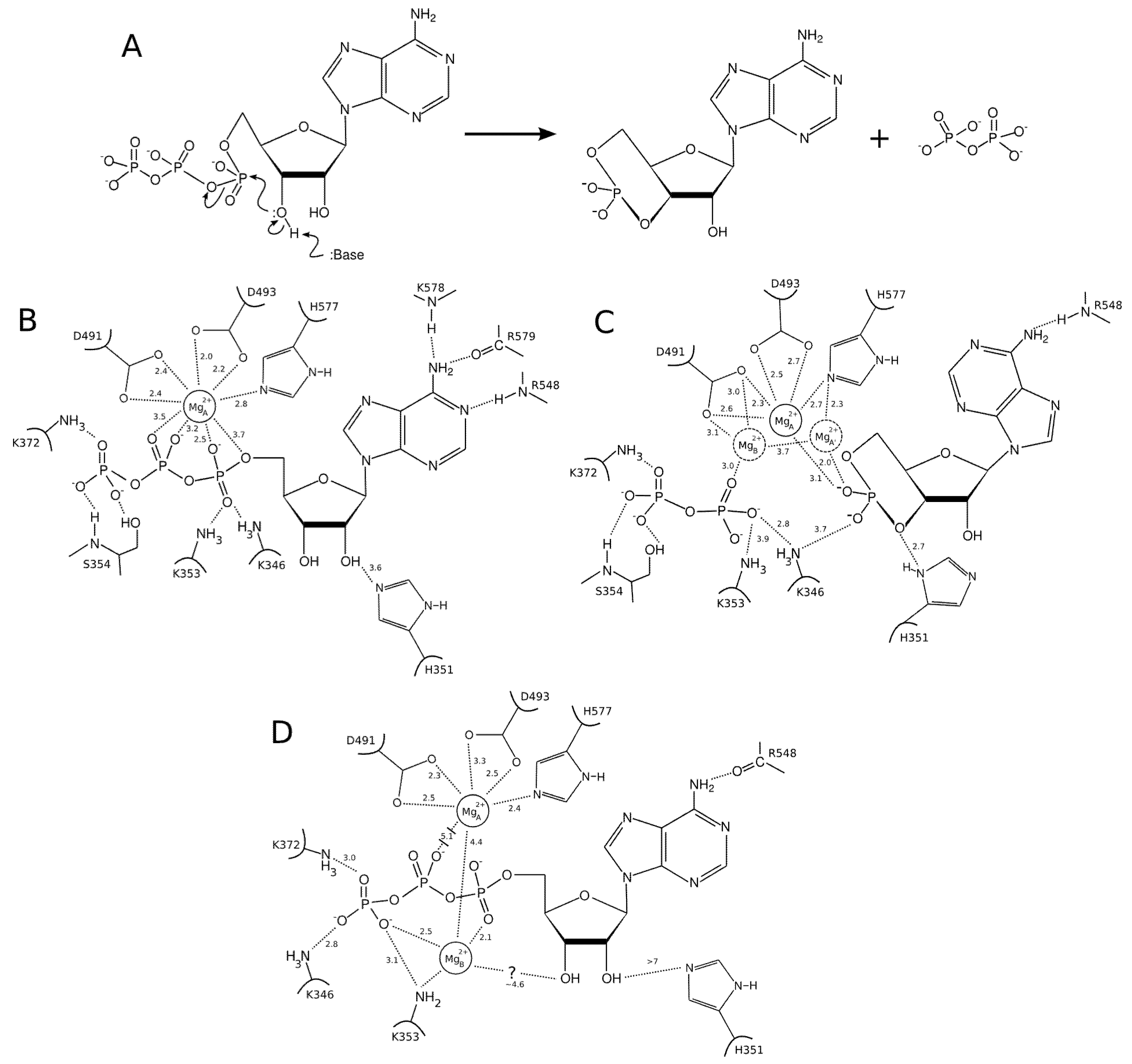
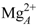 ion in Figure 4C). By contrast, the two-metal-ion binding mode suggests a weaker binding of the ions to the protein; one being mostly coordinated by D491 and PPi and the other mostly coordinated by H577 and cAMP. This arrangement resembles the two-metal-ion binding mode of MACs [60,61]. is coordinated by the protein in a single-ion mode like in Figure 4B, whereas the other ion,
ion in Figure 4C). By contrast, the two-metal-ion binding mode suggests a weaker binding of the ions to the protein; one being mostly coordinated by D491 and PPi and the other mostly coordinated by H577 and cAMP. This arrangement resembles the two-metal-ion binding mode of MACs [60,61]. is coordinated by the protein in a single-ion mode like in Figure 4B, whereas the other ion, 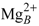 is mostly coordinated by ATP and makes poor contacts with the protein (Figure 4D).
is mostly coordinated by ATP and makes poor contacts with the protein (Figure 4D).3.2. Insights from Computational Modeling
ion is tightly attached to the protein, but loosely to ATP, while is essentially only coordinated by the substrate. Accordingly, ATP displays a larger mobility and flexibility in the active site of this conformation in MD relative to the one-ion binding mode; (3) The presence of two-ions in the product-bound active site firmly attach PPi and cAMP (Figure 4C). The global effect of this strong binding is ambiguous, as it may accelerate the reaction by reducing the transition state energy, but it could also slow down the release of products and thus the reaction. These simulations support that structure 1XFV (substrate and two-ions) would not correspond to a functional state. like ion, thus restoring a 1K90 like conformation competent for catalysis (Figure 4B,D). This reshuffling of Mg2+ ions in the active site may favor transient catalytic competent conformers of ATP [61]. Indeed, paradoxically, the 1K90 binding mode resembles to that found in MACs structure despite the presence of one ion only. Alternatively, Mg2+ free ATP molecules, present in low concentration could also enter the site, directly leading to the 1K90 like conformation. After (iii) cyclization; (iv) the dissociation of products could be facilitated by reshuffling of Mg2+ ions within their multiple possible positions and the entry of new Mg2+ ions to replace those that could have left with one of the products. This mechanism, based on a one-ion catalytic step with possible transient presence of two ions would be different from that of MACs. They suggest a rather plastic catalytic site allowing Mg2+ reshuffling. Obviously, further experiments and simulations are necessary to validate or refute this mechanism.4. Conformational Transition and Inhibitors Discovery
4.1. Challenges in Rational Inhibitor Design
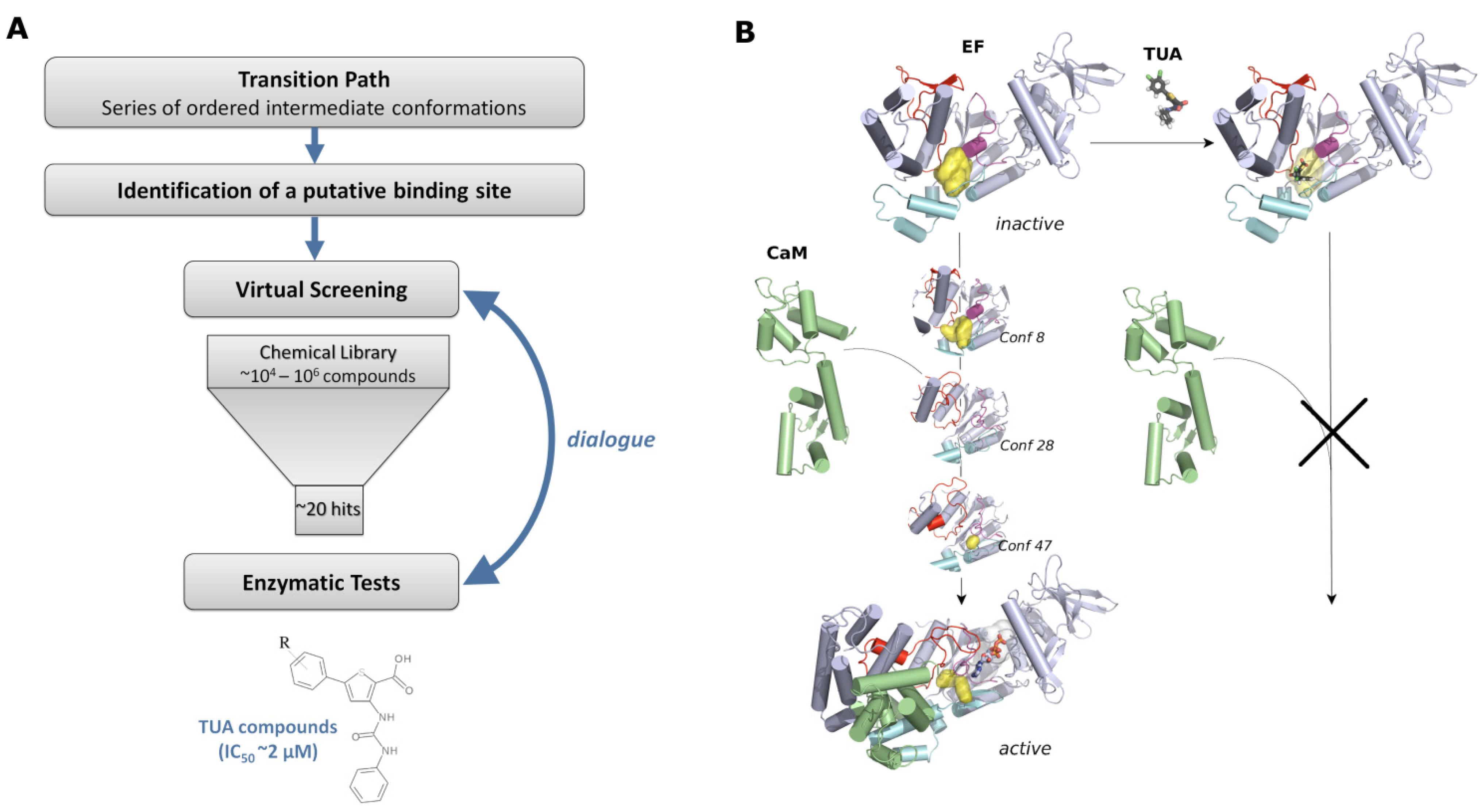
4.2. Design Strategy to Target EF Activation Mechanism
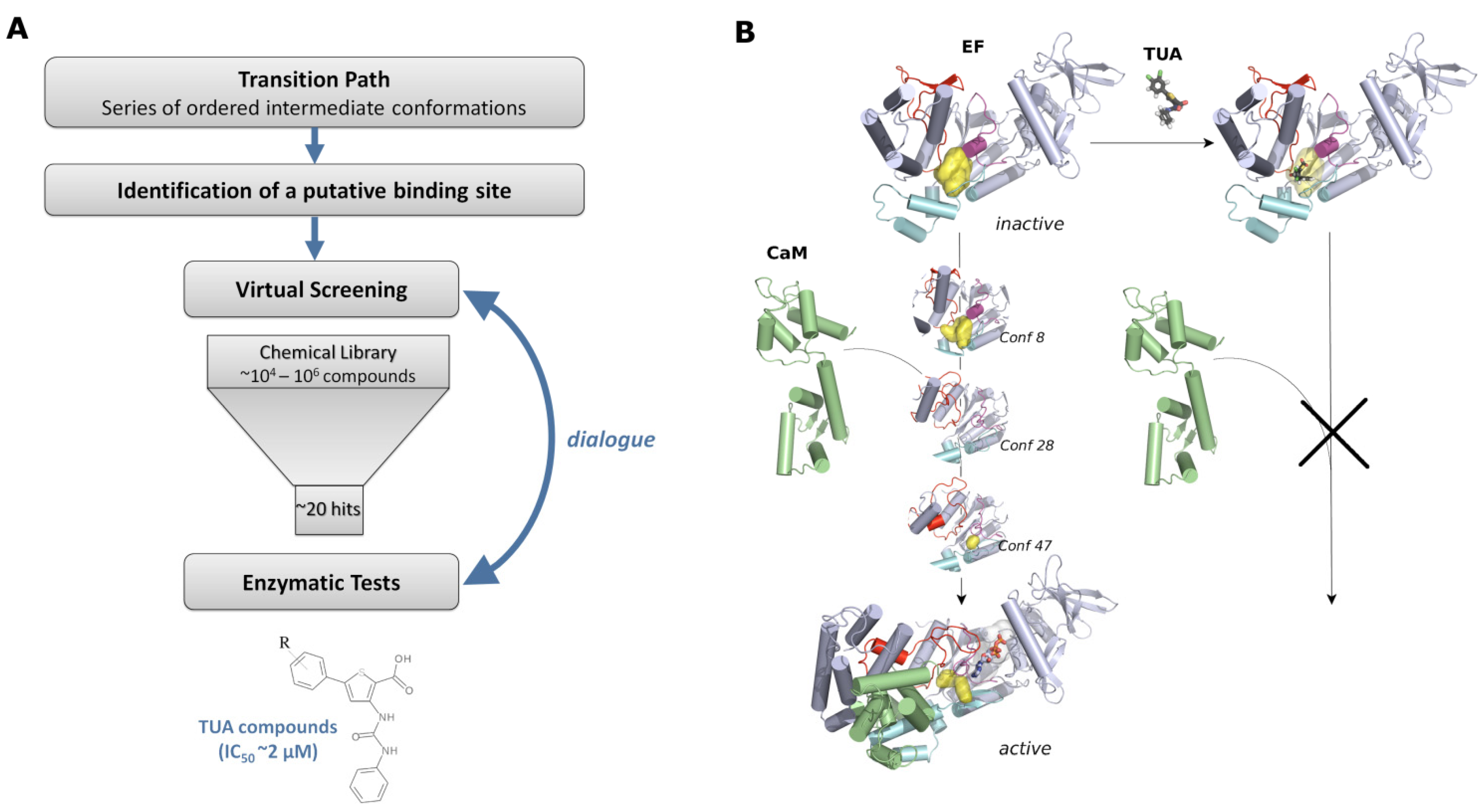
4.3. Identification of the Potential Binding Site
4.4. Identification of a New Family of Inhibitors
5. Conclusions
Acknowledgments
Conflict of Interest
References
- Tournier, J.N.; Rossi Paccani, S.; Quesnel-Hellmann, A.; Baldari, C.T. Anthrax toxins: A weapon to systematically dismantle the host immune defenses. Mol. Aspects Med. 2009, 30, 456–466. [Google Scholar] [CrossRef]
- Göttle, M.; Dove, S.; Kees, F.; Schlossmann, J.; Geduhn, J.; König, B.; Shen, Y.; Tang, W.; Kaever, V.; Seifert, R. Cytidylyl and uridylyl cyclase activity of Bacillus anthracis edema factor 12 and Bordetella pertussis CyaA. Biochemistry 2010, 49, 5494–5503. [Google Scholar]
- Raymond, B.; Leduc, D.; Ravaux, L.; Goffic, R.L.; Candela, T.; Raymondjean, M.; Goossens, P.; Touqui, L. Edema toxin impairs anthracidal phospholipase A2 expression by alveolar macrophages. PLoS Pathog. 2007, 3, e187. [Google Scholar] [CrossRef]
- Dumetz, F.; Jouvon, G.; Khun, H.; Glomski, I.; Corre, J.P.; Rougeaux, C.; Tang, W.J.; Mock, M.; Huerre, M.; Goossens, P.L. Noninvasive imaging technologies reveal edema toxin as a key virulence factor in anthrax. Am. J. Pathol. 2011, 178, 2523–2535. [Google Scholar] [CrossRef]
- Hicks, C.; Cui, X.; Sweeney, D.; Li, Y.; Barochia, A.; Eichacker, P. The potential contributions of lethal and edema toxins to the pathogenesis of anthrax associated shock. Toxins 2011, 3, 1185–1202. [Google Scholar] [CrossRef]
- Guichard, A.; Nizet, V.; Bier, E. New insights into the biological effects of anthrax toxins: Linking cellular to organismal responses. Microbes Infect. 2012, 14, 97–118. [Google Scholar] [CrossRef]
- Watson, A.; Keir, D. Information on which to base assessments of risk from environments contaminated with anthrax spores. Epidemiol. Infect. 1994, 113, 479–490. [Google Scholar] [CrossRef]
- Whitney, E.A.S.; Beatty, M.E.; Taylor, T.H.; Weyant, R.; Sobel, J.; Arduino, M.J.; Ashford, D.A. Inactivation of Bacillus anthracis spores. Emerging Infect. 2003, 9, 623–627. [Google Scholar] [CrossRef]
- U.S. Department of Health & Human services, Centers for Disease Control and Prevention. Update: Investigation of bioterrorism-related anthrax—Connecticut. Morb. Mortal. Wkly. Rep. 2001, 50, 1077–1079.
- Rotz, L.D.; Khan, A.S.; Lillibridge, S.R.; Ostro, S.M.; Hughes, J.M. Public health assessment of potential biological terrorism agents. Emerging Infect. Dis. 2002, 8, 225–230. [Google Scholar] [CrossRef]
- Beierlein, J.; Anderson, A. New developments in vaccines, inhibitors of anthrax toxins, and antibiotic therapeutics for Bacillus anthracis. Curr. Med. Chem. 2011, 18, 5083–5094. [Google Scholar] [CrossRef]
- Sterne, M. Avirulent anthrax vaccine. OnderstepoortJ. Vet. Sci. Anim. Ind. 1946, 21, 41–43. [Google Scholar]
- Spencer, R.C. Bacillus anthracis. J. Clin. Pathol. 2003, 56, 182–187. [Google Scholar] [CrossRef]
- Gauthier, Y.; Tournier, J.; Paucod, J.; Corre, J.; Mock, M.; Goossens, P.; Vidal, D. Efficacy of a vaccine based on protective antigen and killed spores against experimental inhalational anthrax. Infect. Immun. 2009, 77, 1197–1207. [Google Scholar] [CrossRef]
- Chabot, D.; Joyce, J.; Caulfield, M.; Cook, J.; Hepler, R.; Wang, S.; Vietri, N.; Ruthel, G.; Shoop, W.; Pitt, L.; et al. Efficacy of a capsule conjugate vaccine against inhalational anthrax in rabbits and monkeys. Vaccine 2012, 30, 846–852. [Google Scholar] [CrossRef]
- Merkel, T.; Perera, P.; Kelly, V.; Verma, A.; Llewellyn, Z.; Waldmann, T.; Mosca, J.; Perera, L. Development of a highly efficacious vaccinia-based dual vaccine against smallpox and anthrax, two important bioterror entities. Proc. Natl. Acad. Sci. USA 2010, 107, 18091–18096. [Google Scholar]
- U.S. Department of Health & Human services, Centers for Disease Control and Prevention. Update: Investigation of bioterrorism-related anthrax and interim guidelines for exposure management and antimicrobial therapy, October 2001. Morb. Mortal. Wkly. Rep. 2001, 50, 909–919.
- Caplan, D.; Ivana, S.; Caplan, M. Susceptibility to antibiotics of Bacillus anthracis strains isolated in Romania. Roum. Arch. Microbiol. Immunol. 2009, 68, 106–110. [Google Scholar]
- Brook, I.; Elliott, T.; Pryor, H.; Sautter, T.; Gnade, B.; Thakar, J.; Knudson, G. In vitro resistance of Bacillus anthracis Sterne to doxycycline, macrolides and quinolones. Int. J. Antimicrob. Agents 2001, 18, 559–562. [Google Scholar] [CrossRef]
- Athamna, A.; Athamna, M.; Abu-Rashed, N.; Medlej, B.; Bast, D.; Rubinstein, E. Selection of Bacillus anthracis isolates resistant to antibiotics. J. Antimicrob. Chemother. 2004, 54, 424–428. [Google Scholar] [CrossRef]
- Price, L.B.; Vogler, A.; Pearson, T.; Busch, J.D.; Schupp, J.M.; Keim, P. In vitro selection and characterization of Bacillus anthracis mutants with high-level resistance to ciprofloxacin. Antimicrob. Agents Chemother. 2003, 47, 2362–2365. [Google Scholar] [CrossRef]
- Drum, C.; Shen, Y.; Rice, P.; Bohm, A.; Tang, W. Crystallization and preliminary X-ray study of the edema factor exotoxin adenylyl cyclase domain from Bacillus anthracis in the presence of its activator, calmodulin. Acta Crystallogr. D Biol. Crystallogr. 2001, 57, 1881–1884. [Google Scholar]
- Drum, C.; Yan, S.; Bard, J.; Shen, Y.; Lu, D.; Soelaiman, S.; Grabarek, Z.; Bohm, A.; Tang, W. Structural basis for the activation of anthrax adenylyl cyclase exotoxin by calmodulin. Nature 2002, 415, 396–402. [Google Scholar]
- Ulmer, T.; Soelaiman, S.; Li, S.; Klee, C.; Tang, W.; Bax, A. Calcium dependence of the interaction between calmodulin and anthrax edema factor. J. Biol. Chem. 2003, 278, 29261–29266. [Google Scholar]
- Shen, Y.; Lee, Y.; Soelaiman, S.; Bergson, P.; Lu, D.; Chen, A.; Beckingham, K.; Graberek, Z.; Mrksich, M.; Tang, W. Physiological calcium concentrations regulate calmodulin binding and catalysis of adenyl cyclase exotoxins. EMBO J. 2002, 21, 6721–6732. [Google Scholar] [CrossRef]
- Shen, Y.; Zhukovskaya, N.; Zimmer, M.; Soelaiman, S.; Wang, C.; Gibbs, C.; Tang, W. Selective inhibition of anthrax edema factor by adefovir, a drug for chronic hepatitis B virus infection. Proc. Natl. Acad. Sci. USA 2004, 101, 3242–3247. [Google Scholar]
- Shen, Y.; Guo, Q.; Zhukovskaya, N.; Drum, C.; Bohm, A.; Tang, W. Structure of anthrax edema factor-calmodulin-adenosine 5'-(α,β-methylene)-triphosphate complex reveals an alternative mode of ATP binding to the catalytic site. Biochem. Biophys. Res. Commun. 2004, 317, 309–314. [Google Scholar] [CrossRef]
- Guo, Q.; Shen, Y.; Zhukovskaya, N.; Florian, J.; Tang, W. Structural and kinetic analyses of the interaction of anthrax adenylyl cyclase toxin with reaction products cAMP and pyrophosphate. J. Biol. Chem. 2004, 279, 29427–29435. [Google Scholar]
- Shen, Y.; Zhukovskaya, N.; Guo, Q.; Florian, J.; Tang, W. Calcium-independent calmodulin binding and two-metal-ion catalytic mechanism of anthrax edema factor. EMBO J. 2005, 24, 929–941. [Google Scholar] [CrossRef]
- Yang, C.; Jas, G.; Kuczera, K. Structure, dynamics and interaction with kinase targets: Computer simulations of calmodulin. Biochim. Biophys. Acta 2004, 1697, 289–300. [Google Scholar] [CrossRef]
- Joshi, A.; Kate, S.; Poon, V.; Mondal, D.; Boggara, M.; Saraph, A.; Martin, J.; McAlpine, R.; Day, R.; Garcia, A.; et al. Structure-based design of a heptavalent anthrax toxin inhibitor. Biomacromolecules 2011, 12, 791–796. [Google Scholar] [CrossRef]
- Soelaiman, S.; Wei, B.; Bergson, P.; Lee, Y.; Shen, Y.; Mrksich, M.; Shoichet, B.; Tang, W. Structure-based inhibitor discovery against adenylyl cyclase toxins from pathogenic bacteria that cause anthrax and whooping cough. J. Biol. Chem. 2003, 278, 25990–25997. [Google Scholar]
- Chen, D.; Misra, M.; Sower, L.; Peterson, J.; Kellogg, G.; Schein, C. Novel inhibitors of anthrax edema factor. Bioorg. Med. Chem. 2008, 16, 7225–7233. [Google Scholar]
- Chen, D.; Ma, L.; Kanalas, J.; Gao, J.; Pawlik, J.; Jimenez, M.; Walter, M.; Peterson, J.; Gilbertson, S.; Schein, C. Structure-based redesign of an edema toxin inhibitor. Bioorg. Med. Chem. 2012, 20, 368–376. [Google Scholar] [CrossRef]
- Taha, H.; Dove, S.; Geduhn, J.; Konig, B.; Shen, Y.; Tang, W.J.; Seifert, R. Inhibition of the adenylyl cyclase toxin, edema factor, from Bacillus anthracis by a series of 18 mono- and bis-(M)ANT-substituted nucleoside 5'-triphosphates. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2012, 385, 57–68. [Google Scholar]
- Lee, Y.; Bergson, P.; He, W.; Mrksich, M.; Tang, W. Discovery of a small molecule that inhibits the interaction of anthrax edema factor with its cellular activator, calmodulin. Chem. Biol. 2004, 8, 1139–1146. [Google Scholar]
- Laine, E.; Goncalves, C.; Karst, J.; Lesnard, A.; Rault, S.; Tang, W.-J.; Malliavin, T.; Ladant, D.; Blondel, A. Use of allostery to identify inhibitors of calmodulin- induced activation of Bacillus anthracis Edema Factor. Proc. Natl. Acad. Sci. USA 2010, 107, 11277–11282. [Google Scholar]
- Tournier, J.; Quesnel-Hellmann, A.; Mathieu, J.; Montecucco, C.; Tang, W.; Mock, M.; Vidal, D.; Goossens, P.L. Anthrax edema toxin cooperates with lethal toxin to impair cytokine secretion during infection of dendritic cells. J. Immunol. 2005, 174, 4934–4941. [Google Scholar]
- Taha, H.M.; Schmidt, J.; Gottle, M.; Suryanarayana, S.; Shen, Y.; Tang, W.J.; Gille, A.; Geduhn, J.; Konig, B.; Dove, S.; et al. Molecular analysis of the interaction of anthrax adenylyl cyclase toxin, edema factor, with 2'(3')-O-(N-(methyl)anthraniloyl)-substituted purine and pyrimidine nucleotides. Mol. Pharmacol. 2009, 75, 693–703. [Google Scholar] [CrossRef]
- Crivici, A.; Ikura, M. Molecular and structural basis of target recognition by calmodulin. Annu. Rev. Biophys. Biomol. Struct. 1995, 24, 85–116. [Google Scholar] [CrossRef]
- Ishida, H.; Vogel, H. Protein–peptide interaction studies demonstrate the versatility of calmodulin target protein binding. Protein Pept. Lett. 2006, 13, 455–465. [Google Scholar] [CrossRef]
- Zhang, M.; Tanaka, T.; Ikura, M. Calcium-induced conformational transition revealed by the solution structure of apocalmodulin. Nat. Struct. Biol. 1995, 2, 758–767. [Google Scholar] [CrossRef]
- Finn, B.; Evenas, J.; Drakenberg, T.; Waltho, J.; Thulin, E.; Forsen, S. Calcium-induced structural changes and domain autonomy in calmodulin. Nat. Struct. Biol. 1995, 2, 777–783. [Google Scholar]
- Kuboniwa, H.; Tjandra, N.; Grzesiek, S.; Ren, H.; Klee, C.; Bax, A. Solution structure of calcium-free calmodulin. Nat. Struct. Biol. 1995, 2, 768–776. [Google Scholar] [CrossRef]
- Laine, E.; Yoneda, J.; Blondel, A.; Malliavin, T. The conformational plasticity of calmodulin upon calcium complexation gives a model of its interaction with the oedema factor of Bacillus anthracis. Proteins 2008, 71, 1813–1829. [Google Scholar] [CrossRef]
- Tobi, D.; Bahar, I. Structural changes involved in protein binding correlate with intrinsic motions of proteins in the unbound state. Proc. Natl. Acad. Sci. USA 2005, 102, 18908–18913. [Google Scholar] [CrossRef]
- Bahar, I.; Lezon, T.; Yang, L.; Eyal, E. Global dynamics of proteins: Bridging between structure and function. Annu. Rev. Biophys. 2010, 39, 23–42. [Google Scholar] [CrossRef]
- Boehr, D.; Nussinov, R.; Wright, P. The role of dynamic conformational ensembles in biomolecular recognition. Nat. Chem. Biol. 2009, 5, 789–796. [Google Scholar]
- Laine, E.; Blondel, A.; Malliavin, T. Dynamics and energetics: A consensus analysis of the impact of calcium on EF-CaM protein complex. Biophys. J. 2009, 96, 1249–1263. [Google Scholar] [CrossRef]
- Lange, O.; Grubmüller, H. Generalized correlation for biomolecular dynamics. Proteins 2006, 62, 1053–1061. [Google Scholar]
- Zhang, Z.; Wriggers, W. Local feature analysis: A statistical theory for reproducible essential dynamics of large macromolecules. Proteins 2006, 64, 391–403. [Google Scholar] [CrossRef]
- Hamacher, K.; Trylska, J.; McCammon, J. Dependency map of proteins in the small ribosomal subunit. PLoS Comput. Biol. 2006, 2, e10. [Google Scholar]
- Chennubhotla, C.; Bahar, I. Signal propagation in proteins and relation to equilibrium fluctuations. PLoS Comput. Biol. 2007, 3, e172. [Google Scholar] [CrossRef] [Green Version]
- Nussinov, R.; Ma, B. Protein dynamics and conformational selection in bidirectional signal transduction. BMC Biol. 2012, 10, 2. [Google Scholar] [CrossRef]
- Meireles, L.; Gur, M.; Bakan, A.; Bahar, I. Pre-existing soft modes of motion uniquely defined by native contact topology facilitate ligand binding to proteins. Protein Sci. 2011, 20, 1645–1658. [Google Scholar] [CrossRef]
- Laine, E.; Martínez, L.; Blondel, A.; Malliavin, T. Activation of the edema factor of Bacillus anthracis by calmodulin: Evidence of an interplay between the EF-calmodulin interaction and calcium binding. Biophys. J. 2010, 99, 2264–2272. [Google Scholar] [CrossRef]
- Gerlt, J.A.; Coderre, J.A.; Wolin, M.S. Mechanism of the adenylate cyclase reaction. J. Biol. Chem. 1980, 255, 331–334. [Google Scholar]
- Liu, Y.; Ruoho, A.E.; Rao, V.D.; Hurley, J.H. Catalytic mechanism of the adenylyl and guanylyl cyclases: Modeling and mutational analysis. Proc. Natl. Acad. Sci. USA 1997, 94, 13414–13419. [Google Scholar] [CrossRef]
- Hurley, J.H. Structure, mechanism, and regulation of mammalian adenylyl cyclase. J. Biol. Chem. 1999, 274, 7599–7602. [Google Scholar] [CrossRef]
- Tesmer, J.J.G.; Sunhara, R.K.; Johnson, R.A.; Gosselin, G.; Gilman, A.G.; Sprang, S.R. Two-metal-ion catalysis in adenylyl cyclase. Science 1999, 285, 756–760. [Google Scholar] [CrossRef]
- Martínez, L.; Laine, E.; Malliavin, T.; Nilges, M.; Blondel, A. ATP conformations and ion binding modes in the active site of anthrax edema factor: A computational analysis. Proteins 2009, 77, 971–983. [Google Scholar] [CrossRef]
- Martínez, L.; Malliavin, T.; Blondel, A. Mechanism of reactant and product dissociation from the Anthrax Edema Factor: A locally enhanced sampling and steered molecular dynamics study. Proteins 2011, 79, 1649–1661. [Google Scholar] [CrossRef] [Green Version]
- Gupta, M.; Alam, S.; Bhatnagar, R. Kinetic characterization and ligand binding studies of His351 mutants of Bacillus anthracis adenylate cyclase. Arch. Biochem. Biophys. 2006, 446, 28–34. [Google Scholar] [CrossRef]
- Erdorf, M.; Mou, T.-C.; Seifert, R. Impact of divalent metal ions on regulation of adenylyl cyclase isoforms by forskolin analogs. Biochem. Pharmacol. 2011, 82, 1673–1681. [Google Scholar] [CrossRef]
- Beste, K.Y.; Burhenne, H.; Kaever, V.; Stasch, J.P.; Seifert, R. Nucleotidyl cyclase activity of soluble guanylyl cyclase α1β1. Biochemistry 2012, 51, 194–204. [Google Scholar] [CrossRef]
- Beis, I.; Newsholme, E. The contents of adenine nucleotides, phosphagens and some glycolytic intermediates in resting muscles from vertebrates and invertebrates. Biochem. J. 1975, 152, 23–32. [Google Scholar]
- Fischer, S.; Karplus, M. Conjugate peak refinement: An algorithm for finding reaction paths and accurate transition states in systems with many degrees of freedom. Chem. Phys. Lett. 1992, 194, 252–261. [Google Scholar] [CrossRef]
- Blondel, A.; Renaud, J.-P.; Fischer, S.; Moras, D.; Karplus, M. Retinoic acid receptor: A simulation analysis of retinoic acid binding and the resulting conformational changes. J. Mol. Biol. 1999, 291, 101–115. [Google Scholar] [CrossRef]
- An, J.; Totrov, M.; Abagyan, R. Comprehensive identification of “druggable protein” ligand binding sites. Genome Inform. 2004, 15, 31–41. [Google Scholar]
- LeFoulon, F.; Braud, E.; Fabis, F.; Lancelot, J.-C.; Rault, S. Synthesis and combinatorial approach of the reactivity of 6-and 7-arylthieno[3,2-d][1,3]oxazine-2,4-diones. Tetrahedron 2003, 59, 10051–10057. [Google Scholar] [CrossRef]
- LeFoulon, F.-X.; Braud, E.; Fabis, F.; Lancelot, J.-C.; Rault, S. Solution-phase parallel synthesis of a 1140-member ureidothiophene carboxylic acid library. J. Comb. Chem. 2005, 7, 253–257. [Google Scholar] [CrossRef]
- Cox, K.; Shomin, C.; Ghosh, I. Tinkering outside the kinase ATP box: Allosteric (type IV) and bivalent (type V) inhibitors of protein kinases. Future Med. Chem. 2011, 3, 29–43. [Google Scholar] [CrossRef]
- Eglen, R.; Reisine, T. GPCRs revisited: New insights lead to novel drugs. Pharmaceuticals 2011, 4, 244–277. [Google Scholar] [CrossRef]
- Eglen, R.; Reisine, T. Drug discovery and the human kinome: Recent trends. Pharmacol. Ther. 2011, 130, 144–156. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Laine, É.; Martínez, L.; Ladant, D.; Malliavin, T.; Blondel, A. Molecular Motions as a Drug Target: Mechanistic Simulations of Anthrax Toxin Edema Factor Function Led to the Discovery of Novel Allosteric Inhibitors. Toxins 2012, 4, 580-604. https://doi.org/10.3390/toxins4080580
Laine É, Martínez L, Ladant D, Malliavin T, Blondel A. Molecular Motions as a Drug Target: Mechanistic Simulations of Anthrax Toxin Edema Factor Function Led to the Discovery of Novel Allosteric Inhibitors. Toxins. 2012; 4(8):580-604. https://doi.org/10.3390/toxins4080580
Chicago/Turabian StyleLaine, Élodie, Leandro Martínez, Daniel Ladant, Thérèse Malliavin, and Arnaud Blondel. 2012. "Molecular Motions as a Drug Target: Mechanistic Simulations of Anthrax Toxin Edema Factor Function Led to the Discovery of Novel Allosteric Inhibitors" Toxins 4, no. 8: 580-604. https://doi.org/10.3390/toxins4080580