Selenium Metabolism in Cancer Cells: The Combined Application of XAS and XFM Techniques to the Problem of Selenium Speciation in Biological Systems

Abstract
:1. Introduction
2. The Selenium Speciation Problem
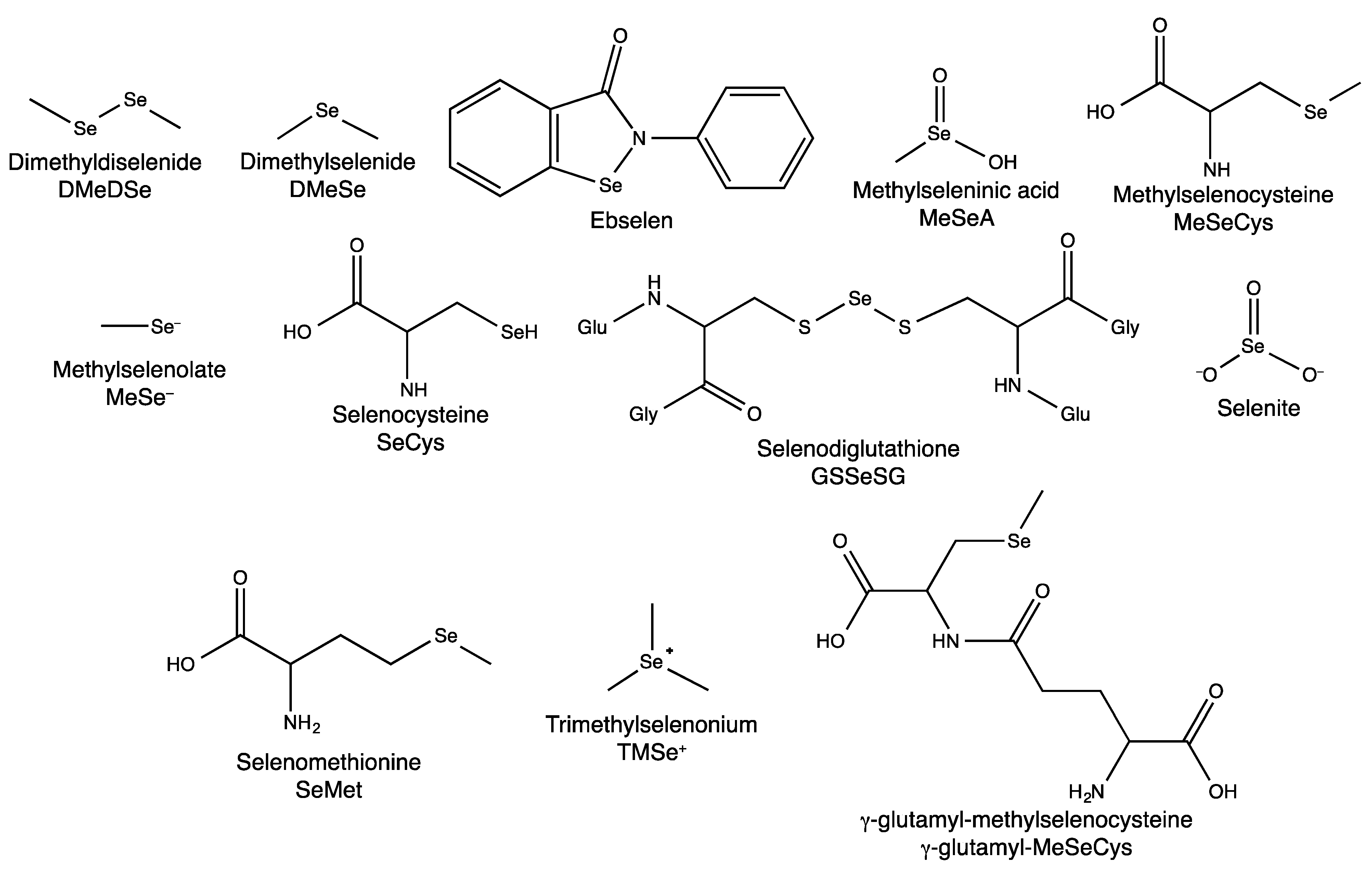
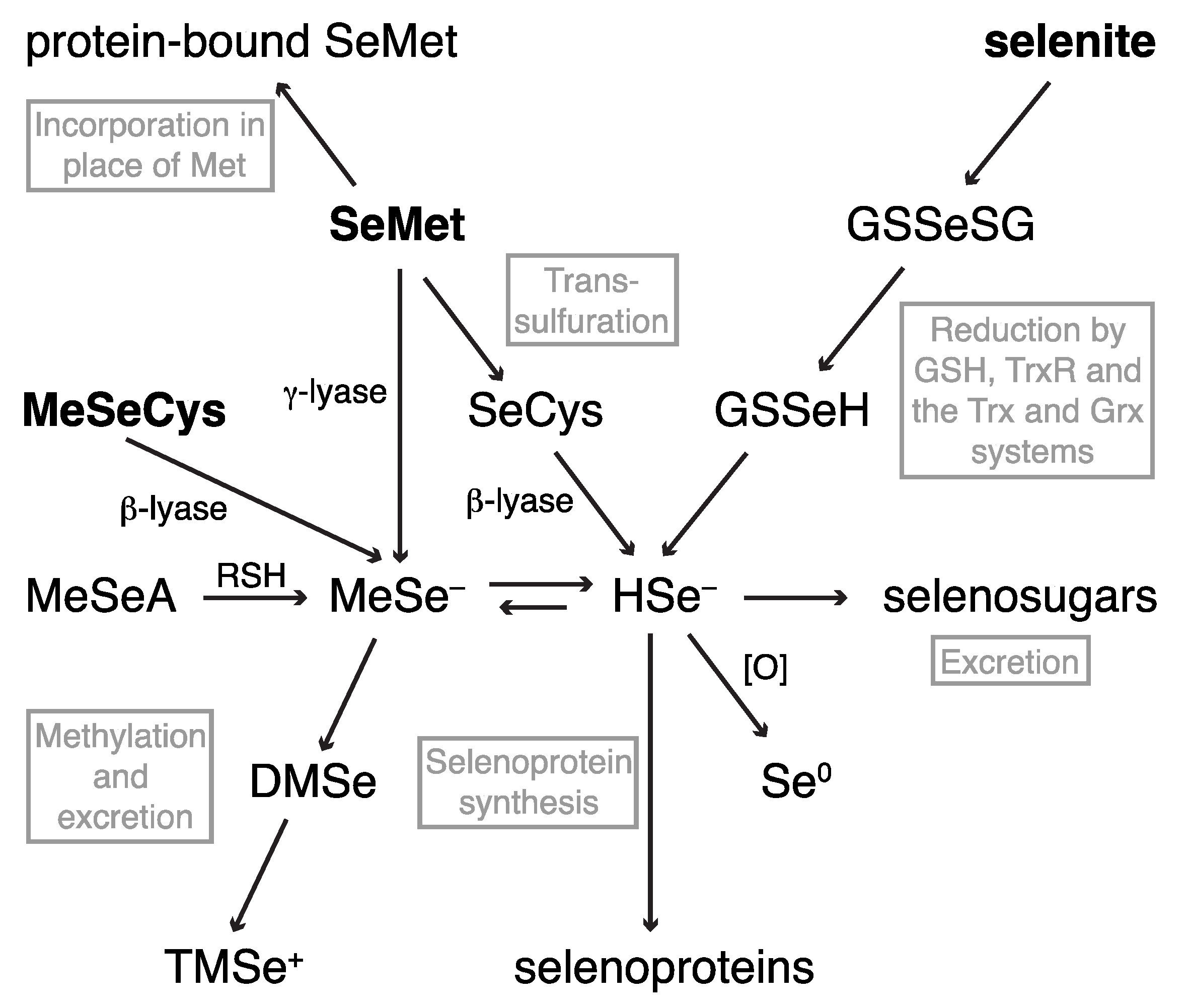
2.1. Dietary Selenium Compounds and Their Metabolites
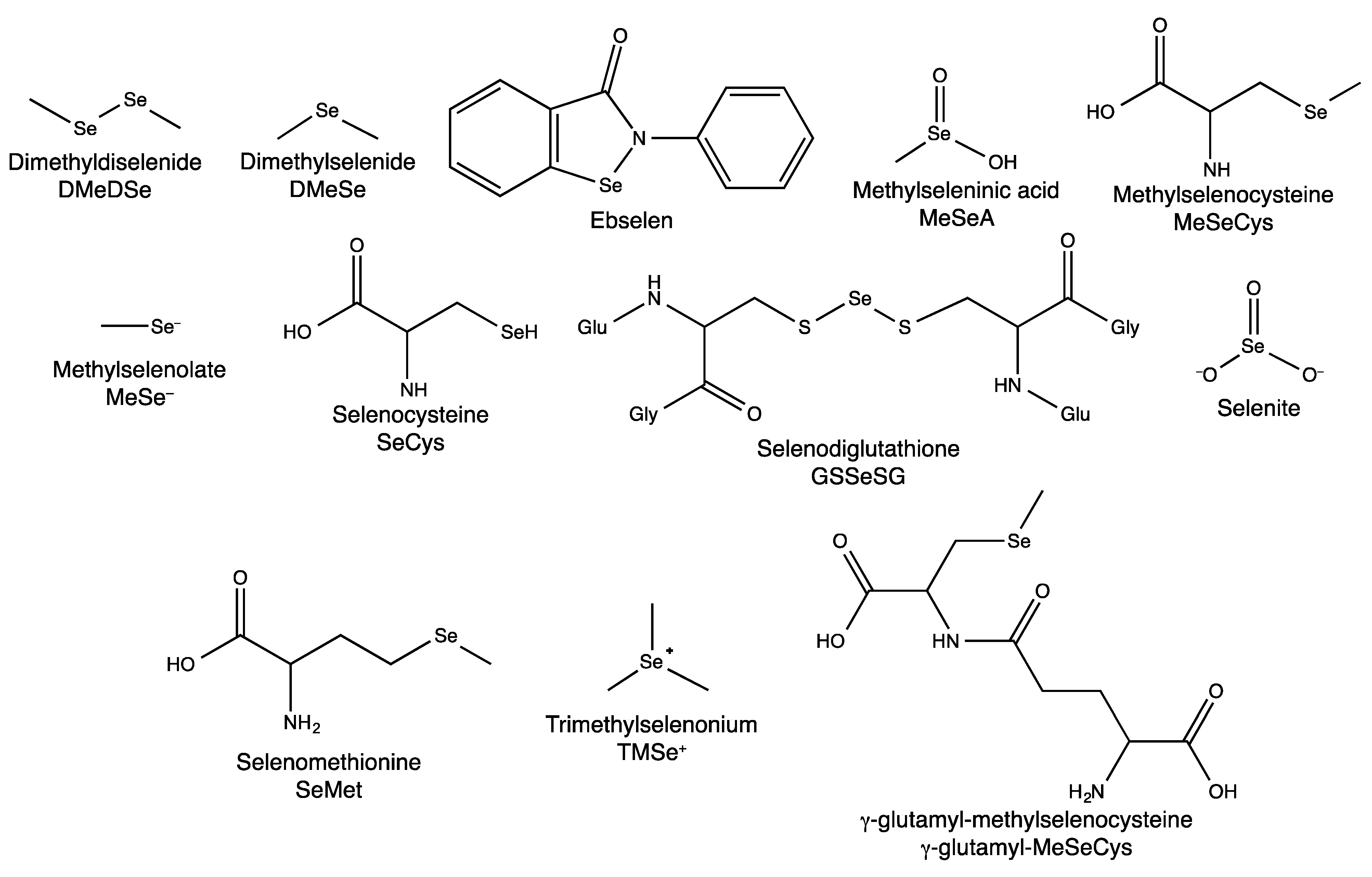
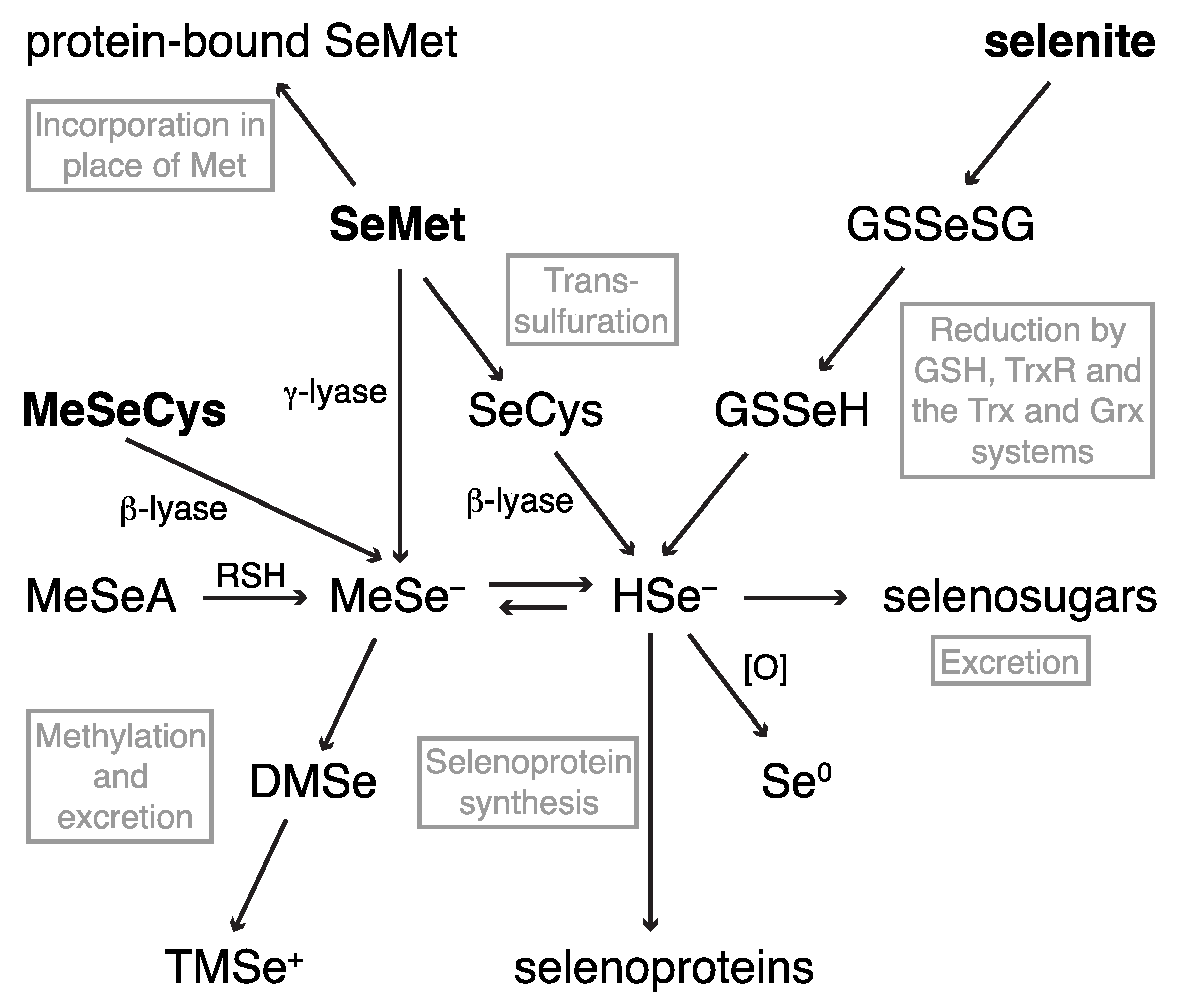
2.1.1. Hyphenated Techniques and Selenium Speciation
3. Synchrotron-Based X-ray Techniques in Elemental Speciation Analysis
The Use of Synchrotron-Based X-Rays to Investigate Selenium in Biology
4. A Top-Down Approach to Investigating Selenium in Biology
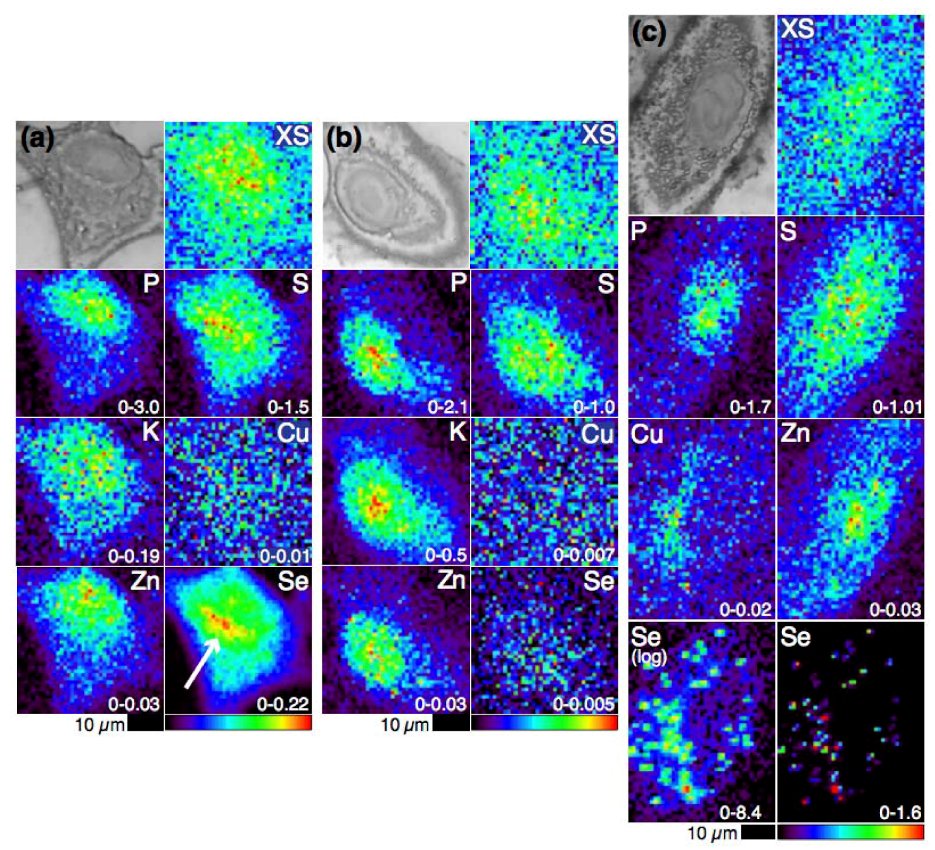
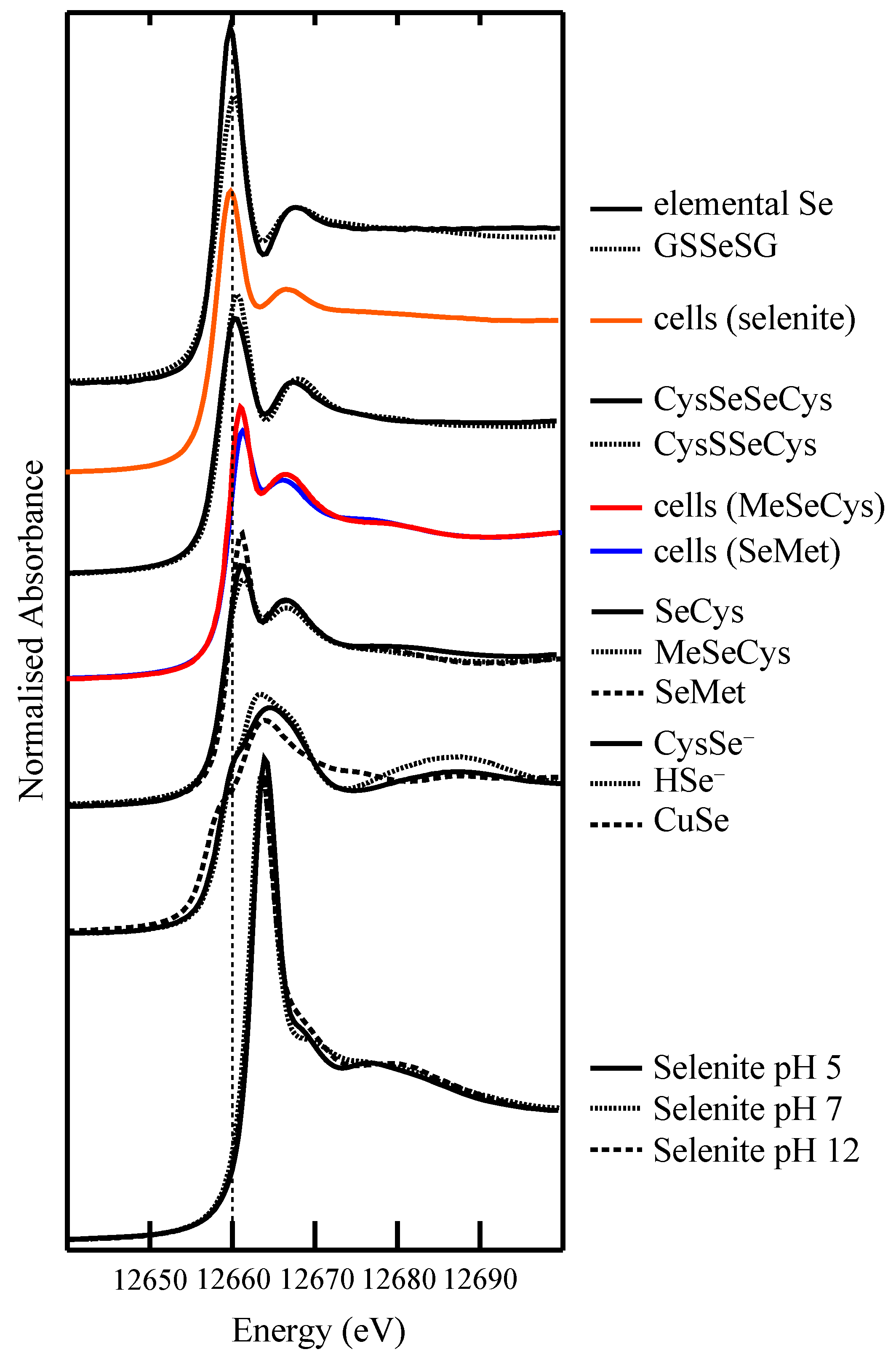
4.1. Selenium Speciation Dictates Distribution in Human Lung Cancer Cells—Findings from XFM
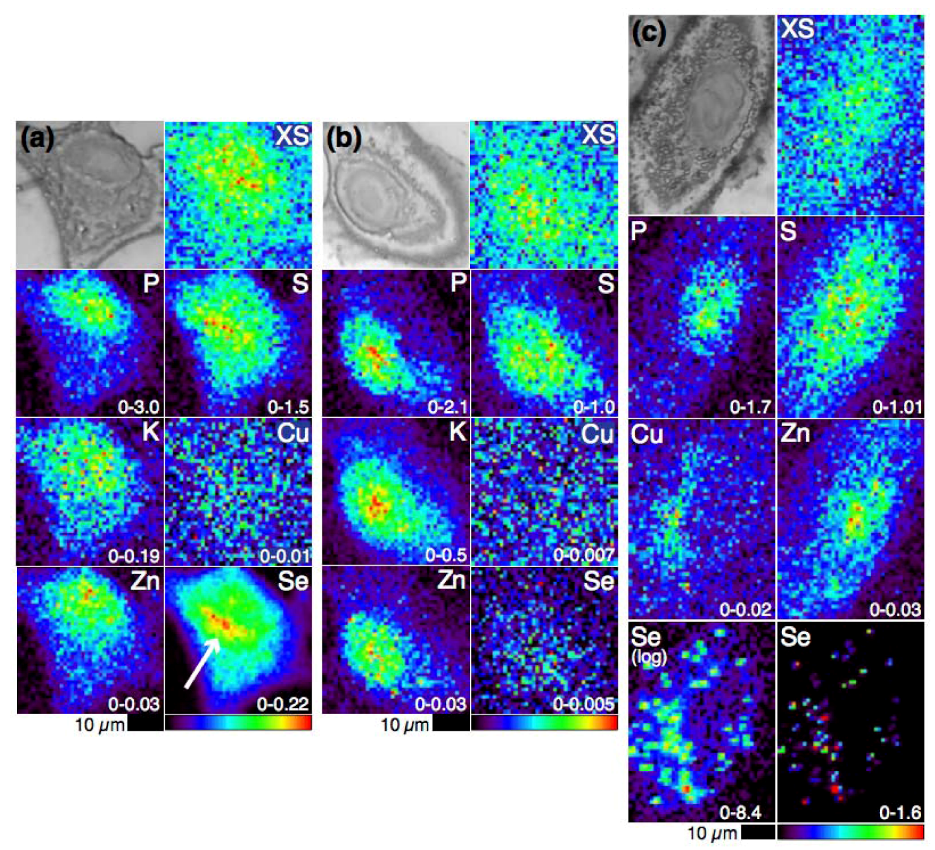
4.2. Selenium Speciation in Bulk Cell Pellets and Single Cancer Cells
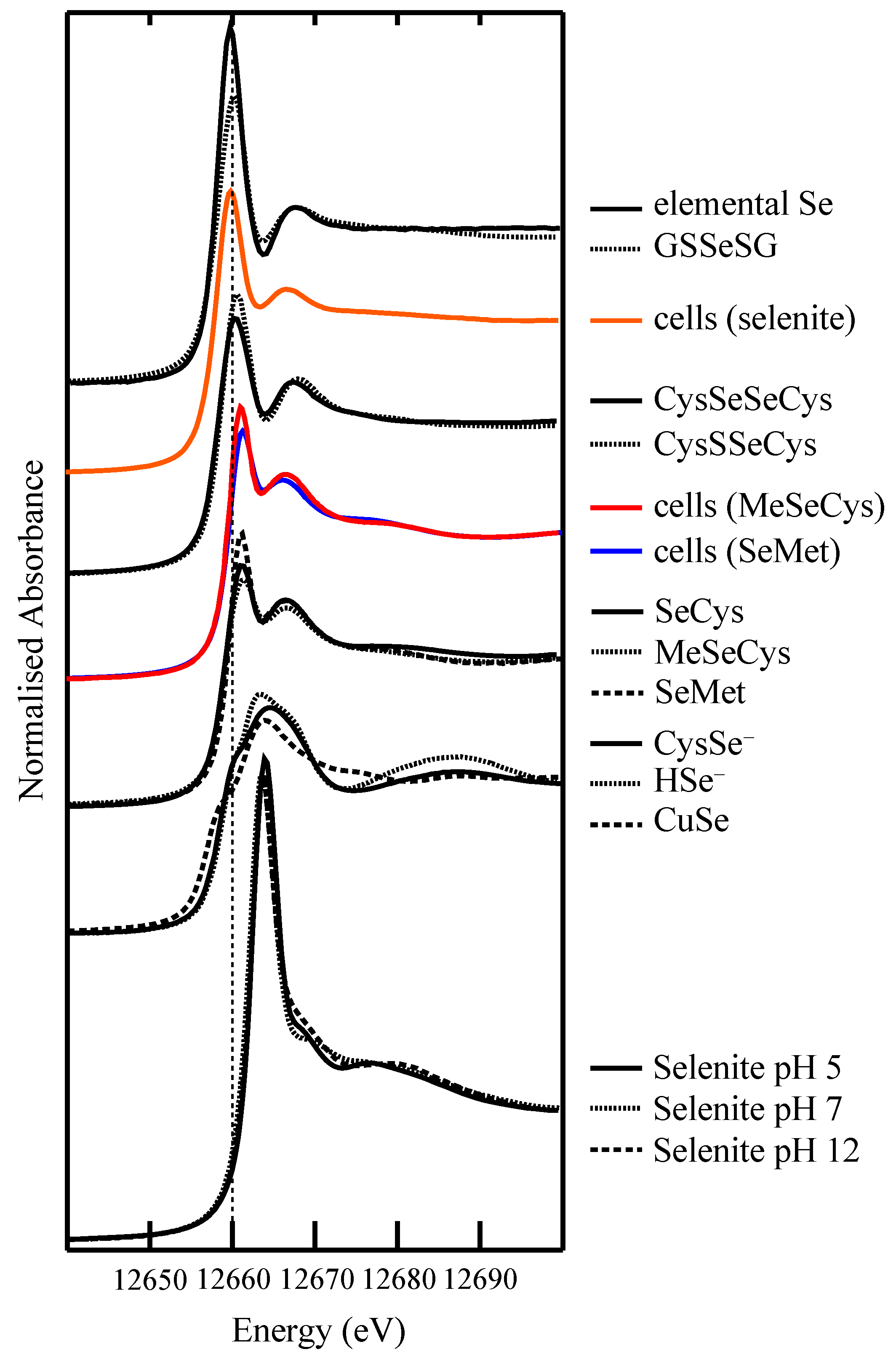
4.3. Extending XFM to Selenium in Electrophoretically-Separated Cell Lysates

5. Conclusions

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Se supplement | Speciation of Se metabolites | Distribution of Se metabolites |
|---|---|---|
| SeMet | R–Se–R | throughout the cells, but particularly concentrated in the perinuclear region bound to proteins |
| MeSeCys | mostly R–Se–R significant R–Se–Se–R component | uncertain |
| Selenite | elemental Se, S–Se–S and R–Se–Se–R some R–Se–R | elemental Se concentrated in small regions throughout the cytosol Se associated with, but not bound to, Cu |
Future Directions
Acknowledgments
Conflict of Interest
References
- Clark, L.C.; Combs, G.F.; Turnbull, B.W.; Slate, E.H.; Chalker, D.K.; Chow, J.; Davis, L.S.; Glover, R.A.; Graham, G.F.; Gross, E.G.; et al. Effects of selenium supplementation for cancer prevention in patients with carcinoma of the skin. A randomized controlled trial. JAMA 1996, 276, 1957–1963. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium and human health. Lancet 2012, 379, 1256–1268. [Google Scholar] [CrossRef]
- Ganther, H.E. Selenotrisulfides. Formation by the reaction of thiols with selenious acid. Biochemistry 1968, 7, 2898–2905. [Google Scholar] [CrossRef]
- Reeves, M.A.; Hoffmann, P.R. The human selenoproteome: Recent insights into functions and regulation. Cell. Mol. Life Sci. 2009, 66, 2457–2478. [Google Scholar] [CrossRef]
- Ganther, H.E. Reduction of the selenotrisulfide derivative of glutathione to a persulfide analog by glutathione reductase. Biochemistry 1971, 10, 4089–4098. [Google Scholar] [CrossRef]
- Hsieh, H.S.; Ganther, H.E. Acid-volatile selenium formation catalyzed by glutathione reductase. Biochemistry 1975, 14, 1632–1636. [Google Scholar] [CrossRef]
- Kumar, S.; Björnstedt, M.; Holmgren, A. Selenite is a substrate for calf thymus thioredoxin reductase and thioredoxin and elicits a large non-stoichiometric oxidation of NADPH in the presence of oxygen. Eur. J. Biochem. 1992, 207, 435–439. [Google Scholar] [CrossRef]
- Wallenberg, M.; Olm, E.; Hebert, C.; Björnstedt, M.; Fernandes, A.P. Selenium compounds are substrates for glutaredoxins: A novel pathway for selenium metabolism and a potential mechanism for selenium-mediated cytotoxicity. Biochem. J. 2010, 429, 85–93. [Google Scholar] [CrossRef]
- Braga, P.; Montes-Bayón, M.; Alvarez, J.; López, J.M.; Sanz-Medel, A. Characterization, biological interactions and in-vivo detection of selenotrisulfide derivatives of glutathion, cysteine and homocysteine by HPLC-ICP-MS. J. Anal. At. Spectrom. 2004, 19, 1128–1133. [Google Scholar]
- Hatfield, D.L.; Yoo, M.-H.; Carlson, B.A.; Gladyshev, V.N. Selenoproteins that function in cancer prevention and promotion. Biochim. Biophys. Acta Gen. Subj. 2009, 1790, 1541–1545. [Google Scholar] [CrossRef]
- Gabel-Jensen, C.; Gammelgaard, B.; Bendahl, L.; Stürup, S.; Jøns, O. Separation and identification of selenotrisulfides in epithelial cell homogenates by LC-ICP-MS and LC-ESI-MS after incubation with selenite. Anal. Bioanal. Chem. 2006, 384, 697–702. [Google Scholar] [CrossRef]
- Irons, R.; Carlson, B.A.; Hatfield, D.L.; Davis, C.D. Both selenoproteins and low molecular weight selenocompounds reduce colon cancer risk in mice with genetically impaired selenoprotein expression. J. Nutr. 2006, 136, 1311–1317. [Google Scholar]
- Lunøe, K.; Gabel-Jensen, C.; Stürup, S.; Andresen, L.; Skov, S.; Gammelgaard, B. Investigation of the selenium metabolism in cancer cell lines. Metallomics 2011, 3, 162–168. [Google Scholar] [CrossRef]
- Duffield-Lillico, A.J.; Reid, M.E.; Turnbull, B.W.; Combs, G.F., Jr.; Slate, E.H.; Fischbach, L.A.; Marshall, J.R.; Clark, L.C. Baseline characteristics and the effect of selenium supplementation on cancer incidence in a randomized clinical trial: A summary report of the Nutritional Prevention of Cancer Trial. Cancer Epidemiol. Biomark. Prev. 2002, 11, 630–639. [Google Scholar]
- Duffield-Lillico, A.J.; Dalkin, B.L.; Reid, M.E.; Turnbull, B.W.; Slate, E.H.; Jacobs, E.T.; Marshall, J.R.; Clark, L.C. Selenium supplementation, baseline plasma selenium status and incidence of prostate cancer: An analysis of the complete treatment period of the Nutritional Prevention of Cancer Trial. BJU Int. 2003, 91, 608–612. [Google Scholar] [CrossRef]
- Esaki, N.; Nakamura, T.; Tanaka, H.; Soda, K. Selenocysteine lyase, a novel enzyme that specifically acts on selenocysteine. Mammalian distribution and purification and properties of pig liver enzyme. J. Biol. Chem. 1982, 257, 4386–4391. [Google Scholar]
- Lippman, S.M.; Goodman, P.J.; Klein, E.A.; Parnes, H.L.; Thompson, I.M.; Kristal, A.R.; Santella, R.M.; Probstfield, J.L.; Moinpour, C.M.; Albanes, D.; et al. Designing the Selenium and Vitamin E Cancer Prevention Trial (SELECT). J. Natl. Cancer Inst. 2005, 97, 94–102. [Google Scholar] [CrossRef]
- Butler, J.A.; Beilstein, M.A.; Whanger, P.D. Influence of dietary methionine on the metabolism of selenomethionine in rats. J. Nutr. 1989, 119, 1001–1009. [Google Scholar]
- Lippman, S.M.; Klein, E.A.; Goodman, P.J.; Lucia, M.S.; Thompson, I.M.; Ford, L.G.; Parnes, H.L.; Minasian, L.M.; Gaziano, J.M.; Hartline, J.A.; et al. Effect of selenium and vitamin e on risk of prostate cancer and other cancers: The Selenium and Vitamin E Cancer Prevention Trial (SELECT). JAMA 2009, 301, 39–51. [Google Scholar] [CrossRef]
- Okuno, T.; Kubota, T.; Kuroda, T.; Ueno, H.; Nakamuro, K. Contribution of enzymic alpha, gamma-elimination reaction in detoxification pathway of selenomethionine in mouse liver. Toxicol. Appl. Pharmacol. 2001, 176, 18–23. [Google Scholar] [CrossRef]
- Klein, E.A. Selenium and Vitamin E: Interesting Biology and Dashed Hope. J. Natl. Cancer Inst. 2009, 101, 283–285. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Kurasaki, K.; Suzuki, N. Selenocysteine beta-lyase and methylselenol demethylase in the metabolism of Se-methylated selenocompounds into selenide. Biochim. Biophys. Acta Gen. Subj. 2007, 1770, 1053–1061. [Google Scholar] [CrossRef]
- Rayman, M.P.; Combs, G.F., Jr.; Waters, D.J. Selenium and vitamin E supplementation for cancer prevention. JAMA 2009, 301, 1876. [Google Scholar] [CrossRef]
- Hatfield, D.L.; Gladyshev, V.N. The outcome of Selenium and Vitamin E Cancer Prevention Trial (SELECT) reveals the need for better understanding of selenium biology. Mol. Interv. 2009, 9, 18–21. [Google Scholar] [CrossRef]
- Goossens, M.E.; Buntinx, F.; Zeegers, M.P. Re: Selenium and Vitamin E: Interesting biology and dashed hope. J. Natl. Cancer Inst. 2009, 101, 1363–1364. [Google Scholar] [CrossRef]
- Ganther, H.E.; Lawrence, J.R. Chemical transformations of selenium in living organisms. Improved forms of selenium for cancer prevention. Tetrahedron 1997, 53, 12299–12310. [Google Scholar] [CrossRef]
- Bird, S.M.; Uden, P.C.; Tyson, J.F.; Block, E.; Denoyer, E. Speciation of selenoamino acids and organoselenium compounds in selenium-enriched yeast using high-performance liquid chromatography-inductively coupled plasma mass spectrometry. J. Anal. At. Spectrom. 1997, 12, 785–788. [Google Scholar] [CrossRef]
- Uden, P.C.; Boakye, H.T.; Kahakachchi, C.L.; Hafezi, R.; Nolibos, P.; Block, E.; Johnson, S.; Tyson, J.F. Element selective characterization of stability and reactivity of selenium species in selenized yeast. J. Anal. At. Spectrom. 2004, 19, 65–73. [Google Scholar] [CrossRef]
- Larsen, E.H.; Hansen, M.; Paulin, H.; Moesgaard, S.; Reid, M.; Rayman, M. Speciation and bioavailability of selenium in yeast-based intervention agents used in cancer chemoprevention studies. J. AOAC Int. 2004, 87, 225–232. [Google Scholar]
- Rayman, M.P.; Infante, H.G.; Sargent, M. Food-chain selenium and human health: Spotlight on speciation. Br. J. Nutr. 2008, 100, 238–253. [Google Scholar]
- Tsuji, Y.; Suzuki, N.; Suzuki, K.T.; Ogra, Y. Selenium metabolism in rats with long-term ingestion of Se-methylselenocysteine using enriched stable isotopes. J. Toxicol. Sci. 2009, 34, 191–200. [Google Scholar] [CrossRef]
- Ogra, Y.; Anan, Y. Selenometabolomics: Identification of selenometabolites and specification of their biological significance by complementary use of elemental and molecular mass spectrometry. J. Anal. At. Spectrom. 2009, 24, 1477–1488. [Google Scholar] [CrossRef]
- Gammelgaard, B.; Jackson, M.I.; Gabel-Jensen, C. Surveying selenium speciation from soil to cell—Forms and transformations. Anal. Bioanal. Chem. 2011, 399, 1743–1763. [Google Scholar] [CrossRef]
- Rayman, M.P. Selenium in cancer prevention: A review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef]
- McConnell, K.P.; Portman, O.W. Excretion of dimethyl selenide by the rat. J. Biol. Chem. 1952, 195, 277–282. [Google Scholar]
- Sinha, R.; Unni, E.; Ganther, H.E.; Medina, D. Methylseleninic acid, a potent growth inhibitor of synchronized mouse mammary epithelial tumor cells in vitro. Biochem. Pharmacol. 2001, 61, 311–317. [Google Scholar] [CrossRef]
- Byard, J.L. Trimethyl selenide. A urinary metabolite of selenite. Arch. Biochem. Biophys. 1969, 130, 556–560. [Google Scholar] [CrossRef]
- Infante, H.G.; Joel, S.P.; Warburton, E.; Hopley, C.; Hearn, R.; Jüliger, S. Investigation of the selenium species distribution in a human B-cell lymphoma line by HPLC-and GC-ICP-MS in combination with HPLC-ESIMS/MS and GC-TOFMS after incubation with methylseleninic acid. J. Anal. At. Spectrom. 2007, 22, 888–896. [Google Scholar] [CrossRef]
- Ogra, Y.; Ishiwata, K.; Takayama, H.; Aimi, N.; Suzuki, K.T. Identification of a novel selenium metabolite, Se-methyl-N-acetylselenohexosamine, in rat urine by high-performance liquid chromatography—Inductively coupled plasma mass spectrometry and—Electrospray ionization tandem mass spectrometry. J. Chromatogr. B 2002, 767, 301–312. [Google Scholar] [CrossRef]
- Francesconi, K.A.; Pannier, F. Selenium metabolites in urine: A critical overview of past work and current status. Clin. Chem. 2004, 50, 2240–2253. [Google Scholar] [CrossRef]
- Ohta, Y.; Suzuki, K.T. Methylation and demethylation of intermediates selenide and methylselenol in the metabolism of selenium. Toxicol. Appl. Pharmacol. 2008, 226, 169–177. [Google Scholar] [CrossRef]
- Jacob, C.; Giles, G.; Giles, N.; Sies, H. Sulfur and selenium: The role of oxidation state in protein structure and function. Angew. Chem. Int. Ed. 2003, 42, 4742–4758. [Google Scholar] [CrossRef]
- Po atajko, A.; Jakubowski, N.; Szpunar, J. State of the art report of selenium speciation in biological samples. J. Anal. At. Spectrom. 2006, 21, 639. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Ohta, Y.; Suzuki, N. Availability and metabolism of 77Se-methylseleninic acid compared simultaneously with those of three related selenocompounds. Toxicol. Appl. Pharmacol. 2006, 217, 51–62. [Google Scholar] [CrossRef]
- Gammelgaard, B.; Gabel-Jensen, C.; Stürup, S.; Hansen, H.R. Complementary use of molecular and element-specific mass spectrometry for identification of selenium compounds related to human selenium metabolism. Anal. Bioanal. Chem. 2008, 390, 1691–1706. [Google Scholar] [CrossRef]
- Ip, C.; Thompson, H.; Zhu, Z.; Ganther, H.E. In vitro and in vivo studies of methylseleninic acid: Evidence that a monomethylated selenium metabolite is critical for cancer chemoprevention. Cancer Res. 2000, 60, 2882–2886. [Google Scholar]
- B’Hymer, C.; Caruso, J.A. Selenium speciation analysis using inductively coupled plasma-mass spectrometry. J. Chromatogr. A 2006, 1114, 1–20. [Google Scholar] [CrossRef]
- Aitken, J.B.; Levina, A.; Lay, P.A. Studies on the biotransformations and biodistributions of metal-containing drugs using X-ray absorption spectroscopy. Curr. Top. Med. Chem. 2011, 11, 553–571. [Google Scholar] [CrossRef]
- George, G.N.; Hedman, B.; Hodgson, K.O. An edge with XAS. Nat. Struct. Biol. 1998, 5, 645–647. [Google Scholar] [CrossRef]
- Nakazawa, E.; Ikemoto, T.; Hokura, A.; Terada, Y.; Kunito, T.; Yamamoto, T.; Yamada, T.K.; Rosas, F.C.W.; Fillmann, G.; Tanabe, S.; Nakai, I. Silver speciation in liver of marine mammals by synchrotron X-ray absorption fine structure and X-ray fluorescence spectroscopies. J. Environ. Monit. 2011, 13, 1678. [Google Scholar] [CrossRef]
- Korbas, M.; O’Donoghue, J.L.; Watson, G.E.; Pickering, I.J.; Singh, S.P.; Myers, G.J.; Clarkson, T.W.; George, G.N. The chemical nature of mercury in human brain following poisoning or environmental exposure. ACS Chem. Neurosci. 2010, 1, 810–818. [Google Scholar] [CrossRef]
- Esaki, N.; Nakamura, T.; Tanaka, H.; Suzuki, T.; Morino, Y.; Soda, K. Enzymatic synthesis of selenocysteine in rat liver. Biochemistry 1981, 20, 4492–4496. [Google Scholar] [CrossRef]
- Ortega, R.; Carmona, A.; Llorens, I.; Solari, P.L. X-ray absorption spectroscopy of biological samples. A tutorial. J. Anal. At. Spectrom. 2012, 27, 2054. [Google Scholar] [CrossRef]
- Pickering, I.J.; George, G.N.; van Fleet-Stalder, V.; Chasteen, T.G.; Prince, R.C. X-ray absorption spectroscopy of selenium-containing amino acids. J. Biol. Inorg. Chem. 1999, 4, 791–794. [Google Scholar]
- George, G.N.; Pickering, I.J.; Pushie, M.J.; Nienaber, K.; Hackett, M.J.; Ascone, I.; Hedman, B.; Hodgson, K.O.; Aitken, J.B.; Levina, A.; et al. X-ray-induced photo-chemistry and X-ray absorption spectroscopy of biological samples. J. Synchrotron Radiat. 2012, 19, 875–886. [Google Scholar] [CrossRef]
- Wang, H.C.; Riahi, M.; Pothen, J.; Bayse, C.A.; Riggs-Gelasco, P.; Brumaghim, J.L. Interactions of Cu(I) with selenium-containing amino acids determined by NMR, XAS, and DFT studies. Inorg. Chem. 2011, 50, 10893–10900. [Google Scholar] [CrossRef]
- Yu-Feng, L.; Xiaoyan, W.; Liming, W.; Bai, L.; Yuxi, G.; Chunying, C. Direct quantitative speciation of selenium in selenium-enriched yeast and yeast-based products by X-ray absorption spectroscopy confirmed by HPLC-ICP-MS. J. Anal. At. Spectrom. 2010, 25, 426–430. [Google Scholar] [CrossRef]
- Aitken, J.B.; Lay, P.A.; Duong, T.T.; Aran, R.; Witting, P.K.; Harris, H.H.; Lai, B.; Vogt, S.; Giles, G.I. Synchrotron radiation induced X-ray emission studies of the antioxidant mechanism of the organoselenium drug ebselen. J. Biol. Inorg. Chem. 2012, 1–10. [Google Scholar]
- Kehr, S.; Malinouski, M.; Finney, L.A.; Vogt, S.; Labunskyy, V.M.; Kasaikina, M.V.; Carlson, B.A.; Zhou, Y.; Hatfield, D.L.; Gladyshev, V.N. X-ray fluorescence microscopy reveals the role of selenium in spermatogenesis. J. Mol. Biol. 2009, 389, 808–818. [Google Scholar] [CrossRef]
- Malinouski, M.; Kehr, S.; Finney, L.A.; Vogt, S.; Carlson, B.A.; Seravalli, J.; Jin, R.; Handy, D.E.; Park, T.J.; Loscalzo, J.; et al. High-resolution imaging of selenium in kidneys: A localized selenium pool associated with glutathione peroxidase 3. Antioxid. Redox Signal. 2012, 16, 185–192. [Google Scholar] [CrossRef]
- Suzuki, K.T.; Doi, C.; Suzuki, N. Metabolism of Se-76-methylselenocysteine compared with that of Se-77-selenomethionine and Se-82-selenite. Toxicol. Appl. Pharmacol. 2006, 217, 185–195. [Google Scholar] [CrossRef]
- Chen, T.; Wong, Y.-S. Selenocystine induces reactive oxygen species-mediated apoptosis in human cancer cells. Biomed. Pharmacother. 2009, 63, 105–113. [Google Scholar] [CrossRef]
- Weekley, C.M.; Aitken, J.B.; Vogt, S.; Finney, L.A.; Paterson, D.J.; de Jonge, M.D.; Howard, D.L.; Musgrave, I.F.; Harris, H.H. Uptake, distribution, and speciation of selenoamino acids by human cancer cells: X-ray absorption and fluorescence methods. Biochemistry 2011, 50, 1641–1650. [Google Scholar]
- Weekley, C.M.; Aitken, J.B.; Vogt, S.; Finney, L.A.; Paterson, D.J.; de Jonge, M.D.; Howard, D.L.; Witting, P.K.; Musgrave, I.F.; Harris, H.H. Metabolism of selenite in human lung cancer cells: X-ray absorption and fluorescence studies. J. Am. Chem. Soc. 2011, 133, 18272–18279. [Google Scholar] [CrossRef]
- Gabel-Jensen, C.; Bak, S.A.; Lauritsen, F.R.; Hansen, H.R.; Badolo, L.; Gammelgaard, B. In situ identification of dimethyl diselenide in hepatocytes treated with methylseleninic acid by membrane inlet mass spectrometry. J. Anal. At. Spectrom. 2009, 24, 949–952. [Google Scholar] [CrossRef]
- Hackett, M.J.; McQuillan, J.A.; El-Assaad, F.; Aitken, J.B.; Levina, A.; Cohen, D.D.; Siegele, R.; Carter, E.A.; Grau, G.E.; Hunt, N.H.; Lay, P.A. Chemical alterations to murine brain tissue induced by formalin fixation: implications for biospectroscopic imaging and mapping studies of disease pathogenesis. Analyst 2011, 136, 2941–2952. [Google Scholar] [CrossRef]
- Shen, C.L.; Song, W.; Pence, B.C. Interactions of selenium compounds with other antioxidants in DNA damage and apoptosis in human normal keratinocytes. Cancer Epidemiol. Biomark. Prev. 2001, 10, 385–390. [Google Scholar]
- Kezhou, W.; Stowe, H.D.; House, A.M.; Chou, K.; Thiel, T. Comparison of cupric and sulfate ion effects on chronic selenosis in rats. J. Anim. Sci. 1987, 64, 1467–1475. [Google Scholar]
- Fahrni, C. Biological applications of X-ray fluorescence microscopy: Exploring the subcellular topography and speciation of transition metals. Curr. Opin. Chem. Biol. 2007, 11, 121–127. [Google Scholar] [CrossRef]
- Misra, S.; Peak, D.; Niyogi, S. Application of XANES spectroscopy in understanding the metabolism of selenium in isolated rainbow trout hepatocytes: Insights into selenium toxicity. Metallomics 2010, 2, 710–717. [Google Scholar] [CrossRef]
- Misra, S.; Peak, D.; Chen, N.; Hamilton, C.; Niyogi, S. Tissue-specific accumulation and speciation of selenium in rainbow trout (Oncorhynchus mykiss) exposed to elevated dietary selenomethionine. Comp. Biochem. Phys. C 2012, 155, 560–565. [Google Scholar]
- Shanu, A.; Groebler, L.; Kim, H.B.; Wood, S.; Weekley, C.M.; Aitken, J.B.; Harris, H.H.; Witting, P.K. Selenium inhibits renal oxidation and inflammation but not acute kidney injury in an animal model of rhabdomyolysis. Antioxid. Redox Signal. 2013, 18, 756–769. [Google Scholar] [CrossRef]
- Harris, H.H.; Levina, A.; Dillon, C.T.; Mulyani, I.; Lai, B.; Cai, Z.; Lay, P.A. Time-dependent uptake, distribution and biotransformation of chromium (VI) in individual and bulk human lung cells: application of synchrotron radiation techniques. J. Biol. Inorg. Chem. 2005, 10, 105–118. [Google Scholar]
- Weekley, C.M.; Aitken, J.B.; Musgrave, I.F.; Harris, H.H. Methylselenocysteine treatment leads to diselenide formation in human cancer cells: Evidence from X-ray absorption spectroscopy studies. Biochemistry 2012, 51, 736–738. [Google Scholar]
- Weekley, C.M.; School of Chemistry and Physics, The University of Adelaide, Adelaide, Australia. Unpublished work. 2013.
- Spallholz, J.E.; Shriver, B.J.; Reid, T.W. Dimethyldiselenide and methylseleninic acid generate superoxide in an in vitro chemiluminescence assay in the presence of glutathione: Implications for the anticarcinogenic activity of l-selenomethionine and l-Se-methylselenocysteine. Nutr. Cancer 2001, 40, 34–41. [Google Scholar] [CrossRef]
- Huber, R.E.; Criddle, R.S. Comparison of the chemical properties of selenocysteine and selenocystine with their sulfur analogs. Arch. Biochem. Biophys. 1967, 122, 164–173. [Google Scholar]
- Gabel-Jensen, C.; Gammelgaard, B. Selenium metabolism in hepatocytes incubated with selenite, selenate, selenomethionine, Se-methylselenocysteine and methylseleninc acid and analysed by LC-ICP-MS. J. Anal. At. Spectrom. 2010, 25, 414–418. [Google Scholar] [CrossRef]
- Shen, H.M.; Yang, C.F.; Ding, W.X.; Liu, J.; Ong, C.N. Superoxide radical-initiated apoptotic signalling pathway in selenite-treated HepG(2) cells: Mitochondria serve as the main target. Free Radic. Biol. Med. 2001, 30, 9–21. [Google Scholar] [CrossRef]
- Xiang, N.; Zhao, R.; Zhong, W. Sodium selenite induces apoptosis by generation of superoxide via the mitochondrial-dependent pathway in human prostate cancer cells. Cancer Chemother. Pharmacol. 2009, 63, 351–362. [Google Scholar] [CrossRef]
- Finney, L.A.; Chishti, Y.; Khare, T.; Giometti, C.; Levina, A.; Lay, P.A.; Vogt, S. Imaging metals in proteins by combining electrophoresis with rapid X-ray fluorescence mapping. ACS Chem. Biol. 2010, 5, 577–587. [Google Scholar] [CrossRef]
- Raimunda, D.; Khare, T.; Giometti, C.; Vogt, S.; Argüello, J.M.; Finney, L.A. Identifying metalloproteins through X-ray fluorescence mapping and mass spectrometry. Metallomics 2012, 4, 921–927. [Google Scholar] [CrossRef]
- Vogt, S. MAPS: A set of software tools for analysis and visualization of 3D X-ray fluorescence data sets. J. Phys. IV 2003, 104, 635–638. [Google Scholar]
- Martínez, J.; Lisa, S.; Sánchez, R.; Kowalczyk, W.; Zurita, E.; Teixidó, M.; Giralt, E.; Andreu, D.; Avila, J.; Gasset, M. Selenomethionine incorporation into amyloid sequences regulates fibrillogenesis and toxicity. PLoS One 2011, 6, e27999. [Google Scholar]
- Pickering, I.J.; Prince, R.C.; Salt, D.E.; George, G.N. Quantitative, chemically specific imaging of selenium transformation in plants. Proc. Natl. Acad. Sci. USA 2000, 97, 10717–10722. [Google Scholar] [CrossRef]
- Pickering, I.J.; Sneeden, E.Y.; Prince, R.C.; Block, E.; Harris, H.H.; Hirsch, G.; George, G.N. Localizing the chemical forms of sulfur in vivo using X-ray fluorescence spectroscopic imaging: application to onion (Allium cepa) tissues. Biochemistry 2009, 48, 6846–6853. [Google Scholar]
Supplementary Files
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Weekley, C.M.; Aitken, J.B.; Finney, L.; Vogt, S.; Witting, P.K.; Harris, H.H. Selenium Metabolism in Cancer Cells: The Combined Application of XAS and XFM Techniques to the Problem of Selenium Speciation in Biological Systems. Nutrients 2013, 5, 1734-1756. https://doi.org/10.3390/nu5051734
Weekley CM, Aitken JB, Finney L, Vogt S, Witting PK, Harris HH. Selenium Metabolism in Cancer Cells: The Combined Application of XAS and XFM Techniques to the Problem of Selenium Speciation in Biological Systems. Nutrients. 2013; 5(5):1734-1756. https://doi.org/10.3390/nu5051734
Chicago/Turabian StyleWeekley, Claire M., Jade B. Aitken, Lydia Finney, Stefan Vogt, Paul K. Witting, and Hugh H. Harris. 2013. "Selenium Metabolism in Cancer Cells: The Combined Application of XAS and XFM Techniques to the Problem of Selenium Speciation in Biological Systems" Nutrients 5, no. 5: 1734-1756. https://doi.org/10.3390/nu5051734