Mucosal Injuries due to Ribosome-Inactivating Stress and the Compensatory Responses of the Intestinal Epithelial Barrier

{kind=link}
Abstract
:1. Introduction
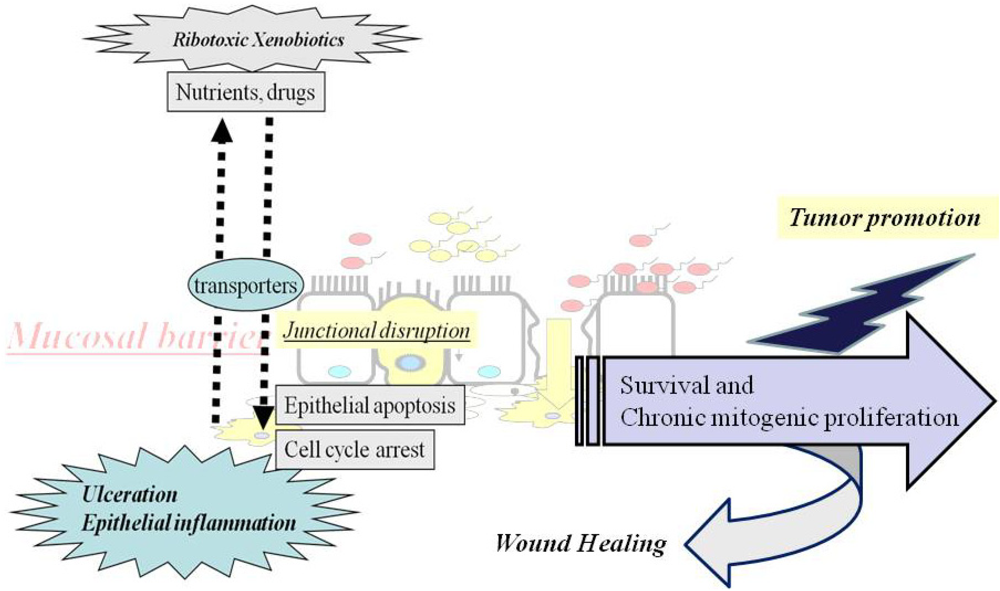
2. Ribotoxic Stress-Mediated Barrier Disruption
2.1. Modulation of the Epithelial Transport System
2.2. Disrupted Epithelial Junctions due to Ribotoxic Stress
3. Mucosal Cell Death due to Ribotoxic Stress and Epithelial Counteractions
3.1. MAPK-Linked Epithelial Cell Death and Reconstitution in Response to Ribotoxin Intoxication
3.2. Other Ribotoxic Mediators of Epithelial Apoptosis
4. Compensatory Responses of Epithelial Cells to Mucosal Ulceration
5. Link of Chronic Mitogenic Stimulation to Tumor Promotion by Chronic Ribotoxic Exposure
6. Conclusion
Acknowledgements
References
- Catalioto, R.M.; Maggi, C.A.; Giuliani, S. Intestinal epithelial barrier dysfunction in disease and possible therapeutical interventions. Curr. Med. Chem. 2011, 18, 398–426. [Google Scholar]
- Laukoetter, M.G.; Nava, P.; Nusrat, A. Role of the intestinal barrier in inflammatory bowel disease. World J. Gastroenterol. 2008, 14, 401–407. [Google Scholar]
- Amasheh, S.; Fromm, M.; Gunzel, D. Claudins of intestine and nephron-A correlation of molecular tight junction structure and barrier function. Acta Physiol. (Oxf.) 2011, 201, 133–140. [Google Scholar] [CrossRef] [PubMed]
- Shimizu, M. Interaction between food substances and the intestinal epithelium. Biosci. Biotechnol. Biochem. 2010, 74, 232–241. [Google Scholar]
- Hofman, P.M. Pathobiology of the neutrophil-intestinal epithelial cell interaction: Role in carcinogenesis. World J. Gastroenterol. 2010, 16, 5790–5800. [Google Scholar]
- Kunisawa, J.; Kiyono, H. Aberrant interaction of the gut immune system with environmental factors in the development of food allergies. Curr. Allergy Asthma Rep. 2010, 10, 215–221. [Google Scholar]
- Medema, J.P.; Vermeulen, L. Microenvironmental regulation of stem cells in intestinal homeostasis and cancer. Nature 2011, 474, 318–326. [Google Scholar]
- Roda, G.; Sartini, A.; Zambon, E.; Calafiore, A.; Marocchi, M.; Caponi, A.; Belluzzi, A.; Roda, E. Intestinal epithelial cells in inflammatory bowel diseases. World J. Gastroenterol. 2010, 16, 4264–4271. [Google Scholar]
- Girbes, T.; Ferreras, J.M.; Arias, F.J.; Stirpe, F. Description, distribution, activity and phylogenetic relationship of ribosome-inactivating proteins in plants, fungi and bacter. Mini. Rev. Med. Chem. 2004, 4, 461–476. [Google Scholar]
- Lacadena, J.; Alvarez-Garcia, E.; Carreras-Sangra, N.; Herrero-Galan, E.; Alegre-Cebollada, J.; Garcia-Ortega, L.; Onaderra, M.; Gavilanes, J.G.; Martinez del Pozo, A. Fungal ribotoxins: Molecular dissection of a family of natural killers. FEMS Microbiol. Rev. 2007, 31, 212–237. [Google Scholar]
- Ng, T.B.; Wong, J.H.; Wang, H. Recent progress in research on ribosome inactivating proteins. Curr. Protein Pept. Sci. 2010, 11, 37–53. [Google Scholar]
- Stirpe, F.; Battelli, M.G. Ribosome-inactivating proteins: Progress and problems. Cell Mol. Life Sci. 2006, 63, 1850–1866. [Google Scholar]
- Iordanov, M.S.; Pribnow, D.; Magun, J.L.; Dinh, T.H.; Pearson, J.A.; Magun, B.E. Ultraviolet radiation triggers the ribotoxic stress response in mammalian cells. J. Biol. Chem. 1998, 273, 15794–15803. [Google Scholar]
- Li, M.; Pestka, J.J. Comparative induction of 28S ribosomal RNA cleavage by ricin and the trichothecenes deoxynivalenol and T-2 toxin in the macrophage. Toxicol. Sci. 2008, 105, 67–78. [Google Scholar]
- Rzymski, T.; Harris, A.L. The unfolded protein response and integrated stress response to anoxia. Clin. Cancer Res. 2007, 13, 2537–2540. [Google Scholar]
- Bunyard, P.; Handley, M.; Pollara, G.; Rutault, K.; Wood, I.; Chaudry, M.; Alderman, C.; Foreman, J.; Katz, D.R.; Chain, B.M. Ribotoxic stress activates p38 and JNK kinases and modulates the antigen-presenting activity of dendritic cells. Mol. Immunol. 2003, 39, 815–827. [Google Scholar]
- Instanes, C.; Hetland, G. Deoxynivalenol (DON) is toxic to human colonic, lung and monocytic cell lines, but does not increase the IgE response in a mouse model for allergy. Toxicology 2004, 204, 13–21. [Google Scholar] [PubMed]
- Smith, W.E.; Kane, A.V.; Campbell, S.T.; Acheson, D.W.; Cochran, B.H.; Thorpe, C.M. Shiga toxin 1 triggers a ribotoxic stress response leading to p38 and JNK activation and induction of apoptosis in intestinal epithelial cells. Infect. Immun. 2003, 71, 1497–1504. [Google Scholar]
- Luo, Y.; Yoshizawa, T.; Katayama, T. Comparative study on the natural occurrence of Fusarium mycotoxins (trichothecenes and zearalenone) in corn and wheat from high- and low-risk areas for human esophageal cancer in China. Appl. Environ. Microbiol. 1990, 56, 3723–3726. [Google Scholar]
- Li, F.Q.; Li, Y.W.; Luo, X.Y.; Yoshizawa, T. Fusarium toxins in wheat from an area in Henan Province, PR China, with a previous human red mould intoxication episode. Food Addit. Contam. 2002, 19, 163–167. [Google Scholar]
- Bhat, R.V.; Beedu, S.R.; Ramakrishna, Y.; Munshi, K.L. Outbreak of trichothecene mycotoxicosis associated with consumption of mould-damaged wheat production in Kashmir Valley, India. Lancet 1989, 1, 35–37. [Google Scholar]
- Bouhet, S.; Oswald, I.P. The effects of mycotoxins, fungal food contaminants, on the intestinal epithelial cell-derived innate immune response. Vet. Immunol. Immunopathol. 2005, 108, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Maresca, M.; Mahfoud, R.; Garmy, N.; Fantini, J. The mycotoxin deoxynivalenol affects nutrient absorption in human intestinal epithelial cells. J. Nutr. 2002, 132, 2723–2731. [Google Scholar]
- Sergent, T.; Parys, M.; Garsou, S.; Pussemier, L.; Schneider, Y.J.; Larondelle, Y. Deoxynivalenol transport across human intestinal Caco-2 cells and its effects on cellular metabolism at realistic intestinal concentrations. Toxicol. Lett. 2006, 164, 167–176. [Google Scholar]
- Stearns-Kurosawa, D.J.; Collins, V.; Freeman, S.; Tesh, V.L.; Kurosawa, S. Distinct physiologic and inflammatory responses elicited in baboons after challenge with Shiga toxin type 1 or 2 from enterohemorrhagic Escherichia coli. Infect. Immun. 2010, 78, 2497–2504. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.M.; Aslam, R.U.; Mantis, N.J. Evidence for widespread epithelial damage and coincident production of monocyte chemotactic protein 1 in a murine model of intestinal ricin intoxication. Infect. Immun. 2007, 75, 1745–1750. [Google Scholar]
- Craddock, V.M.; Hill, R.J.; Henderson, A.R. Acute and chronic effects of diacetoxyscirpenol on cell replication in rat esophagus and stomach. Cancer Lett. 1988, 41, 287–294. [Google Scholar]
- Hsia, C.C.; Wu, J.L.; Lu, X.Q.; Li, Y.S. Natural occurrence and clastogenic effects of nivalenol, deoxynivalenol, 3-acetyl-deoxynivalenol, 15-acetyl-deoxynivalenol, and zearalenone in corn from a high-risk area of esophageal cancer. Cancer Detect. Prev. 1988, 13, 79–86. [Google Scholar] [PubMed]
- Hsia, C.C.; Wu, Z.Y.; Li, Y.S.; Zhang, F.; Sun, Z.T. Nivalenol, a main Fusarium toxin in dietary foods from high-risk areas of cancer of esophagus and gastric cardia in China, induced benign and malignant tumors in mice. Oncol. Rep. 2004, 12, 449–456. [Google Scholar] [PubMed]
- Wattenberg, E.V. Palytoxin: Exploiting a novel skin tumor promoter to explore signal transduction and carcinogenesis. Am. J. Physiol. Cell Physiol. 2007, 292, C24–C32. [Google Scholar]
- Tep, J.; Videmann, B.; Mazallon, M.; Balleydier, S.; Cavret, S.; Lecoeur, S. Transepithelial transport of fusariotoxin nivalenol: Mediation of secretion by ABC transporters. Toxicol. Lett. 2007, 170, 248–258. [Google Scholar]
- Videmann, B.; Tep, J.; Cavret, S.; Lecoeur, S. Epithelial transport of deoxynivalenol: Involvement of human P-glycoprotein (ABCB1) and multidrug resistance-associated protein 2 (ABCC2). Food Chem. Toxicol. 2007, 45, 1938–1947. [Google Scholar]
- Lala, P.; Ito, S.; Lingwood, C.A. Retroviral transfection of Madin-Darby canine kidney cells with human MDR1 results in a major increase in globotriaosylceramide and 10(5)- to 10(6)-fold increased cell sensitivity to verocytotoxin. Role of p-glycoprotein in glycolipid synthesis. J. Biol. Chem. 2000, 275, 6246–6251. [Google Scholar] [PubMed]
- Ho, G.T.; Soranzo, N.; Nimmo, E.R.; Tenesa, A.; Goldstein, D.B.; Satsangi, J. ABCB1/MDR1 gene determines susceptibility and phenotype in ulcerative colitis: Discrimination of critical variants using a gene-wide haplotype tagging approach. Hum. Mol. Genet. 2006, 15, 797–805. [Google Scholar]
- Staley, E.M.; Schoeb, T.R.; Lorenz, R.G. Differential susceptibility of P-glycoprotein deficient mice to colitis induction by environmental insults. Inflamm. Bowel Dis. 2009, 15, 684–696. [Google Scholar]
- Awad, W.A.; Aschenbach, J.R.; Setyabudi, F.M.; Razzazi-Fazeli, E.; Bohm, J.; Zentek, J. In vitro effects of deoxynivalenol on small intestinal D-glucose uptake and absorption of deoxynivalenol across the isolated jejunal epithelium of laying hens. Poult. Sci. 2007, 86, 15–20. [Google Scholar]
- Muise, A.M.; Walters, T.D.; Glowacka, W.K.; Griffiths, A.M.; Ngan, B.Y.; Lan, H.; Xu, W.; Silverberg, M.S.; Rotin, D. Polymorphisms in E-cadherin (CDH1) result in a mis-localised cytoplasmic protein that is associated with Crohn’s disease. Gut 2009, 58, 1121–1127. [Google Scholar]
- Edelblum, K.L.; Turner, J.R. The tight junction in inflammatory disease: Communication breakdown. Curr. Opin. Pharmacol. 2009, 9, 715–720. [Google Scholar]
- Schwarz, B.T.; Wang, F.; Shen, L.; Clayburgh, D.R.; Su, L.; Wang, Y.; Fu, Y.X.; Turner, J.R. LIGHT signals directly to intestinal epithelia to cause barrier dysfunction via cytoskeletal and endocytic mechanisms. Gastroenterology 2007, 132, 2383–2394. [Google Scholar]
- Suenaert, P.; Bulteel, V.; Lemmens, L.; Noman, M.; Geypens, B.; Van Assche, G.; Geboes, K.; Ceuppens, J.L.; Rutgeerts, P. Anti-tumor necrosis factor treatment restores the gut barrier in Crohn’s disease. Am. J. Gastroenterol. 2002, 97, 2000–2004. [Google Scholar]
- Planchon, S.; Fiocchi, C.; Takafuji, V.; Roche, J.K. Transforming growth factor-beta1 preserves epithelial barrier function: Identification of receptors, biochemical intermediates, and cytokine antagonist. J. Cell Physiol. 1999, 181, 55–66. [Google Scholar]
- Schulzke, J.D.; Ploeger, S.; Amasheh, M.; Fromm, A.; Zeissig, S.; Troeger, H.; Richter, J.; Bojarski, C.; Schumann, M.; Fromm, M. Epithelial tight junctions in intestinal inflammation. Ann. N. Y. Acad. Sci. 2009, 1165, 294–300. [Google Scholar]
- De Walle, J.V.; Sergent, T.; Piront, N.; Toussaint, O.; Schneider, Y.J.; Larondelle, Y. Deoxynivalenol affects in vitro intestinal epithelial cell barrier integrity through inhibition of protein synthesis. Toxicol. Appl. Pharmacol. 2010, 245, 291–298. [Google Scholar] [CrossRef] [PubMed]
- Diesing, A.K.; Nossol, C.; Danicke, S.; Walk, N.; Post, A.; Kahlert, S.; Rothkotter, H.J.; Kluess, J. Vulnerability of polarised intestinal porcine epithelial cells to mycotoxin deoxynivalenol depends on the route of application. PLoS One 2011, 6. [Google Scholar] [CrossRef]
- Diesing, A.K.; Nossol, C.; Panther, P.; Walk, N.; Post, A.; Kluess, J.; Kreutzmann, P.; Danicke, S.; Rothkotter, H.J.; Kahlert, S. Mycotoxin deoxynivalenol (DON) mediates biphasic cellular response in intestinal porcine epithelial cell lines IPEC-1 and IPEC-J2. Toxicol. Lett. 2011, 200, 8–18. [Google Scholar]
- Liu, L.; Gao, H.; Li, J.; Dong, Y.; Liu, N.; Wan, J.; Liu, W.; Sun, Y.; Xu, M. Analysis of intestinal injuries induced by ricin in vitro using SPR technology and MS identification. Int. J. Mol. Sci. 2009, 10, 2431–2439. [Google Scholar] [CrossRef] [PubMed]
- Pinton, P.; Braicu, C.; Nougayrede, J.P.; Laffitte, J.; Taranu, I.; Oswald, I.P. Deoxynivalenol impairs porcine intestinal barrier function and decreases the protein expression of claudin-4 through a mitogen-activated protein kinase-dependent mechanism. J. Nutr. 2010, 140, 1956–1962. [Google Scholar]
- Pinton, P.; Nougayrede, J.P.; Del Rio, J.C.; Moreno, C.; Marin, D.E.; Ferrier, L.; Bracarense, A.P.; Kolf-Clauw, M.; Oswald, I.P. The food contaminant deoxynivalenol, decreases intestinal barrier permeability and reduces claudin expression. Toxicol. Appl. Pharmacol. 2009, 237, 41–48. [Google Scholar] [Green Version]
- Van de Walle, J.; During, A.; Piront, N.; Toussaint, O.; Schneider, Y.J.; Larondelle, Y. Physio-pathological parameters affect the activation of inflammatory pathways by deoxynivalenol in Caco-2 cells. Toxicol. in Vitro 2010, 24, 1890–1898. [Google Scholar]
- Chen, L.; Park, S.M.; Turner, J.R.; Peter, M.E. Cell death in the colonic epithelium during inflammatory bowel diseases: CD95/Fas and beyond. Inflamm. Bowel Dis. 2010, 16, 1071–1076. [Google Scholar]
- Fischbeck, A.; Leucht, K.; Frey-Wagner, I.; Bentz, S.; Pesch, T.; Kellermeier, S.; Krebs, M.; Fried, M.; Rogler, G.; Hausmann, M.; Humpf, H.U. Sphingomyelin induces cathepsin D-mediated apoptosis in intestinal epithelial cells and increases inflammation in DSS colitis. Gut 2011, 60, 55–65. [Google Scholar]
- Qiu, W.; Wu, B.; Wang, X.; Buchanan, M.E.; Regueiro, M.D.; Hartman, D.J.; Schoen, R.E.; Yu, J.; Zhang, L. PUMA-mediated intestinal epithelial apoptosis contributes to ulcerative colitis in humans and mice. J. Clin. Invest. 2011, 121, 1722–1732. [Google Scholar]
- Tambuwala, M.M.; Cummins, E.P.; Lenihan, C.R.; Kiss, J.; Stauch, M.; Scholz, C.C.; Fraisl, P.; Lasitschka, F.; Mollenhauer, M.; Saunders, S.P.; et al. Loss of prolyl hydroxylase-1 protects against colitis through reduced epithelial cell apoptosis and increased barrier function. Gastroenterology 2010, 139, 2093–2101. [Google Scholar] [PubMed]
- Gitter, A.H.; Bendfeldt, K.; Schulzke, J.D.; Fromm, M. Leaks in the epithelial barrier caused by spontaneous and TNF-alpha-induced single-cell apoptosis. FASEB J. 2000, 14, 1749–1753. [Google Scholar]
- Araki, Y.; Mukaisyo, K.; Sugihara, H.; Fujiyama, Y.; Hattori, T. Increased apoptosis and decreased proliferation of colonic epithelium in dextran sulfate sodium-induced colitis in mice. Oncol. Rep. 2010, 24, 869–874. [Google Scholar]
- Leek, M.D.; Griffiths, G.D.; Green, M.A. Intestinal pathology following intramuscular ricin poisoning. J. Pathol. 1989, 159, 329–334. [Google Scholar]
- Shifrin, V.I.; Anderson, P. Trichothecene mycotoxins trigger a ribotoxic stress response that activates c-Jun N-terminal kinase and p38 mitogen-activated protein kinase and induces apoptosis. J. Biol. Chem. 1999, 274, 13985–13992. [Google Scholar] [CrossRef] [PubMed]
- Allan, L.A.; Clarke, P.R. Apoptosis and autophagy: Regulation of caspase-9 by phosphorylation. FEBS J. 2009, 276, 6063–6073. [Google Scholar]
- Moon, Y.; Yang, H.; Kim, Y.B. Up-regulation of early growth response gene 1 (EGR-1) via ERK1/2 signals attenuates sulindac sulfide-mediated cytotoxicity in the human intestinal epithelial cells. Toxicol. Appl. Pharmacol. 2007, 223, 155–163. [Google Scholar]
- Moon, Y.; Yang, H.; Lee, S.H. Modulation of early growth response gene 1 and interleukin-8 expression by ribotoxin deoxynivalenol (vomitoxin) via ERK1/2 in human epithelial intestine 407 cells. Biochem. Biophys. Res. Commun. 2007, 362, 256–262. [Google Scholar]
- Lee, S.Y.; Lee, M.S.; Cherla, R.P.; Tesh, V.L. Shiga toxin 1 induces apoptosis through the endoplasmic reticulum stress response in human monocytic cells. Cell Microbiol. 2008, 10, 770–780. [Google Scholar]
- Yang, H.; Park, S.H.; Choi, H.J.; Do, K.H.; Kim, J.; An, T.J.; Lee, S.H.; Moon, Y. Mechanism-based alternative monitoring of endoplasmic reticulum stress by 8-keto-trichothecene mycotoxins using human intestinal epithelial cell line. Toxicol. Lett. 2010, 198, 317–323. [Google Scholar]
- Park, S.H.; Choi, H.J.; Yang, H.; Do, K.H.; Kim, J.; Moon, Y. Repression of peroxisome proliferator-activated receptor gamma by mucosal ribotoxic insult-activated CCAAT/enhancer-binding protein homologous protein. J. Immunol. 2010, 185, 5522–5530. [Google Scholar]
- Yang, H.; Choi, H.J.; Park, S.H.; Kim, J.S.; Moon, Y. Macrophage inhibitory cytokine-1 (MIC-1) and subsequent urokinase-type plasminogen activator mediate cell death responses by ribotoxic anisomycin in HCT-116 colon cancer cells. Biochem. Pharmacol. 2009, 78, 1205–1213. [Google Scholar]
- Brown, D.A.; Ward, R.L.; Buckhaults, P.; Liu, T.; Romans, K.E.; Hawkins, N.J.; Bauskin, A.R.; Kinzler, K.W.; Vogelstein, B.; Breit, S.N. MIC-1 serum level and genotype: Associations with progress and prognosis of colorectal carcinoma. Clin. Cancer Res. 2003, 9, 2642–2650. [Google Scholar]
- Johnen, H.; Lin, S.; Kuffner, T.; Brown, D.A.; Tsai, V.W.; Bauskin, A.R.; Wu, L.; Pankhurst, G.; Jiang, L.; Junankar, S.; et al. Tumor-induced anorexia and weight loss are mediated by the TGF-beta superfamily cytokine MIC-1. Nat. Med. 2007, 13, 1333–1340. [Google Scholar] [PubMed]
- Nakamura, T.; Scorilas, A.; Stephan, C.; Yousef, G.M.; Kristiansen, G.; Jung, K.; Diamandis, E.P. Quantitative analysis of macrophage inhibitory cytokine-1 (MIC-1) gene expression in human prostatic tissues. Br. J. Cancer 2003, 88, 1101–1104. [Google Scholar]
- Baek, S.J.; Wilson, L.C.; Eling, T.E. Resveratrol enhances the expression of non-steroidal anti-inflammatory drug-activated gene (NAG-1) by increasing the expression of p53. Carcinogenesis 2002, 23, 425–434. [Google Scholar]
- Martinez, J.M.; Sali, T.; Okazaki, R.; Anna, C.; Hollingshead, M.; Hose, C.; Monks, A.; Walker, N.J.; Baek, S.J.; Eling, T.E. Drug-induced expression of nonsteroidal anti-inflammatory drug-activated gene/macrophage inhibitory cytokine-1/prostate-derived factor, a putative tumor suppressor, inhibits tumor growth. J. Pharmacol. Exp. Ther. 2006, 318, 899–906. [Google Scholar] [CrossRef] [PubMed]
- Yang, H.; Park, S.H.; Choi, H.J.; Moon, Y. The integrated stress response-associated signals modulates intestinal tumor cell growth by NSAID-activated gene 1 (NAG-1/MIC-1/PTGF-beta). Carcinogenesis 2010, 31, 703–711. [Google Scholar]
- Bauskin, A.R.; Zhang, H.P.; Fairlie, W.D.; He, X.Y.; Russell, P.K.; Moore, A.G.; Brown, D.A.; Stanley, K.K.; Breit, S.N. The propeptide of macrophage inhibitory cytokine (MIC-1), a TGF-beta superfamily member, acts as a quality control determinant for correctly folded MIC-1. EMBO J. 2000, 19, 2212–2220. [Google Scholar] [PubMed]
- Khuu, C.H.; Barrozo, R.M.; Hai, T.; Weinstein, S.L. Activating transcription factor 3 (ATF3) represses the expression of CCL4 in murine macrophages. Mol. Immunol. 2007, 44, 1598–1605. [Google Scholar]
- Baek, S.J.; Kim, J.S.; Moore, S.M.; Lee, S.H.; Martinez, J.; Eling, T.E. Cyclooxygenase inhibitors induce the expression of the tumor suppressor gene EGR-1, which results in the up-regulation of NAG-1, an antitumorigenic protein. Mol. Pharmacol. 2005, 67, 356–364. [Google Scholar] [PubMed]
- Senapati, S.; Rachagani, S.; Chaudhary, K.; Johansson, S.L.; Singh, R.K.; Batra, S.K. Overexpression of macrophage inhibitory cytokine-1 induces metastasis of human prostate cancer cells through the FAK-RhoA signaling pathway. Oncogene 2010, 29, 1293–1302. [Google Scholar]
- Araki, Y.; Sugihara, H.; Hattori, T. In vitro effects of dextran sulfate sodium on a Caco-2 cell line and plausible mechanisms for dextran sulfate sodium-induced colitis. Oncol. Rep. 2006, 16, 1357–1362. [Google Scholar] [PubMed]
- Vetuschi, A.; Latella, G.; Sferra, R.; Caprilli, R.; Gaudio, E. Increased proliferation and apoptosis of colonic epithelial cells in dextran sulfate sodium-induced colitis in rats. Dig. Dis. Sci. 2002, 47, 1447–1457. [Google Scholar]
- Arai, N.; Mitomi, H.; Ohtani, Y.; Igarashi, M.; Kakita, A.; Okayasu, I. Enhanced epithelial cell turnover associated with p53 accumulation and high p21WAF1/CIP1 expression in ulcerative colitis. Mod. Pathol. 1999, 12, 604–611. [Google Scholar]
- Shinozaki, M.; Watanabe, T.; Kubota, Y.; Sawada, T.; Nagawa, H.; Muto, T. High proliferative activity is associated with dysplasia in ulcerative colitis. Dis Colon Rectum. 2000, 43, S34–S39. [Google Scholar]
- Yang, H.; Park, S.H.; Choi, H.J.; Moon, Y. Epithelial cell survival by activating transcription factor 3 (ATF3) in response to chemical ribosome-inactivating stress. Biochem. Pharmacol. 2009, 77, 1105–1115. [Google Scholar]
- Liang, G.; Wolfgang, C.D.; Chen, B.P.; Chen, T.H.; Hai, T. ATF3 gene. Genomic organization, promoter, and regulation. J. Biol. Chem. 1996, 271, 1695–1701. [Google Scholar] [PubMed]
- Jiang, H.Y.; Wek, S.A.; McGrath, B.C.; Lu, D.; Hai, T.; Harding, H.P.; Wang, X.; Ron, D.; Cavener, D.R.; Wek, R.C. Activating transcription factor 3 is integral to the eukaryotic initiation factor 2 kinase stress response. Mol. Cell Biol. 2004, 24, 1365–1377. [Google Scholar]
- Doller, A.; Akool el, S.; Huwiler, A.; Muller, R.; Radeke, H.H.; Pfeilschifter, J.; Eberhardt, W. Posttranslational modification of the AU-rich element binding protein HuR by protein kinase Cdelta elicits angiotensin II-induced stabilization and nuclear export of cyclooxygenase 2 mRNA. Mol. Cell Biol. 2008, 28, 2608–2625. [Google Scholar]
- Bush, K.T.; George, S.K.; Zhang, P.L.; Nigam, S.K. Pretreatment with inducers of ER molecular chaperones protects epithelial cells subjected to ATP depletion. Am. J. Physiol. 1999, 277, F211–F218. [Google Scholar]
- Pluquet, O.; Qu, L.K.; Baltzis, D.; Koromilas, A.E. Endoplasmic reticulum stress accelerates p53 degradation by the cooperative actions of Hdm2 and glycogen synthase kinase 3beta. Mol. Cell Biol. 2005, 25, 9392–9405. [Google Scholar]
- Reimertz, C.; Kogel, D.; Rami, A.; Chittenden, T.; Prehn, J.H. Gene expression during ER stress-induced apoptosis in neurons: Induction of the BH3-only protein Bbc3/PUMA and activation of the mitochondrial apoptosis pathway. J. Cell Biol. 2003, 162, 587–597. [Google Scholar]
- Zhang, F.; Hamanaka, R.B.; Bobrovnikova-Marjon, E.; Gordan, J.D.; Dai, M.S.; Lu, H.; Simon, M.C.; Diehl, J.A. Ribosomal stress couples the unfolded protein response to p53-dependent cell cycle arrest. J. Biol. Chem. 2006, 281, 30036–30045. [Google Scholar]
- Yang, H.; Chung, D.H.; Kim, Y.B.; Choi, Y.H.; Moon, Y. Ribotoxic mycotoxin deoxynivalenol induces G2/M cell cycle arrest via p21Cip/WAF1 mRNA stabilization in human epithelial cells. Toxicology 2008, 243, 145–154. [Google Scholar]
- el-Deiry, W.S.; Harper, J.W.; O’Connor, P.M.; Velculescu, V.E.; Canman, C.E.; Jackman, J.; Pietenpol, J.A.; Burrell, M.; Hill, D.E.; Wang, Y.; et al. WAF1/CIP1 is induced in p53-mediated G1 arrest and apoptosis. Cancer Res. 1994, 54, 1169–1174. [Google Scholar] [PubMed]
- el-Deiry, W.S.; Tokino, T.; Velculescu, V.E.; Levy, D.B.; Parsons, R.; Trent, J.M.; Lin, D.; Mercer, W.E.; Kinzler, K.W.; Vogelstein, B. WAF1, a potential mediator of p53 tumor suppression. Cell 1993, 75, 817–825. [Google Scholar]
- Jess, T.; Simonsen, J.; Nielsen, N.M.; Jorgensen, K.T.; Bager, P.; Ethelberg, S.; Frisch, M. Enteric Salmonella or Campylobacter infections and the risk of inflammatory bowel disease. Gut 2011, 60, 318–324. [Google Scholar] [PubMed]
- Subramanian, S.; Campbell, B.J.; Rhodes, J.M. Bacteria in the pathogenesis of inflammatory bowel disease. Curr. Opin. Infect. Dis. 2006, 19, 475–484. [Google Scholar]
- Johannes, L.; Romer, W. Shiga toxins-From cell biology to biomedical applications. Nat. Rev. Microbiol. 2010, 8, 105–116. [Google Scholar]
- Juranic, Z.; Stojiljkovic, M.P.; Bocarov-Stancic, A.; Kilibarda, V.; Milovanovic, S.R.; Juranic, I.; Bijelogrlic, S.; Vuletic, N.; Radulovic, S. T-2 toxin affects proliferation of three different neoplastic cell lines. J. Exp. Clin. Cancer Res. 1998, 17, 33–40. [Google Scholar]
- Zhou, X.X.; Ji, F.; Zhao, J.L.; Cheng, L.F.; Xu, C.F. Anti-cancer activity of anti-p185HER-2 ricin A chain immunotoxin on gastric cancer cells. J. Gastroenterol. Hepatol. 2010, 25, 1266–1275. [Google Scholar]
- Lin, J.Y.; Tserng, K.Y.; Chen, C.C.; Lin, L.T.; Tung, T.C. Abrin and ricin: New anti-tumour substances. Nature 1970, 227, 292–293. [Google Scholar]
- Iordanov, M.S.; Choi, R.J.; Ryabinina, O.P.; Dinh, T.H.; Bright, R.K.; Magun, B.E. The UV (Ribotoxic) stress response of human keratinocytes involves the unexpected uncoupling of the Ras-extracellular signal-regulated kinase signaling cascade from the activated epidermal growth factor receptor. Mol. Cell Biol. 2002, 22, 5380–5394. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Moon, Y. Mucosal Injuries due to Ribosome-Inactivating Stress and the Compensatory Responses of the Intestinal Epithelial Barrier. Toxins 2011, 3, 1263-1277. https://doi.org/10.3390/toxins3101263
Moon Y. Mucosal Injuries due to Ribosome-Inactivating Stress and the Compensatory Responses of the Intestinal Epithelial Barrier. Toxins. 2011; 3(10):1263-1277. https://doi.org/10.3390/toxins3101263
Chicago/Turabian StyleMoon, Yuseok. 2011. "Mucosal Injuries due to Ribosome-Inactivating Stress and the Compensatory Responses of the Intestinal Epithelial Barrier" Toxins 3, no. 10: 1263-1277. https://doi.org/10.3390/toxins3101263