Multi-Copper Oxidases and Human Iron Metabolism

Abstract
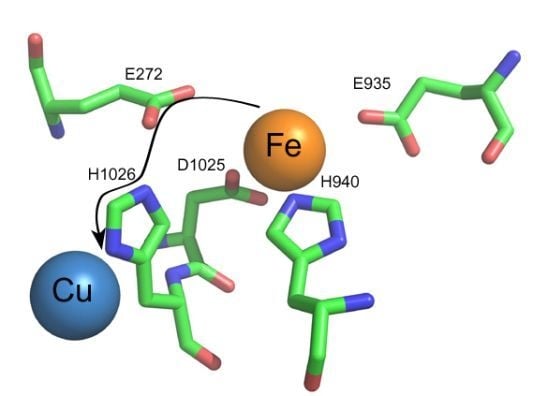
:
1. Introduction
1.1. Iron in Biology
1.2. Iron Metabolism in Humans
1.2.1. Iron Absorption in the Small Intestine
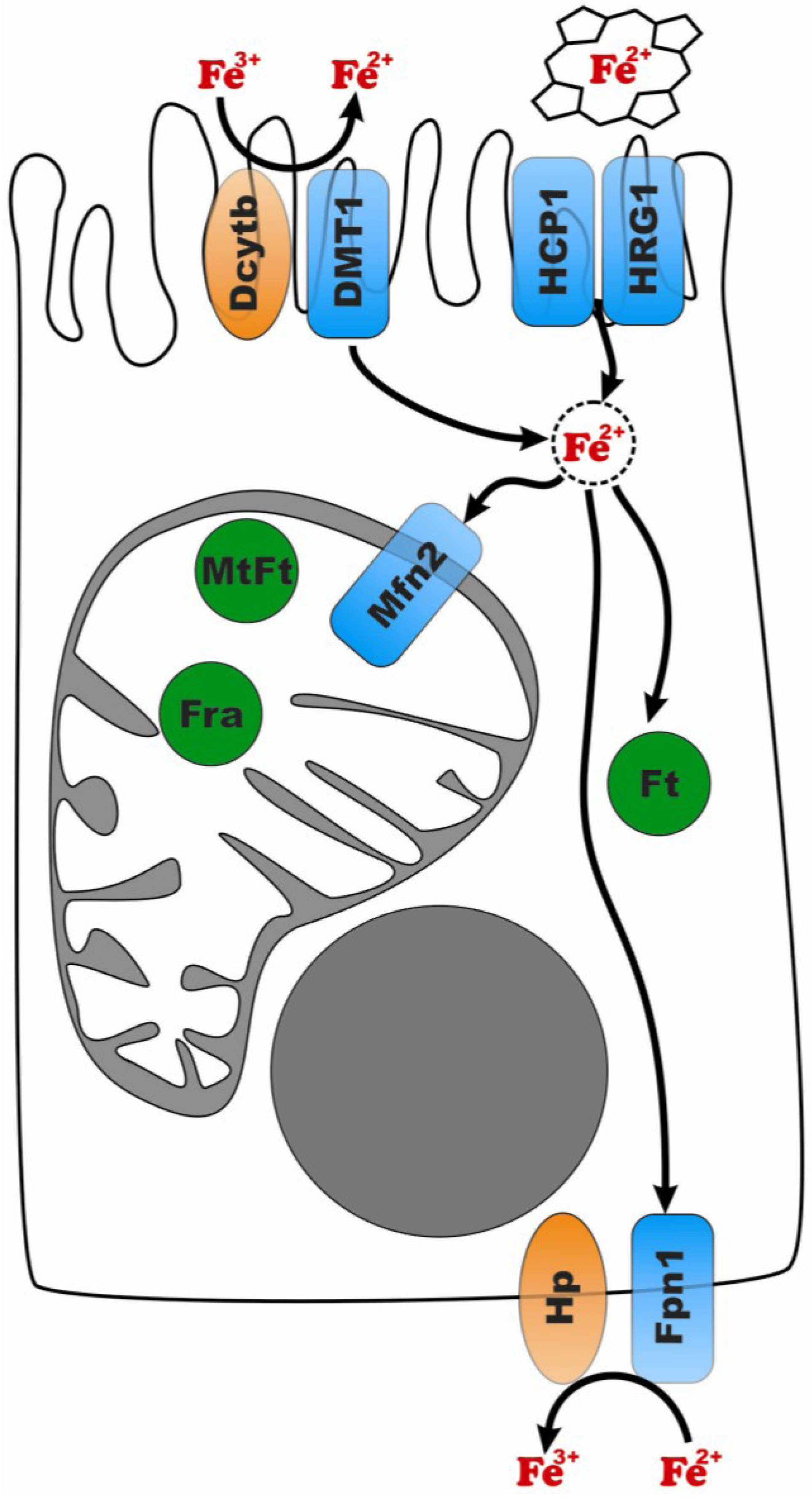
1.2.2. Iron Uptake in Different Cell Types
1.2.3. Regulation of Iron Homeostasis
1.2.4. Inherited Disorders of Human Iron Metabolism

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Function of the protein | Disorder | Phenotype | References |
|---|---|---|---|---|
| DMT1 | Ferrous iron transporter | Multiple missense mutations | Iron deficiency anaemia | [63,64,65] |
| H-ferritin | Iron storage | Mutation in 5′ UTR | Iron loading | [66] |
| L-ferritin | Iron storage | Neuroferritinopathia
Hyperferritinaemia | Brain iron overload
Cataract | [67,68,69] |
| Frataxin | Iron chaperone | Freidreich ataxia | Mitochondrial iron overloading | [70] |
| Ferroportin | Ferrous iron exporter | Hemochromatosis type 4 | Plasma hypoferraemia with tissue iron loading | [71] |
| Ceruloplasmin | Systemic iron oxidase | Aceruloplasminaemia | Plasma hypoferraemia with tissue iron loading | [72] |
| Transferrin | Plasma iron transport protein | Atransferrinaemia | Anaemia refractory to iron therapy | [73,74] |
| TfR2 | Uptake of transferrin
Regulator of iron homeostasis | Hemochromatosis type 3 | Iron loading | [75] |
| HFE | Regulator of iron homeostasis | Hemochromatosis type 1 | Iron loading | [76] |
| Hemojuvelin | Regulator of iron homeostasis | Juvenile hemochromatosis (type 2A) | Iron loading | [77] |
| Hepcidin | Regulator of iron homeostasis | Juvenile hemochromatosis (type 2B) | Iron loading | [77] |
1.3. Structure and Catalytic Mechanism of Multi-Copper Oxidases
1.3.1. Type 1 Copper Sites
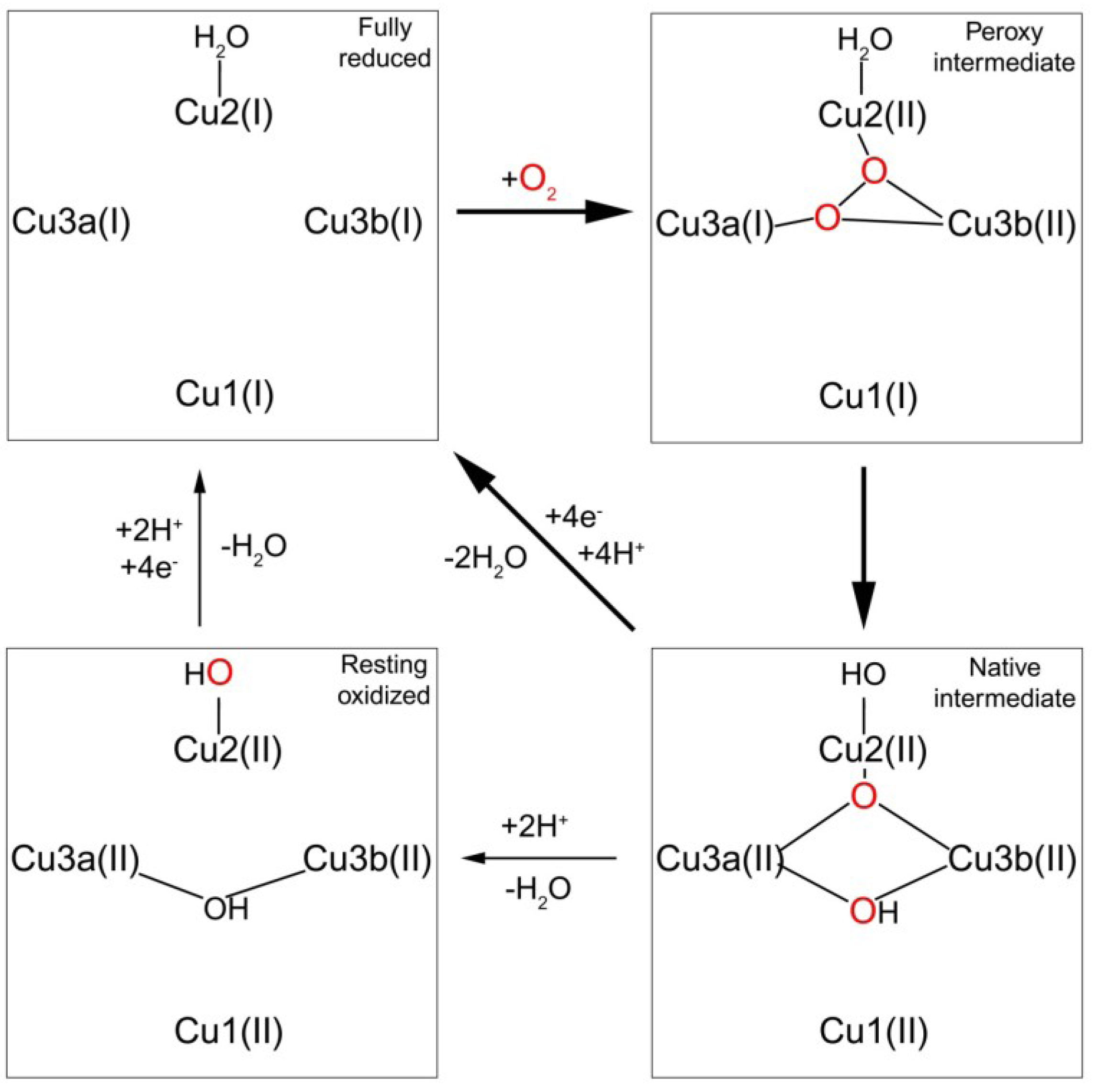
1.3.2. Transfer of Electrons to the Trinuclear Cluster and Dioxygen Reduction
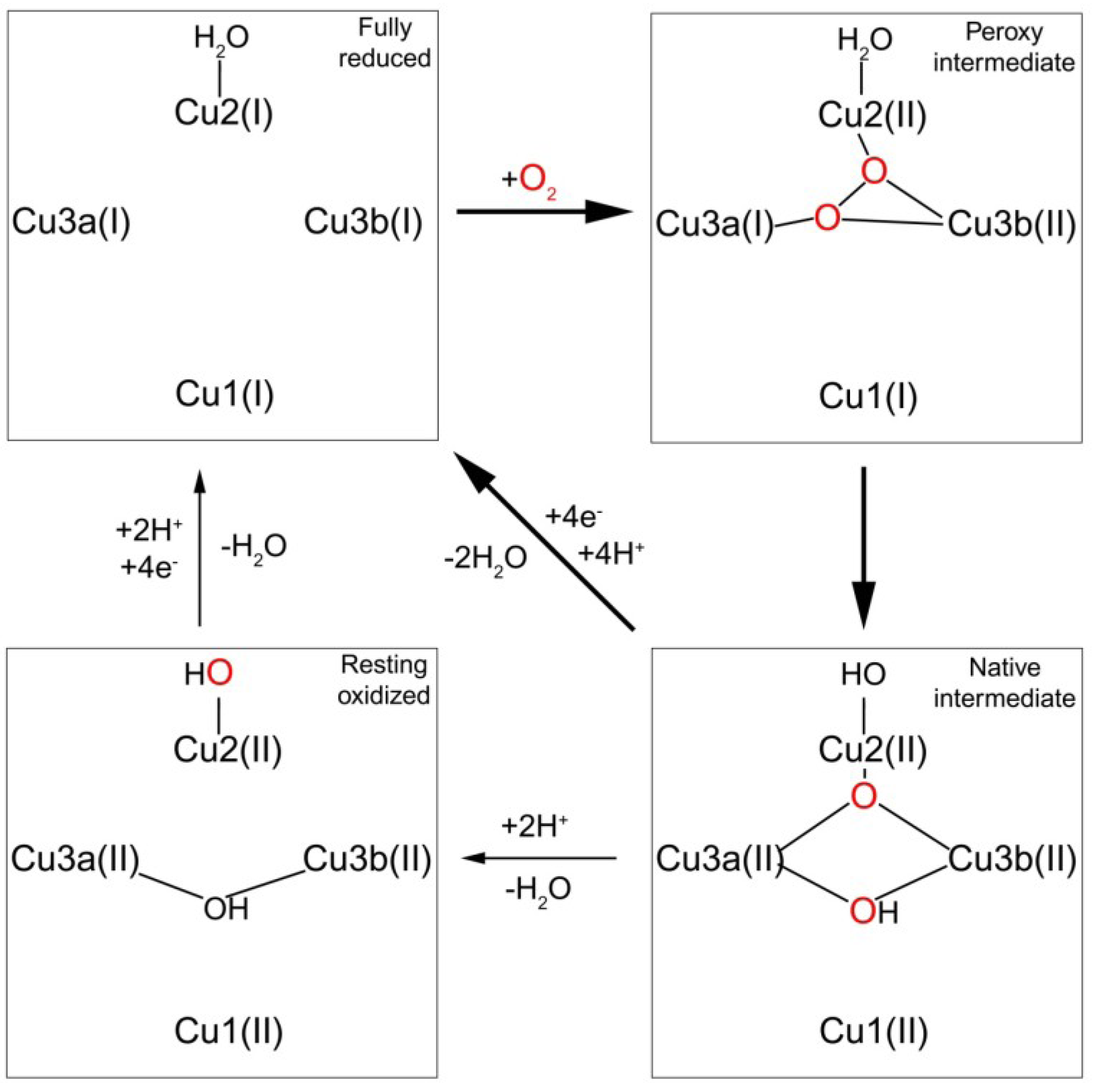
2. Human Multi-Copper Oxidases
2.1. Ceruloplasmin


2.2. Hephaestin
2.3. Zyklopen
2.4. The Interplay between Human Multi-Copper Ferroxidases
3. Conclusions
Acknowledgements
Conflict of Interest
References
- Crichton, R.R.; Pierre, J.L. Old iron, young copper: From Mars to Venus. Biometals 2001, 14, 99–112. [Google Scholar] [CrossRef]
- Ponka, P. Cell biology of heme. Am. J. Med. Sci. 1999, 318, 241–256. [Google Scholar] [CrossRef]
- Danielson, P.B. The cytochrome P450 superfamily: Biochemistry, evolution and drug metabolism in humans. Curr. Drug Metab. 2002, 3, 561–597. [Google Scholar] [CrossRef]
- Lill, R.; Muhlenhoff, U. Iron-sulfur protein biogenesis in eukaryotes: Components and mechanisms. Annu. Rev. Cell Dev. Biol. 2006, 22, 457–486. [Google Scholar] [CrossRef]
- Eklund, H.; Uhlin, U.; Farnegardh, M.; Logan, D.T.; Nordlund, P. Structure and function of the radical enzyme ribonucleotide reductase. Prog. Biophys. Mol. Biol. 2001, 77, 177–268. [Google Scholar] [CrossRef]
- Wink, D.A.; Hines, H.B.; Cheng, R.Y.; Switzer, C.H.; Flores-Santana, W.; Vitek, M.P.; Ridnour, L.A.; Colton, C.A. Nitric oxide and redox mechanisms in the immune response. J. Leukoc. Biol. 2011, 89, 873–891. [Google Scholar] [CrossRef]
- Pierre, J.L.; Fontecave, M. Iron and activated oxygen species in biology: The basic chemistry. Biometals 1999, 12, 195–199. [Google Scholar] [CrossRef]
- Taketani, S. Aquisition, mobilization and utilization of cellular iron and heme: Endless findings and growing evidence of tight regulation. Tohoku J. Exp. Med. 2005, 205, 297–318. [Google Scholar] [CrossRef]
- Micronutrient Deficiencies. Available online: http://www.who.int/nutrition/topics/ida/en/ (accessed 1 March 2013).
- Theil, E.C. Iron homeostasis and nutritional iron deficiency. J. Nutr. 2011, 141, 724S–728S. [Google Scholar] [CrossRef]
- Shayeghi, M.; Latunde-Dada, G.O.; Oakhill, J.S.; Laftah, A.H.; Takeuchi, K.; Halliday, N.; Khan, Y.; Warley, A.; McCann, F.E.; Hider, R.C.; et al. Identification of an intestinal heme transporter. Cell 2005, 122, 789–801. [Google Scholar] [CrossRef]
- Rajagopal, A.; Rao, A.U.; Amigo, J.; Tian, M.; Upadhyay, S.K.; Hall, C.; Uhm, S.; Mathew, M.K.; Fleming, M.D.; Paw, B.H.; et al. Haem homeostasis is regulated by the conserved and concerted functions of HRG-1 proteins. Nature 2008, 453, 1127–1131. [Google Scholar] [CrossRef]
- Latunde-Dada, G.O.; Takeuchi, K.; Simpson, R.J.; McKie, A.T. Haem carrier protein 1 (HCP1): Expression and functional studies in cultured cells. FEBS Lett. 2006, 580, 6865–6870. [Google Scholar] [CrossRef]
- Gunshin, H.; Mackenzie, B.; Berger, U.V.; Gunshin, Y.; Romero, M.F.; Boron, W.F.; Nussberger, S.; Gollan, J.L.; Hediger, M.A. Cloning and characterization of a mammalian proton-coupled metal-ion transporter. Nature 1997, 388, 482–488. [Google Scholar] [CrossRef]
- Gunshin, H.; Fujiwara, Y.; Custodio, A.O.; Direnzo, C.; Robine, S.; Andrews, N.C. Slc11a2 is required for intestinal iron absorption and erythropoiesis but dispensable in placenta and liver. J. Clin. Investig. 2005, 115, 1258–1266. [Google Scholar]
- McKie, A.T.; Barrow, D.; Latunde-Dada, G.O.; Rolfs, A.; Sager, G.; Mudaly, E.; Mudaly, M.; Richardson, C.; Barlow, D.; Bomford, A.; et al. An iron-regulated ferric reductase associated with the absorption of dietary iron. Science 2001, 291, 1755–1759. [Google Scholar] [CrossRef]
- McKie, A.T. The role of Dcytb in iron metabolism: An update. Biochem. Soc. Trans. 2008, 36, 1239–1241. [Google Scholar] [CrossRef]
- Wyman, S.; Simpson, R.J.; McKie, A.T.; Sharp, P.A. Dcytb (Cybrd1) functions as both a ferric and a cupric reductase in vitro. FEBS Lett. 2008, 582, 1901–1906. [Google Scholar] [CrossRef]
- Scheers, N. Regulatory effects of Cu, Zn, and Ca on Fe absorption: The intricate play between nutrient transporters. Nutrients 2013, 5, 957–970. [Google Scholar] [CrossRef]
- Gunshin, H.; Starr, C.N.; Direnzo, C.; Fleming, M.D.; Jin, J.; Greer, E.L.; Sellers, V.M.; Galica, S.M.; Andrews, N.C. Cybrd1 (duodenal cytochrome b) is not necessary for dietary iron absorption in mice. Blood 2005, 106, 2879–2883. [Google Scholar] [CrossRef]
- Chasteen, N.D.; Harrison, P.M. Mineralization in ferritin: An efficient means of iron storage. J. Struct. Biol. 1999, 126, 182–194. [Google Scholar] [CrossRef]
- Miller, L.L.; Miller, S.C.; Torti, S.V.; Tsuji, Y.; Torti, F.M. Iron-independent induction of ferritin H chain by tumor necrosis factor. Proc. Natl. Acad. Sci. USA 1991, 88, 4946–4950. [Google Scholar] [CrossRef]
- Leggett, B.A.; Fletcher, L.M.; Ramm, G.A.; Powell, L.W.; Halliday, J.W. Differential regulation of ferritin H and L subunit mRNA during inflammation and long-term iron overload. J. Gastroenterol. Hepatol. 1993, 8, 21–27. [Google Scholar] [CrossRef]
- Shaw, G.C.; Cope, J.J.; Li, L.; Corson, K.; Hersey, C.; Ackermann, G.E.; Gwynn, B.; Lambert, A.J.; Wingert, R.A.; Traver, D.; et al. Mitoferrin is essential for erythroid iron assimilation. Nature 2006, 440, 96–100. [Google Scholar] [CrossRef]
- Li, F.Y.; Nikali, K.; Gregan, J.; Leibiger, I.; Leibiger, B.; Schweyen, R.; Larsson, C.; Suomalainen, A. Characterization of a novel human putative mitochondrial transporter homologous to the yeast mitochondrial RNA splicing proteins 3 and 4. FEBS Lett. 2001, 494, 79–84. [Google Scholar] [CrossRef]
- Bencze, K.Z.; Kondapalli, K.C.; Cook, J.D.; McMahon, S.; Millan-Pacheco, C.; Pastor, N.; Stemmler, T.L. The structure and function of frataxin. Crit. Rev. Biochem. Mol. Biol. 2006, 41, 269–291. [Google Scholar] [CrossRef]
- Levi, S.; Arosio, P. Mitochondrial ferritin. Int. J. Biochem. Cell Biol. 2004, 36, 1887–1889. [Google Scholar] [CrossRef]
- Donovan, A.; Lima, C.A.; Pinkus, J.L.; Pinkus, G.S.; Zon, L.I.; Robine, S.; Andrews, N.C. The iron exporter ferroportin/Slc40a1 is essential for iron homeostasis. Cell Metab. 2005, 1, 191–200. [Google Scholar] [CrossRef]
- Jeong, S.Y.; David, S. Glycosylphosphatidylinositol-anchored ceruloplasmin is required for iron efflux from cells in the central nervous system. J. Biol. Chem. 2003, 278, 27144–27148. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; di Patti, M.C.; Jeong, S.Y.; David, S.; Musci, G.; Kaplan, J. Ferroxidase activity is required for the stability of cell surface ferroportin in cells expressing GPI-ceruloplasmin. EMBO J. 2007, 26, 2823–2831. [Google Scholar] [CrossRef]
- Kono, S.; Yoshida, K.; Tomosugi, N.; Terada, T.; Hamaya, Y.; Kanaoka, S.; Miyajima, H. Biological effects of mutant ceruloplasmin on hepcidin-mediated internalization of ferroportin. Biochim. Biophys. Acta 2010, 1802, 968–975. [Google Scholar] [CrossRef]
- Han, O.; Kim, E.Y. Colocalization of ferroportin-1 with hephaestin on the basolateral membrane of human intestinal absorptive cells. J. Cell. Biochem. 2007, 101, 1000–1010. [Google Scholar] [CrossRef]
- Yeh, K.Y.; Yeh, M.; Mims, L.; Glass, J. Iron feeding induces ferroportin 1 and hephaestin migration and interaction in rat duodenal epithelium. Am. J. Physiol. Gastrointest. Liver Physiol. 2009, 296, G55–G65. [Google Scholar]
- Aisen, P.; Leibman, A.; Zweier, J. Stoichiometric and site characteristics of the binding of iron to human transferrin. J. Biol. Chem. 1978, 253, 1930–1937. [Google Scholar]
- Ha-Duong, N.T.; Eid, C.; Hemadi, M.; El Hage Chahine, J.M. In vitro interaction between ceruloplasmin and human serum transferrin. Biochemistry 2010, 49, 10261–10263. [Google Scholar] [CrossRef]
- Zakharova, E.T.; Shavlovski, M.M.; Bass, M.G.; Gridasova, A.A.; Pulina, M.O.; de Filippis, V.; Beltramini, M.; di Muro, P.; Salvato, B.; Fontana, A.; et al. Interaction of lactoferrin with ceruloplasmin. Arch. Biochem. Biophys. 2000, 374, 222–228. [Google Scholar] [CrossRef]
- Hudson, D.M.; Krisinger, M.J.; Griffiths, T.A.; MacGillivray, R.T. Neither human hephaestin nor ceruloplasmin forms a stable complex with transferrin. J. Cell. Biochem. 2008, 103, 1849–1855. [Google Scholar] [CrossRef]
- Dautry-Varsat, A.; Ciechanover, A.; Lodish, H.F. pH and the recycling of transferrin during receptor-mediated endocytosis. Proc. Natl. Acad. Sci. USA 1983, 80, 2258–2262. [Google Scholar] [CrossRef]
- Kawabata, H.; Germain, R.S.; Vuong, P.T.; Nakamaki, T.; Said, J.W.; Koeffler, H.P. Transferrin receptor 2-alpha supports cell growth both in iron-chelated cultured cells and in vivo. J. Biol. Chem. 2000, 275, 16618–16625. [Google Scholar]
- Trinder, D.; Baker, E. Transferrin receptor 2: A new molecule in iron metabolism. Int. J. Biochem. Cell Biol. 2003, 35, 292–296. [Google Scholar] [CrossRef]
- Ohgami, R.S.; Campagna, D.R.; Greer, E.L.; Antiochos, B.; McDonald, A.; Chen, J.; Sharp, J.J.; Fujiwara, Y.; Barker, J.E.; Fleming, M.D. Identification of a ferrireductase required for efficient transferrin-dependent iron uptake in erythroid cells. Nat. Genet. 2005, 37, 1264–1269. [Google Scholar] [CrossRef]
- Knutson, M.D. Steap proteins: Implications for iron and copper metabolism. Nutr. Rev. 2007, 65, 335–340. [Google Scholar] [CrossRef]
- Liuzzi, J.P.; Aydemir, F.; Nam, H.; Knutson, M.D.; Cousins, R.J. Zip14 (Slc39a14) mediates non-transferrin-bound iron uptake into cells. Proc. Natl. Acad. Sci. USA 2006, 103, 13612–13617. [Google Scholar] [CrossRef]
- Christensen, E.I.; Birn, H. Megalin and cubilin: Multifunctional endocytic receptors. Nat. Rev. Mol. Cell Biol. 2002, 3, 256–266. [Google Scholar]
- Hvidberg, V.; Jacobsen, C.; Strong, R.K.; Cowland, J.B.; Moestrup, S.K.; Borregaard, N. The endocytic receptor megalin binds the iron transporting neutrophil-gelatinase-associated lipocalin with high affinity and mediates its cellular uptake. FEBS Lett. 2005, 579, 773–777. [Google Scholar] [CrossRef]
- Smith, K.D. Iron metabolism at the host pathogen interface: Lipocalin 2 and the pathogen-associated iroA gene cluster. Int. J. Biochem. Cell Biol. 2007, 39, 1776–1780. [Google Scholar] [CrossRef]
- Devireddy, L.R.; Gazin, C.; Zhu, X.; Green, M.R. A cell-surface receptor for lipocalin 24p3 selectively mediates apoptosis and iron uptake. Cell 2005, 123, 1293–1305. [Google Scholar] [CrossRef]
- Knutson, M.; Wessling-Resnick, M. Iron metabolism in the reticuloendothelial system. Crit. Rev. Biochem. Mol. Biol. 2003, 38, 61–88. [Google Scholar] [CrossRef]
- Nielsen, M.J.; Moller, H.J.; Moestrup, S.K. Hemoglobin and heme scavenger receptors. Antioxid. Redox. Signal. 2010, 12, 261–273. [Google Scholar] [CrossRef]
- Troadec, M.B.; Ward, D.M.; Kaplan, J. A Tf-independent iron transport system required for organogenesis. Dev. Cell 2009, 16, 3–4. [Google Scholar] [CrossRef]
- Chen, T.T.; Li, L.; Chung, D.H.; Allen, C.D.; Torti, S.V.; Torti, F.M.; Cyster, J.G.; Chen, C.Y.; Brodsky, F.M.; Niemi, E.C.; et al. TIM-2 is expressed on B cells and in liver and kidney and is a receptor for H-ferritin endocytosis. J. Exp. Med. 2005, 202, 955–965. [Google Scholar] [CrossRef]
- Li, L.; Fang, C.J.; Ryan, J.C.; Niemi, E.C.; Lebron, J.A.; Bjorkman, P.J.; Arase, H.; Torti, F.M.; Torti, S.V.; Nakamura, M.C.; et al. Binding and uptake of H-ferritin are mediated by human transferrin receptor-1. Proc. Natl. Acad. Sci. USA 2010, 107, 3505–3510. [Google Scholar] [CrossRef]
- Mukhopadhyay, C.K.; Attieh, Z.K.; Fox, P.L. Role of ceruloplasmin in cellular iron uptake. Science 1998, 279, 714–717. [Google Scholar] [CrossRef]
- Hentze, M.W.; Muckenthaler, M.U.; Andrews, N.C. Balancing acts: Molecular control of mammalian iron metabolism. Cell 2004, 117, 285–297. [Google Scholar] [CrossRef]
- Eisenstein, R.S.; Ross, K.L. Novel roles for iron regulatory proteins in the adaptive response to iron deficiency. J. Nutr. 2003, 133, 1510S–1516S. [Google Scholar]
- McKie, A.T.; Marciani, P.; Rolfs, A.; Brennan, K.; Wehr, K.; Barrow, D.; Miret, S.; Bomford, A.; Peters, T.J.; Farzaneh, F.; et al. A novel duodenal iron-regulated transporter, IREG1, implicated in the basolateral transfer of iron to the circulation. Mol. Cell 2000, 5, 299–309. [Google Scholar] [CrossRef]
- Zhang, D.L.; Hughes, R.M.; Ollivierre-Wilson, H.; Ghosh, M.C.; Rouault, T.A. A ferroportin transcript that lacks an iron-responsive element enables duodenal and erythroid precursor cells to evade translational repression. Cell Metab. 2009, 9, 461–473. [Google Scholar] [CrossRef]
- Lee, P.L.; Gelbart, T.; West, C.; Halloran, C.; Beutler, E. The human Nramp2 gene: Characterization of the gene structure, alternative splicing, promoter region and polymorphisms. Blood Cells Mol. Dis. 1998, 24, 199–215. [Google Scholar] [CrossRef]
- Meyron-Holtz, E.G.; Ghosh, M.C.; Iwai, K.; LaVaute, T.; Brazzolotto, X.; Berger, U.V.; Land, W.; Ollivierre-Wilson, H.; Grinberg, A.; Love, P.; et al. Genetic ablations of iron regulatory proteins 1 and 2 reveal why iron regulatory protein 2 dominates iron homeostasis. EMBO J. 2004, 23, 386–395. [Google Scholar] [CrossRef]
- Smith, S.R.; Ghosh, M.C.; Ollivierre-Wilson, H.; Tong, W.H.; Rouault, T.A. Complete loss of iron regulatory proteins 1 and 2 prevents viability of murine zygotes beyond the blastocyst stage of embryonic development. Blood Cells Mol. Dis. 2006, 36, 283–287. [Google Scholar] [CrossRef]
- Fleming, R.E.; Sly, W.S. Hepcidin: A putative iron-regulatory hormone relevant to hereditary hemochromatosis and the anemia of chronic disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8160–8162. [Google Scholar] [CrossRef]
- Nemeth, E.; Tuttle, M.S.; Powelson, J.; Vaughn, M.B.; Donovan, A.; Ward, D.M.; Ganz, T.; Kaplan, J. Hepcidin regulates cellular iron efflux by binding to ferroportin and inducing its internalization. Science 2004, 306, 2090–2093. [Google Scholar] [CrossRef]
- Bardou-Jacquet, E.; Island, M.L.; Jouanolle, A.M.; Detivaud, L.; Fatih, N.; Ropert, M.; Brissot, E.; Mosser, A.; Maisonneuve, H.; Brissot, P.; et al. A novel N491S mutation in the human SLC11A2 gene impairs protein trafficking and in association with the G212V mutation leads to microcytic anemia and liver iron overload. Blood Cells Mol. Dis. 2011, 47, 243–248. [Google Scholar] [CrossRef]
- Beaumont, C.; Delaunay, J.; Hetet, G.; Grandchamp, B.; de Montalembert, M.; Tchernia, G. Two new human DMT1 gene mutations in a patient with microcytic anemia, low ferritinemia, and liver iron overload. Blood 2006, 107, 4168–4170. [Google Scholar] [CrossRef]
- Blanco, E.; Kannengiesser, C.; Grandchamp, B.; Tasso, M.; Beaumont, C. Not all DMT1 mutations lead to iron overload. Blood Cells Mol. Dis. 2009, 43, 199–201. [Google Scholar] [CrossRef]
- Kato, J.; Fujikawa, K.; Kanda, M.; Fukuda, N.; Sasaki, K.; Takayama, T.; Kobune, M.; Takada, K.; Takimoto, R.; Hamada, H.; et al. A mutation, in the iron-responsive element of H ferritin mRNA, causing autosomal dominant iron overload. Am. J. Hum. Genet. 2001, 69, 191–197. [Google Scholar] [CrossRef]
- Curtis, A.R.; Fey, C.; Morris, C.M.; Bindoff, L.A.; Ince, P.G.; Chinnery, P.F.; Coulthard, A.; Jackson, M.J.; Jackson, A.P.; McHale, D.P.; et al. Mutation in the gene encoding ferritin light polypeptide causes dominant adult-onset basal ganglia disease. Nat. Genet. 2001, 28, 350–354. [Google Scholar] [CrossRef]
- Levi, S.; Cozzi, A.; Arosio, P. Neuroferritinopathy: A neurodegenerative disorder associated with l-ferritin mutation. Best Pract. Res. Clin. Haematol. 2005, 18, 265–276. [Google Scholar] [CrossRef]
- Cazzola, M.; Bergamaschi, G.; Tonon, L.; Arbustini, E.; Grasso, M.; Vercesi, E.; Barosi, G.; Bianchi, P.E.; Cairo, G.; Arosio, P. Hereditary hyperferritinemia-cataract syndrome: Relationship between phenotypes and specific mutations in the iron-responsive element of ferritin light-chain mRNA. Blood 1997, 90, 814–821. [Google Scholar]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef]
- De Domenico, I.; Ward, D.M.; Nemeth, E.; Vaughn, M.B.; Musci, G.; Ganz, T.; Kaplan, J. The molecular basis of ferroportin-linked hemochromatosis. Proc. Natl. Acad. Sci. USA 2005, 102, 8955–8960. [Google Scholar]
- Xu, X.; Pin, S.; Gathinji, M.; Fuchs, R.; Harris, Z.L. Aceruloplasminemia: An inherited neurodegenerative disease with impairment of iron homeostasis. Ann. N. Y. Acad. Sci. 2004, 1012, 299–305. [Google Scholar] [CrossRef]
- Hamill, R.L.; Woods, J.C.; Cook, B.A. Congenital atransferrinemia. A case report and review of the literature. Am. J. Clin. Pathol. 1991, 96, 215–218. [Google Scholar]
- Shamsian, B.S.; Rezaei, N.; Arzanian, M.T.; Alavi, S.; Khojasteh, O.; Eghbali, A. Severe hypochromic microcytic anemia in a patient with congenital atransferrinemia. Pediatr. Hematol. Oncol. 2009, 26, 356–362. [Google Scholar] [CrossRef]
- Chen, J.; Enns, C.A. Hereditary hemochromatosis and transferrin receptor 2. Biochim. Biophys. Acta 2011, 1820, 256–263. [Google Scholar] [CrossRef]
- Rochette, J.; le Gac, G.; Lassoued, K.; Ferec, C.; Robson, K.J. Factors influencing disease phenotype and penetrance in HFE haemochromatosis. Hum. Genet. 2010, 128, 233–248. [Google Scholar] [CrossRef]
- Goldberg, Y.P. Juvenile Hereditary Hemochromatosis. In GeneReviews™ [Internet]; Pagon, R.A., Bird, T.D., Dolan, C.R., Stephens, K., Adam, M.P., Eds.; University of Washington: Seattle, WA, USA, 2011. [Google Scholar]
- Sakurai, T.; Kataoka, K. Basic and applied features of multicopper oxidases, CueO, bilirubin oxidase, and laccase. Chem. Rec. 2007, 7, 220–229. [Google Scholar] [CrossRef]
- Kosman, D.J. Multicopper oxidases: A workshop on copper coordination chemistry, electron transfer, and metallophysiology. J. Biol. Inorg. Chem. 2010, 15, 15–28. [Google Scholar] [CrossRef]
- Nakamura, K.; Kawabata, T.; Yura, K.; Go, N. Novel types of two-domain multi-copper oxidases: Possible missing links in the evolution. FEBS Lett. 2003, 553, 239–244. [Google Scholar] [CrossRef]
- Skalova, T.; Dohnalek, J.; Ostergaard, L.H.; Ostergaard, P.R.; Kolenko, P.; Duskova, J.; Stepankova, A.; Hasek, J. The structure of the small laccase from Streptomyces coelicolor reveals a link between laccases and nitrite reductases. J. Mol. Biol. 2009, 385, 1165–1178. [Google Scholar] [CrossRef]
- Nakamura, K.; Go, N. Function and molecular evolution of multicopper blue proteins. Cell. Mol. Life Sci. 2005, 62, 2050–2066. [Google Scholar] [CrossRef]
- Giardina, P.; Faraco, V.; Pezzella, C.; Piscitelli, A.; Vanhulle, S.; Sannia, G. Laccases: A never-ending story. Cell. Mol. Life Sci. 2009, 67, 369–385. [Google Scholar]
- Hirose, J.; Sakurai, T.; Imamura, K.; Watanabe, H.; Iwamoto, H.; Hiromi, K.; Itoh, H.; Shin, T.; Murao, S. Characterization of ascorbate oxidase from Acremonium sp. HI-25. J. Biochem. 1994, 115, 811–813. [Google Scholar]
- Terzulli, A.; Kosman, D.J. Analysis of the high-affinity iron uptake system at the Chlamydomonas reinhardtii plasma membrane. Eukaryot. Cell 2010, 9, 815–826. [Google Scholar] [CrossRef]
- Roberts, S.A.; Weichsel, A.; Grass, G.; Thakali, K.; Hazzard, J.T.; Tollin, G.; Rensing, C.; Montfort, W.R. Crystal structure and electron transfer kinetics of CueO, a multicopper oxidase required for copper homeostasis in Escherichia coli. Proc. Natl. Acad. Sci. USA 2002, 99, 2766–2771. [Google Scholar] [CrossRef]
- Dick, G.J.; Torpey, J.W.; Beveridge, T.J.; Tebo, B.M. Direct identification of a bacterial manganese(II) oxidase, the multicopper oxidase MnxG, from spores of several different marine Bacillus species. Appl. Environ. Microbiol. 2008, 74, 1527–1534. [Google Scholar] [CrossRef]
- Palmer, A.E.; Szilagyi, R.K.; Cherry, J.R.; Jones, A.; Xu, F.; Solomon, E.I. Spectroscopic characterization of the Leu513His variant of fungal laccase: Effect of increased axial ligand interaction on the geometric and electronic structure of the type 1 Cu site. Inorg. Chem. 2003, 42, 4006–4017. [Google Scholar] [CrossRef]
- Xu, F.; Palmer, A.E.; Yaver, D.S.; Berka, R.M.; Gambetta, G.A.; Brown, S.H.; Solomon, E.I. Targeted mutations in a Trametes villosa laccase. Axial perturbations of the T1 copper. J. Biol. Chem. 1999, 274, 12372–12375. [Google Scholar] [CrossRef]
- Solomon, E.I.; Szilagyi, R.K.; DeBeer George, S.; Basumallick, L. Electronic structures of metal sites in proteins and models: Contributions to function in blue copper proteins. Chem. Rev. 2004, 104, 419–458. [Google Scholar] [CrossRef]
- Quintanar, L.; Stoj, C.; Taylor, A.B.; Hart, P.J.; Kosman, D.J.; Solomon, E.I. Shall we dance? How a multicopper oxidase chooses its electron transfer partner. Acc. Chem. Res. 2007, 40, 445–452. [Google Scholar] [CrossRef]
- Solomon, E.I. Spectroscopic methods in bioinorganic chemistry: Blue to green to red copper sites. Inorg. Chem. 2006, 45, 8012–8025. [Google Scholar] [CrossRef]
- Sakurai, T.; Kataoka, K. Structure and function of type I copper in multicopper oxidases. Cell. Mol. Life Sci. 2007, 64, 2642–2656. [Google Scholar] [CrossRef]
- Quintanar, L.; Stoj, C.; Wang, T.P.; Kosman, D.J.; Solomon, E.I. Role of aspartate 94 in the decay of the peroxide intermediate in the multicopper oxidase Fet3p. Biochemistry 2005, 44, 6081–6091. [Google Scholar] [CrossRef]
- Yoon, J.; Solomon, E.I. Electronic structure of the peroxy intermediate and its correlation to the native intermediate in the multicopper oxidases: Insights into the reductive cleavage of the O–O bond. J. Am. Chem. Soc. 2007, 129, 13127–13136. [Google Scholar] [CrossRef]
- Yoon, J.; Liboiron, B.D.; Sarangi, R.; Hodgson, K.O.; Hedman, B.; Solomon, E.I. The two oxidized forms of the trinuclear Cu cluster in the multicopper oxidases and mechanism for the decay of the native intermediate. Proc. Natl. Acad. Sci. USA 2007, 104, 13609–13614. [Google Scholar]
- Solomon, E.I.; Augustine, A.J.; Yoon, J. O2 reduction to H2O by the multicopper oxidases. Dalton Trans. 2008, 3921–3932. [Google Scholar] [CrossRef]
- Holmberg, C.G.; Laurell, C.B. Investigations in serum copper. II. Isolation of copper containing protein, and a description of some of its properties. Acta Chem. Scand. 1948, 2, 550–556. [Google Scholar] [CrossRef]
- Osaki, S.; Johnson, D.A.; Frieden, E. The possible significance of the ferrous oxidase activity of ceruloplasmin in normal human serum. J. Biol. Chem. 1966, 241, 2746–2751. [Google Scholar]
- Healy, J.; Tipton, K. Ceruloplasmin and what it might do. J. Neural Transm. 2007, 114, 777–781. [Google Scholar] [CrossRef]
- Patel, B.N.; David, S. A novel glycosylphosphatidylinositol-anchored form of ceruloplasmin is expressed by mammalian astrocytes. J. Biol. Chem. 1997, 272, 20185–20190. [Google Scholar] [CrossRef]
- Hahn, P.; Qian, Y.; Dentchev, T.; Chen, L.; Beard, J.; Harris, Z.L.; Dunaief, J.L. Disruption of ceruloplasmin and hephaestin in mice causes retinal iron overload and retinal degeneration with features of age-related macular degeneration. Proc. Natl. Acad. Sci. USA 2004, 101, 13850–13855. [Google Scholar] [CrossRef]
- Fortna, R.R.; Watson, H.A.; Nyquist, S.E. Glycosyl phosphatidylinositol-anchored ceruloplasmin is expressed by rat Sertoli cells and is concentrated in detergent-insoluble membrane fractions. Biol. Reprod. 1999, 61, 1042–1049. [Google Scholar] [CrossRef]
- Vachette, P.; Dainese, E.; Vasyliev, V.B.; di Muro, P.; Beltramini, M.; Svergun, D.I.; de Filippis, V.; Salvato, B. A key structural role for active site type 3 copper ions in human ceruloplasmin. J. Biol. Chem. 2002, 277, 40823–40831. [Google Scholar] [CrossRef]
- Lindley, P.F.; Card, G.; Zaitseva, I.; Zaitsev, V.; Reinhammar, B.; Selin-Lindgren, E.; Yoshida, K. An X-ray structural study of human ceruloplasmin in relation to ferroxidase activity. J. Biol. Inorg. Chem. 1997, 2, 454–463. [Google Scholar] [CrossRef]
- Quintanar, L.; Gebhard, M.; Wang, T.P.; Kosman, D.J.; Solomon, E.I. Ferrous binding to the multicopper oxidases Saccharomyces cerevisiae Fet3p and human ceruloplasmin: Contributions to ferroxidase activity. J. Am. Chem. Soc. 2004, 126, 6579–6589. [Google Scholar] [CrossRef]
- Machonkin, T.E.; Zhang, H.H.; Hedman, B.; Hodgson, K.O.; Solomon, E.I. Spectroscopic and magnetic studies of human ceruloplasmin: Identification of a redox-inactive reduced type 1 copper site. Biochemistry 1998, 37, 9570–9578. [Google Scholar] [CrossRef]
- Brown, M.A.; Stenberg, L.M.; Mauk, A.G. Identification of catalytically important amino acids in human ceruloplasmin by site-directed mutagenesis. FEBS Lett. 2002, 520, 8–12. [Google Scholar] [CrossRef]
- Inoue, K.; Akaike, T.; Miyamoto, Y.; Okamoto, T.; Sawa, T.; Otagiri, M.; Suzuki, S.; Yoshimura, T.; Maeda, H. Nitrosothiol formation catalyzed by ceruloplasmin. Implication for cytoprotective mechanism in vivo. J. Biol. Chem. 1999, 274, 27069–27075. [Google Scholar] [CrossRef]
- Cha, M.K.; Kim, I.H. Ceruloplasmin has a distinct active site for the catalyzing glutathione-dependent reduction of alkyl hydroperoxide. Biochemistry 1999, 38, 12104–12110. [Google Scholar] [CrossRef]
- Stoj, C.; Kosman, D.J. Cuprous oxidase activity of yeast Fet3p and human ceruloplasmin: Implication for function. FEBS Lett. 2003, 554, 422–426. [Google Scholar] [CrossRef]
- Mukhopadhyay, C.K.; Mazumder, B.; Lindley, P.F.; Fox, P.L. Identification of the prooxidant site of human ceruloplasmin: A model for oxidative damage by copper bound to protein surfaces. Proc. Natl. Acad. Sci. USA 1997, 94, 11546–11551. [Google Scholar] [CrossRef]
- Young, S.N.; Curzon, G. A method for obtaining linear reciprocal plots with caeruloplasmin and its application in a study of the kinetic parameters of caeruloplasmin substrates. Biochem. J. 1972, 129, 273–283. [Google Scholar]
- McDermott, J.A.; Huber, C.T.; Osaki, S.; Frieden, E. Role of iron in the oxidase activity of ceruloplasmin. Biochim. Biophys. Acta. 1968, 151, 541–557. [Google Scholar] [CrossRef]
- Zaitsev, V.N.; Zaitseva, I.; Papiz, M.; Lindley, P.F. An X-ray crystallographic study of the binding sites of the azide inhibitor and organic substrates to ceruloplasmin, a multi-copper oxidase in the plasma. J. Biol. Inorg. Chem. 1999, 4, 579–587. [Google Scholar] [CrossRef]
- Harris, Z.L.; Takahashi, Y.; Miyajima, H.; Serizawa, M.; MacGillivray, R.T.; Gitlin, J.D. Aceruloplasminemia: Molecular characterization of this disorder of iron metabolism. Proc. Natl. Acad. Sci. USA 1995, 92, 2539–2543. [Google Scholar]
- Yazaki, M.; Yoshida, K.; Nakamura, A.; Furihata, K.; Yonekawa, M.; Okabe, T.; Yamashita, N.; Ohta, M.; Ikeda, S. A novel splicing mutation in the ceruloplasmin gene responsible for hereditary ceruloplasmin deficiency with hemosiderosis. J. Neurol. Sci. 1998, 156, 30–34. [Google Scholar] [CrossRef]
- Kono, S.; Suzuki, H.; Oda, T.; Shirakawa, K.; Takahashi, Y.; Kitagawa, M.; Miyajima, H. Cys-881 is essential for the trafficking and secretion of truncated mutant ceruloplasmin in aceruloplasminemia. J. Hepatol. 2007, 47, 844–850. [Google Scholar] [CrossRef]
- Hellman, N.E.; Kono, S.; Miyajima, H.; Gitlin, J.D. Biochemical analysis of a missense mutation in aceruloplasminemia. J. Biol. Chem. 2002, 277, 1375–1380. [Google Scholar]
- Kono, S.; Suzuki, H.; Takahashi, K.; Takahashi, Y.; Shirakawa, K.; Murakawa, Y.; Yamaguchi, S.; Miyajima, H. Hepatic iron overload associated with a decreased serum ceruloplasmin level in a novel clinical type of aceruloplasminemia. Gastroenterology 2006, 131, 240–245. [Google Scholar] [CrossRef]
- Patel, B.N.; Dunn, R.J.; Jeong, S.Y.; Zhu, Q.; Julien, J.P.; David, S. Ceruloplasmin regulates iron levels in the CNS and prevents free radical injury. J. Neurosci. 2002, 22, 6578–6586. [Google Scholar]
- Vulpe, C.D.; Kuo, Y.M.; Murphy, T.L.; Cowley, L.; Askwith, C.; Libina, N.; Gitschier, J.; Anderson, G.J. Hephaestin, a ceruloplasmin homologue implicated in intestinal iron transport, is defective in the sla mouse. Nat. Genet. 1999, 21, 195–199. [Google Scholar] [CrossRef]
- Edwards, J.A.; Bannerman, R.M. Hereditary defect of intestinal iron transport in mice with sex-linked anemia. J. Clin. Investig. 1970, 49, 1869–1871. [Google Scholar] [CrossRef]
- Syed, B.A.; Beaumont, N.J.; Patel, A.; Naylor, C.E.; Bayele, H.K.; Joannou, C.L.; Rowe, P.S.; Evans, R.W.; Srai, S.K. Analysis of the human hephaestin gene and protein: Comparative modelling of the N-terminus ecto-domain based upon ceruloplasmin. Protein Eng. 2002, 15, 205–214. [Google Scholar] [CrossRef]
- Frazer, D.M.; Vulpe, C.D.; McKie, A.T.; Wilkins, S.J.; Trinder, D.; Cleghorn, G.J.; Anderson, G.J. Cloning and gastrointestinal expression of rat hephaestin: Relationship to other iron transport proteins. Am. J. Physiol. Gastrointest. Liver Physiol. 2001, 281, G931–G939. [Google Scholar]
- Hudson, D.M.; Curtis, S.B.; Smith, V.C.; Griffiths, T.A.; Wong, A.Y.; Scudamore, C.H.; Buchan, A.M.; MacGillivray, R.T. Human hephaestin expression is not limited to enterocytes of the gastrointestinal tract but is also found in the antrum, the enteric nervous system, and pancreatic β-cells. Am. J. Physiol. Gastrointest. Liver Physiol. 2010, 298, G425–G432. [Google Scholar] [CrossRef]
- Qian, Z.M.; Chang, Y.Z.; Leung, G.; Du, J.R.; Zhu, L.; Wang, Q.; Niu, L.; Xu, Y.J.; Yang, L.; Ho, K.P.; et al. Expression of ferroportin1, hephaestin and ceruloplasmin in rat heart. Biochim. Biophys. Acta 2007, 1772, 527–532. [Google Scholar] [CrossRef]
- Qian, Z.M.; Chang, Y.Z.; Zhu, L.; Yang, L.; Du, J.R.; Ho, K.P.; Wang, Q.; Li, L.Z.; Wang, C.Y.; Ge, X.; et al. Development and iron-dependent expression of hephaestin in different brain regions of rats. J. Cell. Biochem. 2007, 102, 1225–1233. [Google Scholar] [CrossRef]
- Kingston, P.J.; Bannerman, C.E.; Bannerman, R.M. Iron deficiency anaemia in newborn sla mice: A genetic defect of placental iron transport. Br. J. Haematol. 1978, 40, 265–276. [Google Scholar] [CrossRef]
- Vashchenko, G.; Bleackley, M.R.; Griffiths, T.A.; MacGillivray, R.T. Oxidation of organic and biogenic amines by recombinant human hephaestin expressed in Pichia pastoris. Arch. Biochem. Biophys. 2011, 514, 50–56. [Google Scholar] [CrossRef]
- Vashchenko, G.; Macgillivray, R.T. Functional role of the putative iron ligands in the ferroxidase activity of recombinant human hephaestin. J. Biol. Inorg. Chem. 2012, 17, 1187–1195. [Google Scholar] [CrossRef]
- Chen, H.; Attieh, Z.K.; Syed, B.A.; Kuo, Y.M.; Stevens, V.; Fuqua, B.K.; Andersen, H.S.; Naylor, C.E.; Evans, R.W.; Gambling, L.; et al. Identification of zyklopen, a new member of the vertebrate multicopper ferroxidase family, and characterization in rodents and human cells. J. Nutr. 2010, 140, 1728–1735. [Google Scholar] [CrossRef]
- Danzeisen, R.; Ponnambalam, S.; Lea, R.G.; Page, K.; Gambling, L.; McArdle, H.J. The effect of ceruloplasmin on iron release from placental (BeWo) cells; evidence for an endogenous Cu oxidase. Placenta 2000, 21, 805–812. [Google Scholar] [CrossRef]
- Danzeisen, R.; Fosset, C.; Chariana, Z.; Page, K.; David, S.; McArdle, H.J. Placental ceruloplasmin homolog is regulated by iron and copper and is implicated in iron metabolism. Am. J. Physiol. Cell Physiol. 2002, 282, C472–C478. [Google Scholar] [CrossRef]
- Cui, R.; Duan, X.L.; Anderson, G.J.; Qiao, Y.T.; Yu, P.; Qian, Z.M.; Yoshida, K.; Takeda, S.; Guo, P.; Yang, Z.L.; et al. Age-dependent expression of hephaestin in the brain of ceruloplasmin-deficient mice. J. Trace Elem. Med. Biol. 2009, 23, 290–299. [Google Scholar] [CrossRef]
- Harris, Z.L.; Durley, A.P.; Man, T.K.; Gitlin, J.D. Targeted gene disruption reveals an essential role for ceruloplasmin in cellular iron efflux. Proc. Natl. Acad. Sci. USA 1999, 96, 10812–10817. [Google Scholar]
- Bannerman, R.M.; Cooper, R.G. Sex-linked anemia: A hypochromic anemia of mice. Science 1966, 151, 581–582. [Google Scholar]
- Li, Y.Q.; Bai, B.; Cao, X.X.; Yan, H.; Zhuang, G.H. Ferroportin 1 and hephaestin expression in BeWo cell line with different iron treatment. Cell Biochem. Funct. 2012, 30, 249–255. [Google Scholar] [CrossRef]
- Cherukuri, S.; Potla, R.; Sarkar, J.; Nurko, S.; Harris, Z.L.; Fox, P.L. Unexpected role of ceruloplasmin in intestinal iron absorption. Cell Metab. 2005, 2, 309–319. [Google Scholar] [CrossRef]
- Mukhopadhyay, C.K.; Mazumder, B.; Fox, P.L. Role of hypoxia-inducible factor-1 in transcriptional activation of ceruloplasmin by iron deficiency. J. Biol. Chem. 2000, 275, 21048–21054. [Google Scholar] [CrossRef]
- Tapryal, N.; Mukhopadhyay, C.; Das, D.; Fox, P.L.; Mukhopadhyay, C.K. Reactive oxygen species regulate ceruloplasmin by a novel mRNA decay mechanism involving its 3′-untranslated region: Implications in neurodegenerative diseases. J. Biol. Chem. 2009, 284, 1873–1883. [Google Scholar]
- Sampath, P.; Mazumder, B.; Seshadri, V.; Fox, P.L. Transcript-selective translational silencing by gamma interferon is directed by a novel structural element in the ceruloplasmin mRNA 3′ untranslated region. Mol. Cell. Biol. 2003, 23, 1509–1519. [Google Scholar] [CrossRef]
- Persichini, T.; Maio, N.; di Patti, M.C.; Rizzo, G.; Toscano, S.; Colasanti, M.; Musci, G. Interleukin-1beta induces ceruloplasmin and ferroportin-1 gene expression via MAP kinases and C/EBPbeta, AP-1, and NF-kappaB activation. Neurosci. Lett. 2010, 484, 133–138. [Google Scholar] [CrossRef]
- Chen, H.; Su, T.; Attieh, Z.K.; Fox, T.C.; McKie, A.T.; Anderson, G.J.; Vulpe, C.D. Systemic regulation of Hephaestin and Ireg1 revealed in studies of genetic and nutritional iron deficiency. Blood 2003, 102, 1893–1899. [Google Scholar] [CrossRef]
- Lee, S.M.; Attieh, Z.K.; Son, H.S.; Chen, H.; Bacouri-Haidar, M.; Vulpe, C.D. Iron repletion relocalizes hephaestin to a proximal basolateral compartment in polarized MDCK and Caco2 cells. Biochem. Biophys. Res. Commun. 2012, 421, 449–455. [Google Scholar] [CrossRef]
- Hinoi, T.; Gesina, G.; Akyol, A.; Kuick, R.; Hanash, S.; Giordano, T.J.; Gruber, S.B.; Fearon, E.R. CDX2-regulated expression of iron transport protein hephaestin in intestinal and colonic epithelium. Gastroenterology 2005, 128, 946–961. [Google Scholar] [CrossRef]
- Nittis, T.; Gitlin, J.D. Role of copper in the proteosome-mediated degradation of the multicopper oxidase hephaestin. J. Biol. Chem. 2004, 279, 25696–25702. [Google Scholar] [CrossRef]
- Chen, H.; Huang, G.; Su, T.; Gao, H.; Attieh, Z.K.; McKie, A.T.; Anderson, G.J.; Vulpe, C.D. Decreased hephaestin activity in the intestine of copper-deficient mice causes systemic iron deficiency. J. Nutr. 2006, 136, 1236–1241. [Google Scholar]
- Gitlin, J.D.; Schroeder, J.J.; Lee-Ambrose, L.M.; Cousins, R.J. Mechanisms of caeruloplasmin biosynthesis in normal and copper-deficient rats. Biochem. J. 1992, 282, 835–839. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Vashchenko, G.; MacGillivray, R.T.A. Multi-Copper Oxidases and Human Iron Metabolism. Nutrients 2013, 5, 2289-2313. https://doi.org/10.3390/nu5072289
Vashchenko G, MacGillivray RTA. Multi-Copper Oxidases and Human Iron Metabolism. Nutrients. 2013; 5(7):2289-2313. https://doi.org/10.3390/nu5072289
Chicago/Turabian StyleVashchenko, Ganna, and Ross T. A. MacGillivray. 2013. "Multi-Copper Oxidases and Human Iron Metabolism" Nutrients 5, no. 7: 2289-2313. https://doi.org/10.3390/nu5072289
APA StyleVashchenko, G., & MacGillivray, R. T. A. (2013). Multi-Copper Oxidases and Human Iron Metabolism. Nutrients, 5(7), 2289-2313. https://doi.org/10.3390/nu5072289