Development of an in Vitro Potency Assay for Anti-anthrax Lethal Toxin Neutralizing Antibodies

Abstract
:1. Introduction
2. Materials and Methods
2.1. Anthrax Lethal Toxin
2.2. Antibody Preparations

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| ID (Specificity) | Source | Species | TN activity [Reference] |
|---|---|---|---|
| IQNLF (anti-LF) | IQ Therapeutics, The Netherlands | Human | 50% at 15 ng/mL [9] |
| IQNPA (anti-PA) | 50% at 50 ng/mL [9] | ||
| IQNLF and IQNPA | >50% at 35 ng/mL [9] | ||
| 9A11 (anti-LF) | Duke University, North Carolina, USA | Mouse | 50% at 200 ng/mL [10] |
2.3. Cell Culture
2.4. Analysis of Gene Expression in HUVEC jr2 Cells
2.5. TN Assay Based on HUVEC jr2 Cells
2.6. Cell Viability Assay
2.7. IL-6 and IL-8 ELISA
2.8. Processing and Analysis of ELISA Data
3. Results
3.1. Lethal Toxin Rapidly Alters Gene Expression in Endothelial Cells
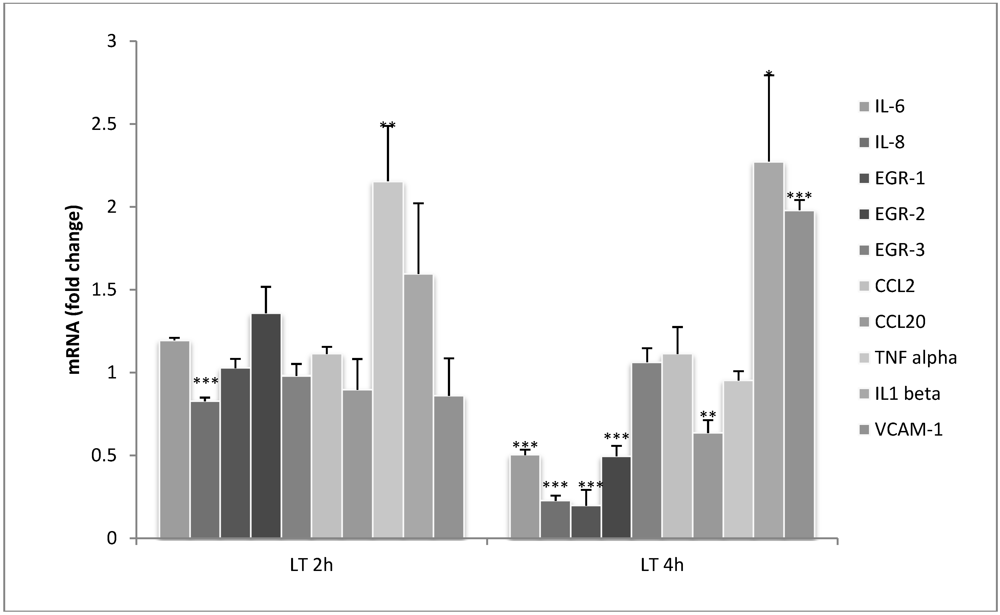
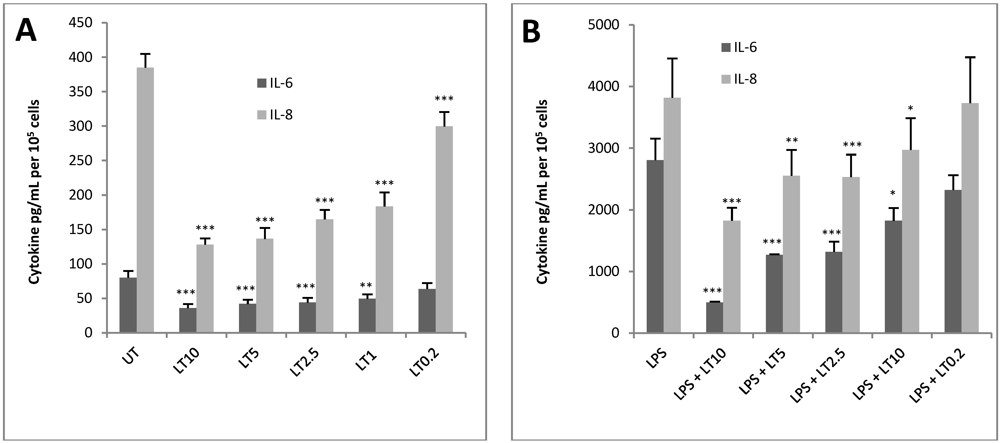
3.2. LT Reduces Cytokine Production in Endothelial Cells
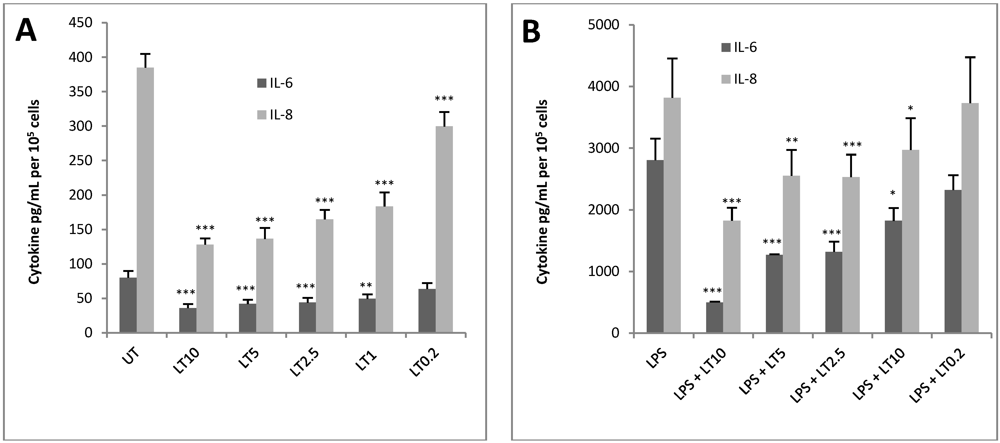
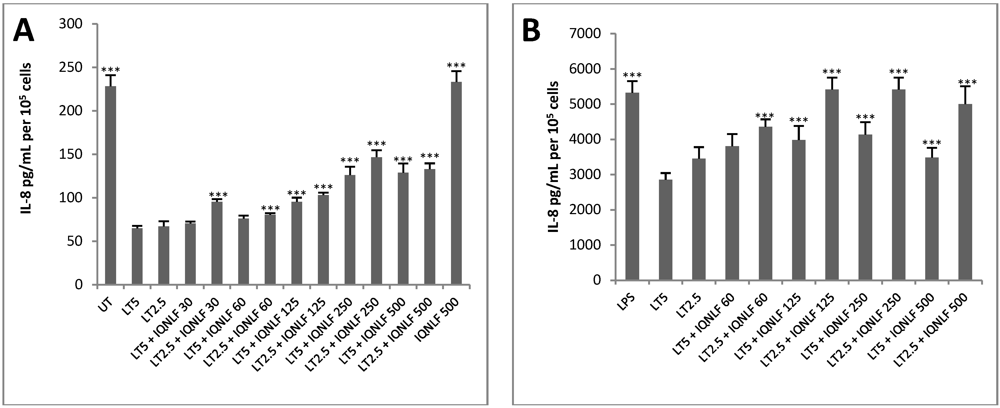
3.3. Application of HUVEC jr2 Cells in Toxin Neutralisation Assay
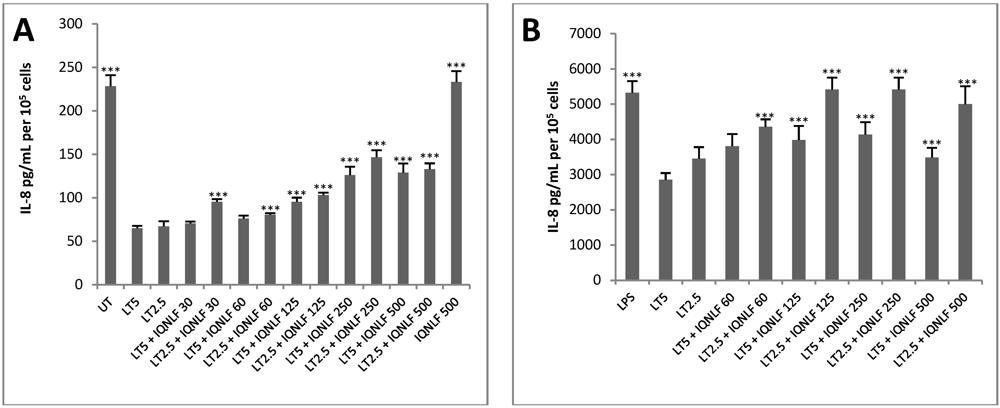
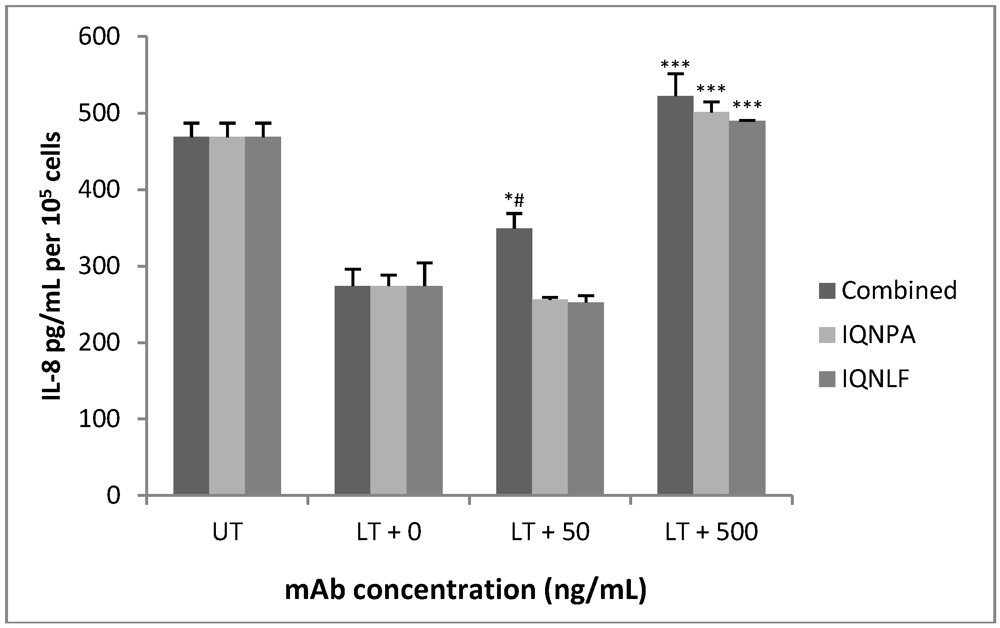
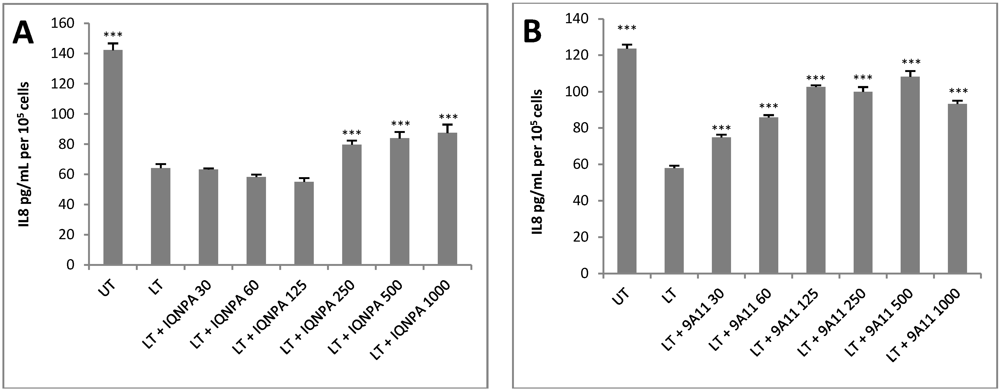
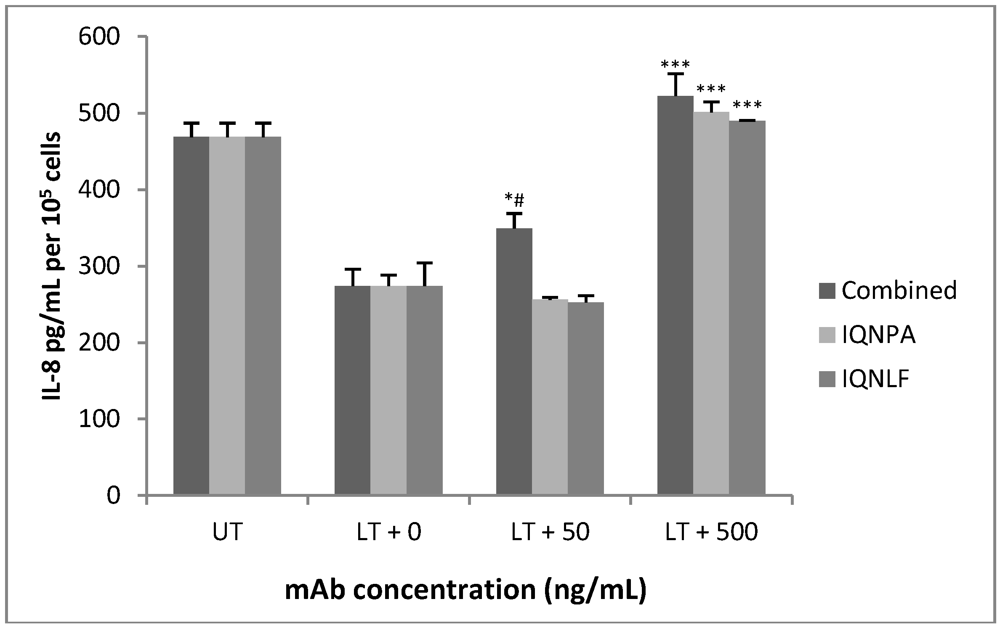
3.4. TN Antibodies Restore IL-8 Production in LT-Treated HUVEC jr2 Cells
4. Discussion
Acknowledgments
Conflict of Interest
References
- Collier, R.J.; Young, J.A. Anthrax toxin. Ann. Rev. Cell Dev. Biol. 2003, 19, 45–70. [Google Scholar] [CrossRef]
- Moayeri, M.; Leppla, S.H. The roles of anthrax toxin in pathogenesis. Curr. Opin. Microbiol. 2004, 7, 19–24. [Google Scholar] [CrossRef] [PubMed]
- Leppla, S. Anthrax toxin edema factor: a bacterial adenylate cyclase that increases cyclic AMP concentrations of eukaryotic cells. Proc. Natl. Acad. Sci. USA 1982, 79, 3162–3166. [Google Scholar] [CrossRef]
- Duesbery, N.S.; Webb, C.P.; Leppla, S.H.; Gordon, V.M.; Klimpel, K.R.; Copeland, T.D.; Ahn, N.G.; Oskarsson, M.K.; Fukasawa, K.; Paul, K.D.; et al. Proteolytic inactivation of MAP-kinase-kinase by anthrax lethal factor. Science 1998, 280, 734–737. [Google Scholar] [PubMed]
- Bardwell, A.J.; Abdollahi, M.; Bardwell, L. Anthrax lethal factor-cleavage products of MAPK (mitogen activated protein kinase) kinases exhibit reduced binding to their cognate MAPKs. Biochem. J. 2004, 378, 569–577. [Google Scholar] [CrossRef] [PubMed]
- Chopra, A.P.; Boone, S.A.; Liang, X.; Duesbery, N.S. Anthrax lethal factor proteolysis and inactivation of MAPK kinase. J. Biol. Chem. 2003, 278, 9402–9406. [Google Scholar] [PubMed]
- Matheny, J.; Mair, M.; Smith, B. Cost/success projections for US biodefense countermeasure development. Nat. Biotechnol. 2008, 26, 981–983. [Google Scholar] [CrossRef] [PubMed]
- Chen, Z.; Moayeri, M.; Purcell, R. Monoclonal antibody therapies against anthrax. Toxins 2011, 3, 1004–1019. [Google Scholar] [CrossRef] [PubMed]
- Albrecht, M.T.; Li, H.; Williamson, E.D.; LeButt, C.S.; Flick-Smith, H.C.; Quinn, C.P.; Westra, H.; Galloway, D.; Mateczun, A.; Goldman, S.; et al. Human monoclonal antibodies against anthrax lethal factor and protective antigen act independently to protect against Bacillus anthracis infection and enhance endogenous immunity to anthrax. Infect. Immun. 2007, 75, 5425–5433. [Google Scholar] [PubMed]
- Staats, H.F.; Alam, S.M.; Scearce, R.M.; Kirwan, S.M.; Zhang, J.X.; Gwinn, W.M.; Haynes, B.F. In vitro and in vivo characterization of anthrax anti-protective antigen and anti-lethal factor monoclonal antibodies after passive transfer in a mouse lethal toxin challenge model to define correlates of immunity. Infect. Immun. 2007, 75, 5443–5452. [Google Scholar] [CrossRef] [PubMed]
- Friedlander, A.M.; Bhatnagar, R.; Leppla, S.H.; Johnson, L.; Singh, Y. Characterization of macrophage sensitivity and resistance to anthrax lethal toxin. Infect. Immun. 1993, 61, 245–252. [Google Scholar] [PubMed]
- Hering, D.; Thompson, W.; Hewetson, J.; Little, S.; Norris, S.; Pace-Templeton, J. Validation of the anthrax lethal toxin neutralization assay. Biologicals 2004, 32, 17–27. [Google Scholar] [CrossRef] [PubMed]
- Pittman, P.R.; Kim-Ahn, G.; Pifat, D.Y.; Coonan, K.; Gibbs, P.; Little, S.; Pace-Templeton, J.G.; Myers, R.; Parker, G.W.; Friedlander, A.M. Anthrax vaccine: immunogenicity and safety of a dose-reduction, route-change comparison study in humans. Vaccine 2002, 20, 1412–1420. [Google Scholar] [CrossRef] [PubMed]
- Zmuda, J.F.; Zhang, L.; Richards, T.; Pham, Q.; Zukauskas, D.; Pierre, J.L.; Laird, M.W.; Askins, J.; Choi, G.H. Development of an edema factor-mediated cAMP-induction bioassay for detecting antibody-mediated neutralization of anthrax protective antigen. J. Immunol. Methods 2005, 298, 47–60. [Google Scholar] [CrossRef] [PubMed]
- Barson, H.V.; Mollenkopf, H.; Kaufmann, S.H.E.; Rijpkema, S. In vitro exposure to anthrax lethal toxin suppresses chemokine production in the human neutrophil-like cell line NB-4. Biochem. Biophy. Res. Commun. 2008, 374, 288–293. [Google Scholar] [CrossRef]
- Wheeler, J.X.; Whiting, G.; Rijpkema, S. Proteomic analysis of the response of the human neutrophil-like cell line NB-4 after exposure to anthrax lethal toxin. Proteomics Clin. Appl. 2007, 1, 1266–1279. [Google Scholar] [CrossRef] [PubMed]
- Batty, S.; Chow, E.M.C.; Kassam, A.; Der, S.D.; Mogridge, J. Inhibition of mitogen-activated protein kinase signaling by Bacillus anthracis lethal toxin causes destabilization of interleukin-8 mRNA. Cell Microbiol. 2006, 8, 130–138. [Google Scholar] [CrossRef] [PubMed]
- During, R.L.; Li, W.; Hao, B.; Koenig, J.M.; Stephens, D.S.; Quinn, C.P.; Southwick, F.S. Anthrax lethal toxin paralyzes neutrophil actin-based motility. J. Infect. Dis. 2005, 192, 837–845. [Google Scholar] [CrossRef] [PubMed]
- During, R.L.; Gibson, B.G.; Li, W.; Bishai, E.A.; Sidhu, G.S.; Landry, J.; Southwick, F.S. Anthrax lethal toxin paralyzes actin-based motility by blocking Hsp27 phosphorylation. EMBO J. 2007, 26, 2240–2250. [Google Scholar] [CrossRef] [PubMed]
- Fang, H.; Cordoba-Rodriguez, R.; Lankford, C.S.; Frucht, D.M.J. Anthrax lethal toxin blocks MAPK kinase-dependent IL-2 production in CD4 + T cells. J. Immunol. 2005, 174, 4966–4971. [Google Scholar] [PubMed]
- Popov, S.G.; Villasmil, R.; Bernardi, J.; Grene, E.; Cardwell, J.; Popova, T.; Wu, A.; Alibek, D.; Bailey, C.; Alibek, K. Effect of Bacillus anthracis lethal toxin on human peripheral blood mononuclear cells. FEBS Lett. 2002, 527, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Rolando, M.; Stefani, C.; Flatau, G.; Auberger, P.; Mettouchi, A.; Mhlanga, M.; Rapp, U.; Galmiche, A.; Lemichez, E. Transcriptome dysregulation by anthrax lethal toxin plays a key role in induction of human endothelial cell cytotoxicity. Cell Microbiol. 2010, 12, 891–905. [Google Scholar] [CrossRef] [PubMed]
- Stebbings, R.; Findlay, L.; Edwards, C.; Eastwood, D.; Bird, C.; North, D.; Mistry, Y.; Dilger, P.; Liefooghe, E.; Cludts, I.; et al. Cytokine storm in the phase I trial of monoclonal antibody TGN1412: Better understanding the causes to improve preclinical testing of immunotherapeutics. J. Immunol. 2007, 179, 3325–3331. [Google Scholar] [PubMed]
- Fleck, R.A.; Athwal, H.; Bygraves, J.A.; Hockley, D.J.; Feavers, I.M.; Stacey, G.N. Optimization of NB-4 and HL-60 differentiation for use in opsonophagocytosis assays. In Vitro Cell Dev. Biol. Anim. 2003, 39, 235–242. [Google Scholar] [CrossRef] [PubMed]
- Combistats computer program for the statistical analysis of data from biological dilution assays or potency assays (EDQM). Available online: http://combistats.edqm.eu/ (accessed on 9 December 2011).
- Erwin, J.J.; DaSilva, L.M.; Bavari, S.; Little, S.F.; Friedlander, A.M.; Chanh, T.C. Macrophage-derived cell lines do not express proinflammatory cytokines after exposure to Bacillus anthracis lethal toxin. Infect. Immun. 2001, 69, 1175–1177. [Google Scholar] [PubMed]
- Warfel, J.M.; D’Agnillo, F. Anthrax lethal toxin enhances TNF-Induced endothelial VCAM-1 expression via an IFN regulatory factor-1-dependent mechanism. J. Immunol. 2008, 180, 7516–7524. [Google Scholar] [PubMed]
- Guarner, J.; Jernigan, J.A.; Shieh, W.J.; Tatti, K.; Flannagan, L.M.; Stephens, D.S.; Popovic, T.; Ashford, D.A.; Perkins, B.A.; Zaki, S.R. Pathology and pathogenesis of bioterrorism-related inhalational anthrax. Am. J. Pathol. 2003, 163, 701–709. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Haines, D.; Young, H.A.; Leppla, S.H. Bacillus anthracis lethal toxin induces TNF-alpha-independent hypoxia-mediated toxicity in mice. J. Clin. Invest. 2003, 112, 670–682. [Google Scholar] [PubMed]
- Park, J.M.; Ng, V.H.; Maeda, S.; Rest, R.F.; Karin, M. Anthrolysin O and other Gram-positive cytolysins are Toll-like receptor 4 agonists. J. Exp. Med. 2004, 200, 1647–1655. [Google Scholar] [CrossRef] [PubMed]
- van Sorge, N.M.; Ebrahimi, C.M.; McGillivray, S.M.; Quach, D.; Sabet, M.; Guiney, D.G.; Doran, K.S. Anthrax toxins inhibit neutrophil signaling pathways in brain endothelium and contribute to the pathogenesis of meningitis. PLoS One 2008, 3, e2964. [Google Scholar] [PubMed]
- Warfel, J.M.; Steele, A.D.; D’Agnillo, F. Anthrax lethal toxin induces endothelial barrier dysfunction. Am. J. Pathol. 2005, 166, 1871–1881. [Google Scholar] [CrossRef] [PubMed]
- Posern, G.; Treisman, R. Actin’ together: Serum response factor, its cofactors and the link to signal transduction. Trends Cell. Biol. 2006, 16, 588–596. [Google Scholar] [CrossRef] [PubMed]
- Steele, A.D.; Warfel, J.M.; D’Agnillo, F. Anthrax lethal toxin enhances cytokine-induced VCAM-1 expression on human endothelial cells. Biochem. Biophys. Res. Commun. 2005, 337, 1249–1256. [Google Scholar] [CrossRef] [PubMed]
- Raymond, B.; Batsche, E.; Boutillon, F.; Wu, Y-Z.; Leduc, D.; Balloy, V.; Raoust, E.; Muchardt, C.; Goossens, P.L.; Touqui, L. Anthrax lethal toxin impairs IL-8 expression in epithelial cells through Inhibition of histone H3 modification. PLoS Pathog. 2009, 5, e1000359. [Google Scholar] [CrossRef] [PubMed]
- Moayeri, M.; Crown, D.; Newman, Z.L.; Okugawa, S.; Eckhaus, M.; Cataisson, C.; Liu, S.; Sastalla, I.; Leppla, S.H. Inflammasome sensor Nlrp1b-dependent resistance to anthrax is mediated by Caspase-1, IL-1 signaling and neutrophil recruitment. PLoS Pathog. 2010, 6, e1001222. [Google Scholar] [CrossRef] [PubMed]
- Newman, Z.L.; Crown, D.; Leppla, S.H.; Moayeri, M. Anthrax lethal toxin activates the inflammasome in sensitive rat macrophages. Biochem. Biophys. Res. Commun. 2010, 398, 785–789. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, M.L.; Terzyan, S.; Ballard, J.D.; James, J.A.; Farris, A.D. The major neutralizing antibody responses to recombinant anthrax lethal and edema factors are directed to non-cross-reactive epitopes. Infect. Immun. 2009, 77, 4714–4723. [Google Scholar] [CrossRef] [PubMed]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Whiting, G.; Baker, M.; Rijpkema, S. Development of an in Vitro Potency Assay for Anti-anthrax Lethal Toxin Neutralizing Antibodies. Toxins 2012, 4, 28-41. https://doi.org/10.3390/toxins4010028
Whiting G, Baker M, Rijpkema S. Development of an in Vitro Potency Assay for Anti-anthrax Lethal Toxin Neutralizing Antibodies. Toxins. 2012; 4(1):28-41. https://doi.org/10.3390/toxins4010028
Chicago/Turabian StyleWhiting, Gail, Michael Baker, and Sjoerd Rijpkema. 2012. "Development of an in Vitro Potency Assay for Anti-anthrax Lethal Toxin Neutralizing Antibodies" Toxins 4, no. 1: 28-41. https://doi.org/10.3390/toxins4010028
APA StyleWhiting, G., Baker, M., & Rijpkema, S. (2012). Development of an in Vitro Potency Assay for Anti-anthrax Lethal Toxin Neutralizing Antibodies. Toxins, 4(1), 28-41. https://doi.org/10.3390/toxins4010028