Dyslipidemia in Obesity: Mechanisms and Potential Targets

{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Overview of Lipoprotein Metabolism
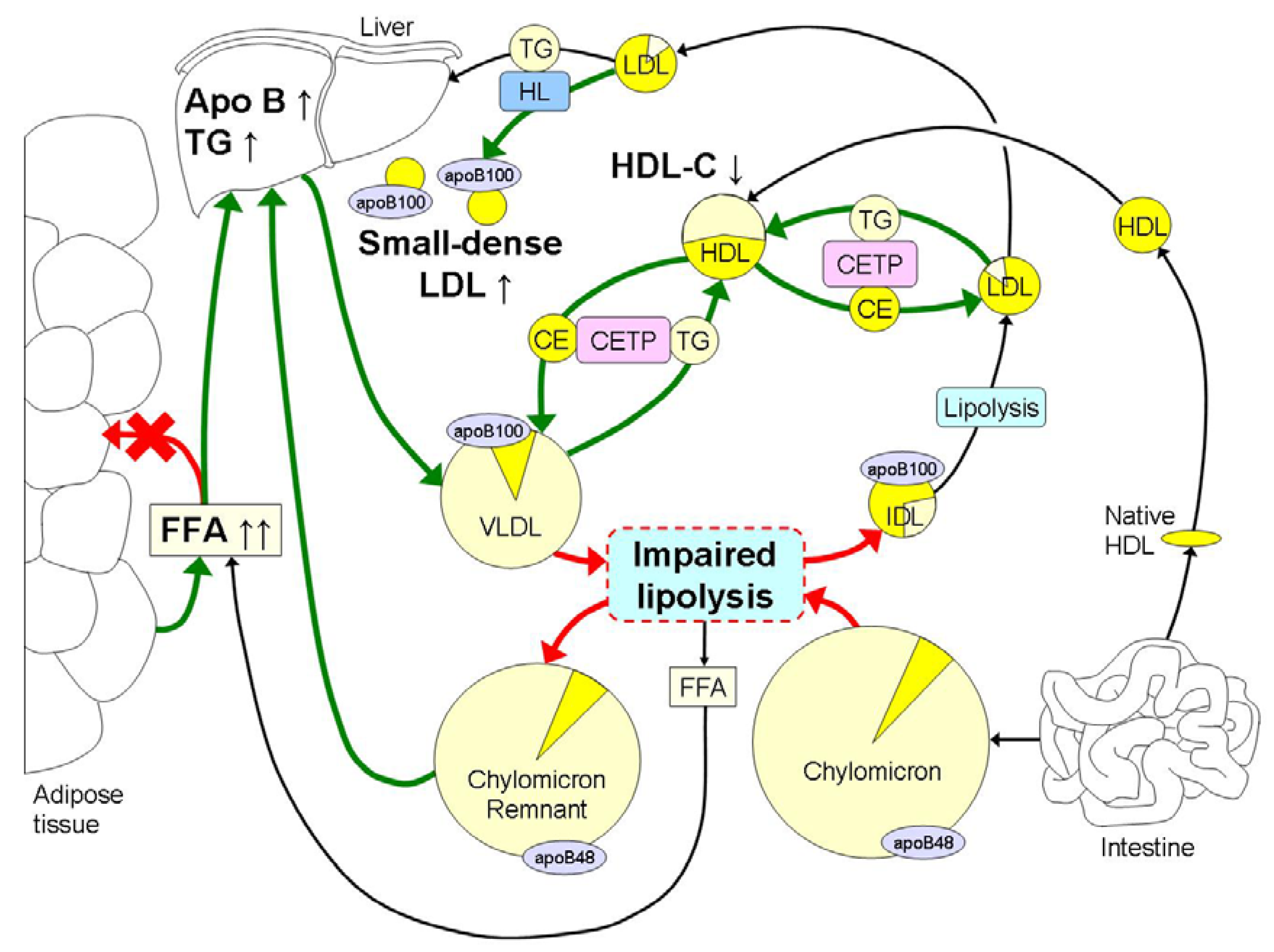
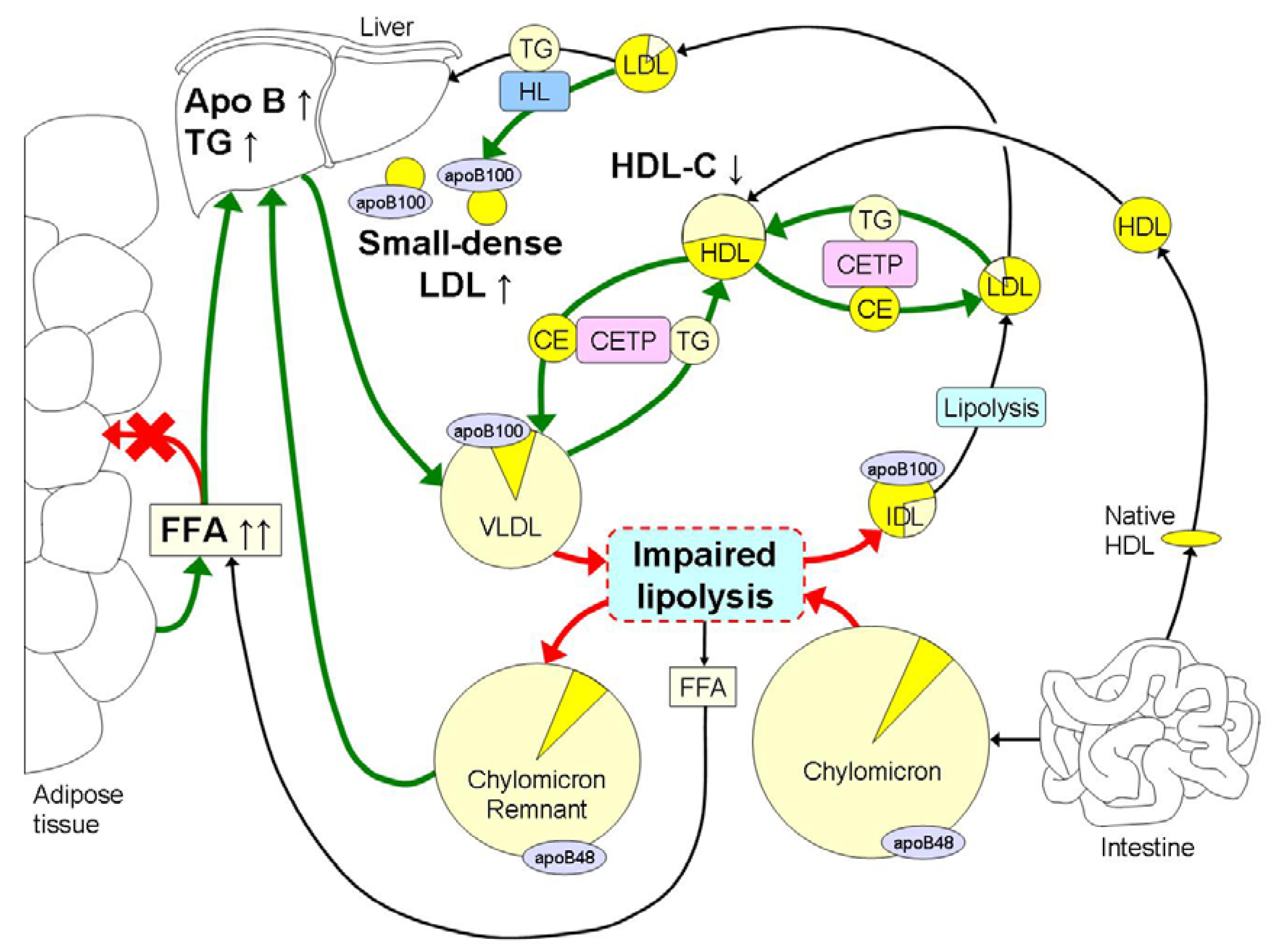
3. Obesity Induced Changes in Lipoprotein Metabolism and Atherogenic Effects
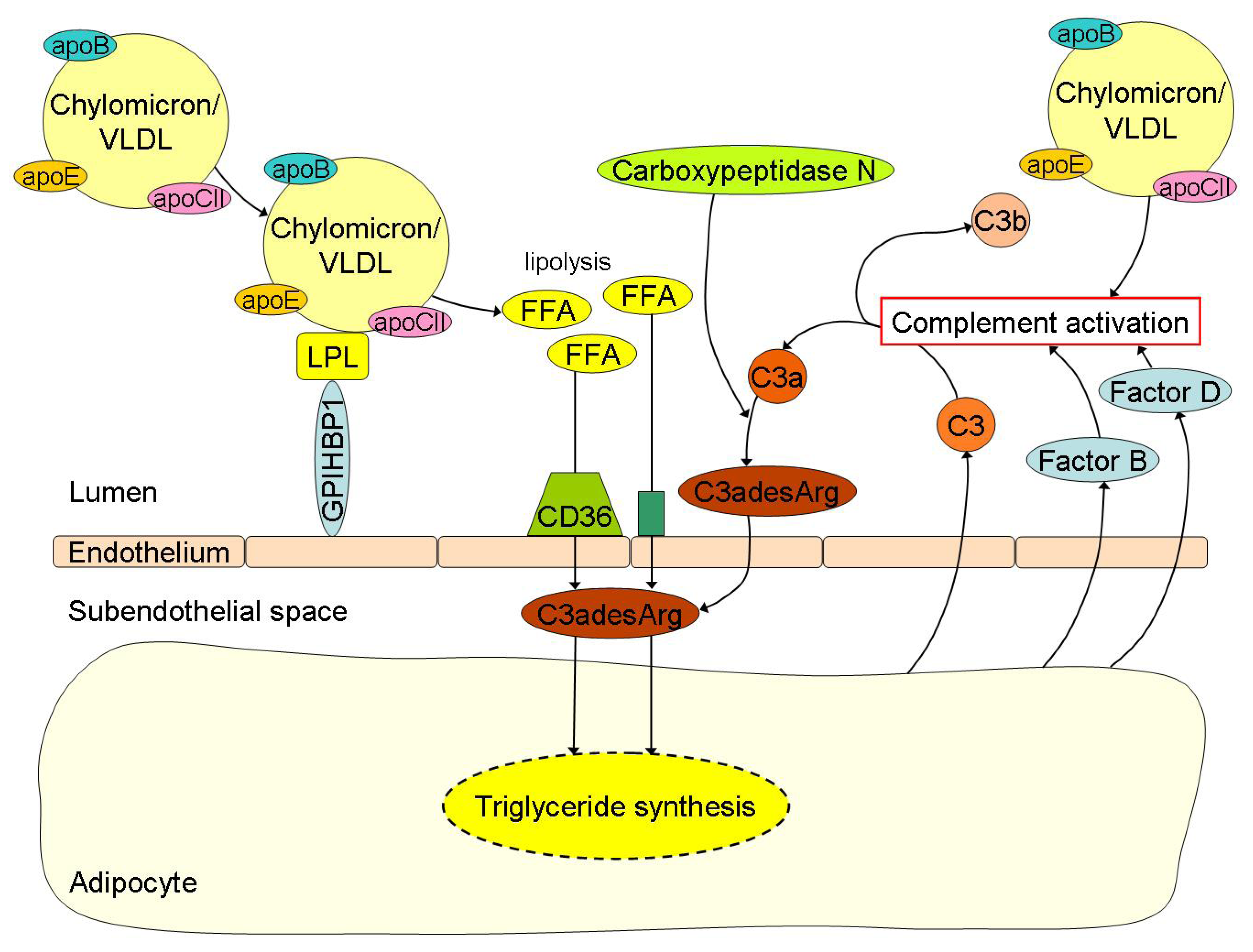
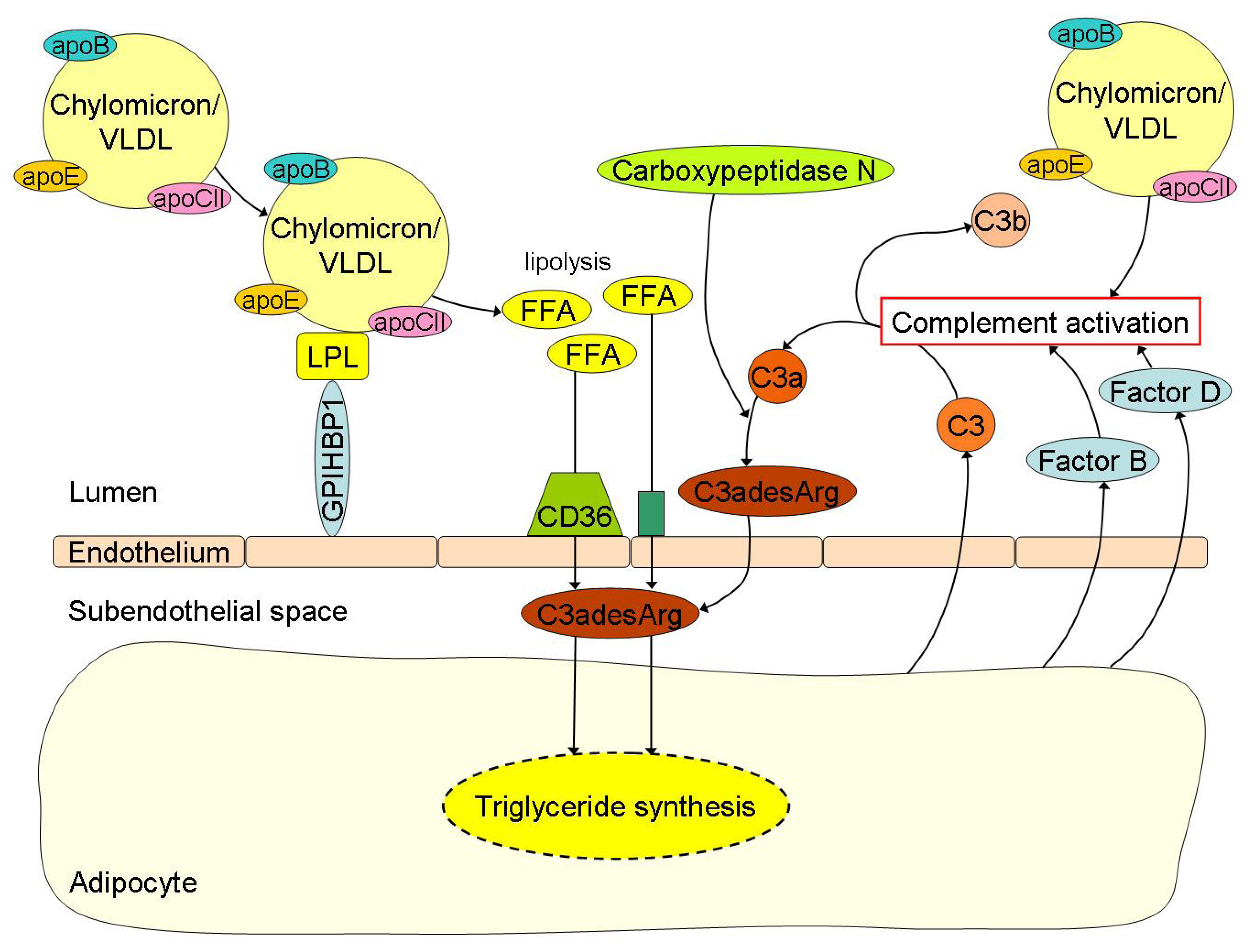
4. Interplay between FFA Metabolism and Inflammation in Obesity: Crossroad between Innate Immunity and Lipid Metabolism
5. Lifestyle Interventions for Dyslipidemia in Obesity
6. Lipid Targets and the Pharmacological Treatment of Dyslipidemia in Obesity
7. Conclusions
Conflict of Interests
References
- Knight, J.A. Diseases and disorders associated with excess body weight. Ann. Clin. Lab Sci. 2011, 41, 107–121. [Google Scholar]
- Flock, M.R.; Green, M.H.; Kris-Etherton, P.M. Effects of adiposity on plasma lipid response to reductions in dietary saturated fatty acids and cholesterol. Adv. Nutr. 2011, 2, 261–274. [Google Scholar] [CrossRef]
- Boden, G. Obesity, insulin resistance and free fatty acids. Curr. Opin. Endocrinol. Diabetes Obes. 2011, 18, 139–143. [Google Scholar] [CrossRef]
- Zalesin, K.C.; Franklin, B.A.; Miller, W.M.; Peterson, E.D.; McCullough, P.A. Impact of obesity on cardiovascular disease. Med. Clin. North. Am. 2011, 95, 919–937. [Google Scholar] [CrossRef]
- Castro Cabezas, M.; Elte, J.W. Farewell to the metabolic syndrome? Not too soon. Atherosclerosis 2009, 204, 348–349; author reply 350–351. [Google Scholar]
- Franssen, R.; Monajemi, H.; Stroes, E.S.; Kastelein, J.J. Obesity and dyslipidemia. Med. Clin. North. Am. 2011, 95, 893–902. [Google Scholar] [CrossRef]
- Wang, H.; Peng, D.Q. New insights into the mechanism of low high-density lipoprotein cholesterol in obesity. Lipids Health Dis. 2011, 10. [Google Scholar] [CrossRef]
- Pan, X.; Hussain, M.M. Gut triglyceride production. Biochim. Biophys. Acta 2011, 1821, 727–735. [Google Scholar]
- Altmann, S.W.; Davis, H.R., Jr.; Zhu, L.J.; Yao, X.; Hoos, L.M.; Tetzloff, G.; Iyer, S.P.; Maguire, M.; Golovko, A.; Zeng, M.; et al. Niemann-Pick C1 Like 1 protein is critical for intestinal cholesterol absorption. Science 2004, 303, 1201–1204. [Google Scholar] [CrossRef]
- Davis, H.R., Jr.; Zhu, L.J.; Hoos, L.M.; Tetzloff, G.; Maguire, M.; Liu, J.; Yao, X.; Iyer, S.P.; Lam, M.H.; Lund, E.G.; et al. Niemann-Pick C1 Like 1 (NPC1L1) is the intestinal phytosterol and cholesterol transporter and a key modulator of whole-body cholesterol homeostasis. J. Biol. Chem. 2004, 279, 33586–33592. [Google Scholar] [CrossRef]
- Klop, B.; Jukema, J.W.; Rabelink, T.J.; Castro Cabezas, M. A physician’s guide for the management of hypertriglyceridemia: The etiology of hypertriglyceridemia determines treatment strategy. Panminerva Med. 2012, 54, 91–103. [Google Scholar]
- Innerarity, T.L.; Young, S.G.; Poksay, K.S.; Mahley, R.W.; Smith, R.S.; Milne, R.W.; Marcel, Y.L.; Weisgraber, K.H. Structural relationship of human apolipoprotein B48 to apolipoprotein B100. J. Clin. Invest. 1987, 80, 1794–1798. [Google Scholar] [CrossRef]
- Goldberg, I.J.; Eckel, R.H.; Abumrad, N.A. Regulation of fatty acid uptake into tissues: lipoprotein lipase- and CD36-mediated pathways. J. Lipid Res. 2009, 50, S86–S90. [Google Scholar] [CrossRef]
- Dallinga-Thie, G.M.; Franssen, R.; Mooij, H.L.; Visser, M.E.; Hassing, H.C.; Peelman, F.; Kastelein, J.J.; Peterfy, M.; Nieuwdorp, M. The metabolism of triglyceride-rich lipoproteins revisited: new players, new insight. Atherosclerosis 2010, 211, 1–8. [Google Scholar] [CrossRef]
- Davies, B.S.; Beigneux, A.P.; Barnes, R.H., II; Tu, Y.; Gin, P.; Weinstein, M.M.; Nobumori, C.; Nyren, R.; Goldberg, I.; Olivecrona, G.; et al. GPIHBP1 is responsible for the entry of lipoprotein lipase into capillaries. Cell Metab. 2010, 12, 42–52. [Google Scholar] [CrossRef]
- Davies, B.S.; Beigneux, A.P.; Fong, L.G.; Young, S.G. New wrinkles in lipoprotein lipase biology. Curr. Opin. Lipidol. 2012, 23, 35–42. [Google Scholar] [CrossRef]
- Karpe, F.; Dickmann, J.R.; Frayn, K.N. Fatty acids, obesity, and insulin resistance: Time for a reevaluation. Diabetes 2011, 60, 2441–2449. [Google Scholar] [CrossRef]
- McQuaid, S.E.; Hodson, L.; Neville, M.J.; Dennis, A.L.; Cheeseman, J.; Humphreys, S.M.; Ruge, T.; Gilbert, M.; Fielding, B.A.; Frayn, K.N.; et al. Downregulation of adipose tissue fatty acid trafficking in obesity: A driver for ectopic fat deposition? Diabetes 2011, 60, 47–55. [Google Scholar] [CrossRef]
- Ooi, E.M.; Barrett, P.H.; Chan, D.C.; Watts, G.F. Apolipoprotein C-III: Understanding an emerging cardiovascular risk factor. Clin. Sci. (Lond.) 2008, 114, 611–624. [Google Scholar] [CrossRef]
- Brunzell, J.D.; Hazzard, W.R.; Porte, D., Jr.; Bierman, E.L. Evidence for a common, saturable, triglyceride removal mechanism for chylomicrons and very low density lipoproteins in man. J. Clin. Invest. 1973, 52, 1578–1585. [Google Scholar] [CrossRef]
- Baldo, A.; Sniderman, A.D.; St-Luce, S.; Avramoglu, R.K.; Maslowska, M.; Hoang, B.; Monge, J.C.; Bell, A.; Mulay, S.; Cianflone, K. The adipsin-acylation stimulating protein system and regulation of intracellular triglyceride synthesis. J. Clin. Invest. 1993, 92, 1543–1547. [Google Scholar] [CrossRef]
- Germinario, R.; Sniderman, A.D.; Manuel, S.; Lefebvre, S.P.; Baldo, A.; Cianflone, K. Coordinate regulation of triacylglycerol synthesis and glucose transport by acylation-stimulating protein. Metabolism 1993, 42, 574–580. [Google Scholar] [CrossRef]
- Abumrad, N.A.; Davidson, N.O. Role of the gut in lipid homeostasis. Physiol. Rev. 2012, 92, 1061–1085. [Google Scholar] [CrossRef]
- Evans, K.; Burdge, G.C.; Wootton, S.A.; Clark, M.L.; Frayn, K.N. Regulation of dietary fatty acid entrapment in subcutaneous adipose tissue and skeletal muscle. Diabetes 2002, 51, 2684–2690. [Google Scholar] [CrossRef]
- Mahley, R.W.; Ji, Z.S. Remnant lipoprotein metabolism: key pathways involving cell-surface heparan sulfate proteoglycans and apolipoprotein E. J. Lipid Res. 1999, 40, 1–16. [Google Scholar]
- Mahley, R.W.; Huang, Y.; Rall, S.C., Jr. Pathogenesis of type III hyperlipoproteinemia (dysbetalipoproteinemia). Questions, quandaries, and paradoxes. J. Lipid Res. 1999, 40, 1933–1949. [Google Scholar]
- Sultan, F.; Lagrange, D.; Jansen, H.; Griglio, S. Inhibition of hepatic lipase activity impairs chylomicron remnant-removal in rats. Biochim. Biophys. Acta 1990, 1042, 150–152. [Google Scholar] [CrossRef]
- Kowal, R.C.; Herz, J.; Goldstein, J.L.; Esser, V.; Brown, M.S. Low density lipoprotein receptor-related protein mediates uptake of cholesteryl esters derived from apoprotein E-enriched lipoproteins. Proc. Natl. Acad. Sci. USA 1989, 86, 5810–5814. [Google Scholar] [CrossRef]
- Hussain, M.M.; Maxfield, F.R.; Mas-Oliva, J.; Tabas, I.; Ji, Z.S.; Innerarity, T.L.; Mahley, R.W. Clearance of chylomicron remnants by the low density lipoprotein receptor-related protein/alpha 2-macroglobulin receptor. J. Biol. Chem. 1991, 266, 13936–13940. [Google Scholar]
- Beisiegel, U.; Weber, W.; Bengtsson-Olivecrona, G. Lipoprotein lipase enhances the binding of chylomicrons to low density lipoprotein receptor-related protein. Proc. Natl. Acad. Sci. USA 1991, 88, 8342–8346. [Google Scholar] [CrossRef]
- Goldstein, J.L.; Brown, M.S. The LDL receptor. Arterioscler. Thromb. Vasc. Biol. 2009, 29, 431–438. [Google Scholar] [CrossRef]
- Lambert, G.; Sjouke, B.; Choque, B.; Kastelein, J.J.; Hovingh, G.K. The PCSK9 decade: Thematic Review Series: New Lipid and Lipoprotein Targets for the Treatment of Cardiometabolic Diseases. J. Lipid Res. 2012, 53, 2515–2524. [Google Scholar] [CrossRef]
- Raal, F.; Scott, R.; Somaratne, R.; Bridges, I.; Li, G.; Wasserman, S.M.; Stein, E.A. Low-density lipoprotein cholesterol-lowering effects of AMG 145, a monoclonal antibody to proprotein convertase subtilisin/kexin type 9 serine protease in patients with heterozygous familial hypercholesterolemia: The reduction of LDL-C with PCSK9 inhibition in heterozygous familial hypercholesterolemia disorder (RUTHERFORD) randomized trial. Circulation 2012, 126, 2408–2417. [Google Scholar] [CrossRef]
- Patsch, J.R.; Miesenbock, G.; Hopferwieser, T.; Muhlberger, V.; Knapp, E.; Dunn, J.K.; Gotto, A.M., Jr.; Patsch, W. Relation of triglyceride metabolism and coronary artery disease. Studies in the postprandial state. Arterioscler. Thromb. 1992, 12, 1336–1345. [Google Scholar] [CrossRef]
- Engelberg, H. Serum lipemia: An overlooked cause of tissue hypoxia. Cardiology 1983, 70, 273–279. [Google Scholar] [CrossRef]
- Simons, L.A.; Dwyer, T.; Simons, J.; Bernstein, L.; Mock, P.; Poonia, N.S.; Balasubramaniam, S.; Baron, D.; Branson, J.; Morgan, J.; et al. Chylomicrons and chylomicron remnants in coronary artery disease: A case-control study. Atherosclerosis 1987, 65, 181–189. [Google Scholar] [CrossRef]
- Ryu, J.E.; Howard, G.; Craven, T.E.; Bond, M.G.; Hagaman, A.P.; Crouse, J.R., III. Postprandial triglyceridemia and carotid atherosclerosis in middle-aged subjects. Stroke 1992, 23, 823–828. [Google Scholar] [CrossRef]
- Karpe, F.; Steiner, G.; Uffelman, K.; Olivecrona, T.; Hamsten, A. Postprandial lipoproteins and progression of coronary atherosclerosis. Atherosclerosis 1994, 106, 83–97. [Google Scholar] [CrossRef]
- Meyer, E.; Westerveld, H.T.; de Ruyter-Meijstek, F.C.; van Greevenbroek, M.M.; Rienks, R.; van Rijn, H.J.; Erkelens, D.W.; de Bruin, T.W. Abnormal postprandial apolipoprotein B-48 and triglyceride responses in normolipidemic women with greater than 70% stenotic coronary artery disease: A case-control study. Atherosclerosis 1996, 124, 221–235. [Google Scholar] [CrossRef]
- Groot, P.H.; van Stiphout, W.A.; Krauss, X.H.; Jansen, H.; van Tol, A.; van Ramshorst, E.; Chin-On, S.; Hofman, A.; Cresswell, S.R.; Havekes, L. Postprandial lipoprotein metabolism in normolipidemic men with and without coronary artery disease. Arterioscler. Thromb. 1991, 11, 653–662. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Jones, J.; Blaner, W.S.; Thomas, A.; Karmally, W.; Fields, L.; Blood, D.; Begg, M.D. Association of postprandial triglyceride and retinyl palmitate responses with newly diagnosed exercise-induced myocardial ischemia in middle-aged men and women. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 1829–1838. [Google Scholar] [CrossRef]
- Sakata, K.; Miho, N.; Shirotani, M.; Yoshida, H.; Takada, Y.; Takada, A. Remnant-like particle cholesterol is a major risk factor for myocardial infarction in vasospastic angina with nearly normal coronary artery. Atherosclerosis 1998, 136, 225–231. [Google Scholar] [CrossRef]
- Ellsworth, J.L.; Fong, L.G.; Kraemer, F.B.; Cooper, A.D. Differences in the processing of chylomicron remnants and β-VLDL by macrophages. J. Lipid Res. 1990, 31, 1399–1411. [Google Scholar]
- Genest, J.; Sniderman, A.; Cianflone, K.; Teng, B.; Wacholder, S.; Marcel, Y.; Kwiterovich, P., Jr. Hyperapobetalipoproteinemia. Plasma lipoprotein responses to oral fat load. Arteriosclerosis 1986, 6, 297–304. [Google Scholar] [CrossRef]
- Castro Cabezas, M.; de Bruin, T.W.; Jansen, H.; Kock, L.A.; Kortlandt, W.; Erkelens, D.W. Impaired chylomicron remnant clearance in familial combined hyperlipidemia. Arterioscler. Thromb. 1993, 13, 804–814. [Google Scholar] [CrossRef]
- Castro Cabezas, M.; de Bruin, T.W.; de Valk, H.W.; Shoulders, C.C.; Jansen, H.; Willem Erkelens, D. Impaired fatty acid metabolism in familial combined hyperlipidemia. A mechanism associating hepatic apolipoprotein B overproduction and insulin resistance. J. Clin. Invest. 1993, 92, 160–168. [Google Scholar] [CrossRef]
- Castro Cabezas, M.; de Bruin, T.W.; Kock, L.A.; Kortlandt, W.; Van Linde-Sibenius Trip, M.; Jansen, H.; Erkelens, D.W. Simvastatin improves chylomicron remnant removal in familial combined hyperlipidemia without changing chylomicron conversion. Metabolism 1993, 42, 497–503. [Google Scholar] [CrossRef]
- Capell, W.H.; Zambon, A.; Austin, M.A.; Brunzell, J.D.; Hokanson, J.E. Compositional differences of LDL particles in normal subjects with LDL subclass phenotype A and LDL subclass phenotype B. Arterioscler Thromb. Vasc. Biol. 1996, 16, 1040–1046. [Google Scholar] [CrossRef]
- Hokanson, J.E.; Krauss, R.M.; Albers, J.J.; Austin, M.A.; Brunzell, J.D. LDL physical and chemical properties in familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 1995, 15, 452–459. [Google Scholar] [CrossRef]
- Clemente-Postigo, M.; Queipo-Ortuno, M.I.; Fernandez-Garcia, D.; Gomez-Huelgas, R.; Tinahones, F.J.; Cardona, F. Adipose tissue gene expression of factors related to lipid processing in obesity. PLoS One 2011, 6, e24783. [Google Scholar] [CrossRef]
- Peterson, J.; Bihain, B.E.; Bengtsson-Olivecrona, G.; Deckelbaum, R.J.; Carpentier, Y.A.; Olivecrona, T. Fatty acid control of lipoprotein lipase: a link between energy metabolism and lipid transport. Proc. Natl. Acad. Sci. USA 1990, 87, 909–913. [Google Scholar] [CrossRef]
- Karpe, F.; Olivecrona, T.; Walldius, G.; Hamsten, A. Lipoprotein lipase in plasma after an oral fat load: Relation to free fatty acids. J. Lipid Res. 1992, 33, 975–984. [Google Scholar]
- Tchernof, A.; Lamarche, B.; Prud’Homme, D.; Nadeau, A.; Moorjani, S.; Labrie, F.; Lupien, P.J.; Despres, J.P. The dense LDL phenotype. Association with plasma lipoprotein levels, visceral obesity, and hyperinsulinemia in men. Diabetes Care 1996, 19, 629–637. [Google Scholar]
- Packard, C.J. Triacylglycerol-rich lipoproteins and the generation of small, dense low-density lipoprotein. Biochem. Soc. Trans. 2003, 31, 1066–1069. [Google Scholar] [CrossRef]
- Klop, B.; Proctor, S.D.; Mamo, J.C.; Botham, K.M.; Castro Cabezas, M. Understanding postprandial inflammation and its relationship to lifestyle behaviour and metabolic diseases. Int. J. Vasc. Med. 2012, 2012, 947417. [Google Scholar] [CrossRef]
- Proctor, S.D.; Mamo, J.C. Intimal retention of cholesterol derived from apolipoprotein B100- and apolipoprotein B48-containing lipoproteins in carotid arteries of Watanabe heritable hyperlipidemic rabbits. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1595–1600. [Google Scholar] [CrossRef]
- Proctor, S.D.; Vine, D.F.; Mamo, J.C. Arterial retention of apolipoprotein B(48)- and B(100)-containing lipoproteins in atherogenesis. Curr. Opin. Lipidol. 2002, 13, 461–470. [Google Scholar] [CrossRef]
- Tabas, I.; Williams, K.J.; Boren, J. Subendothelial lipoprotein retention as the initiating process in atherosclerosis: Update and therapeutic implications. Circulation 2007, 116, 1832–1844. [Google Scholar] [CrossRef]
- Klop, B.; Castro Cabezas, M. Chylomicrons: A key biomarker and risk factor for cardiovascular disease and for the understanding of obesity. Curr. Cardiovasc. Risk. Rep. 2012, 6, 27–34. [Google Scholar] [CrossRef]
- Subramanian, S.; Chait, A. Hypertriglyceridemia secondary to obesity and diabetes. Biochim. Biophys. Acta 2012, 1821, 819–825. [Google Scholar] [CrossRef]
- Pacifico, L.; Anania, C.; Osborn, J.F.; Ferraro, F.; Bonci, E.; Olivero, E.; Chiesa, C. Low 25(OH)D3 levels are associated with total adiposity, metabolic syndrome, and hypertension in Caucasian children and adolescents. Eur. J. Endocrinol. 2011, 165, 603–611. [Google Scholar] [CrossRef]
- Proctor, S.D.; Vine, D.F.; Mamo, J.C. Arterial permeability and efflux of apolipoprotein B-containing lipoproteins assessed by in situ perfusion and three-dimensional quantitative confocal microscopy. Arterioscler. Thromb. Vasc. Biol. 2004, 24, 2162–2167. [Google Scholar] [CrossRef]
- Watts, G.F.; Chan, D.C.; Barrett, P.H.; Martins, I.J.; Redgrave, T.G. Preliminary experience with a new stable isotope breath test for chylomicron remnant metabolism: A study in central obesity. Clin. Sci. (Lond.) 2001, 101, 683–690. [Google Scholar] [CrossRef]
- Taskinen, M.R.; Adiels, M.; Westerbacka, J.; Soderlund, S.; Kahri, J.; Lundbom, N.; Lundbom, J.; Hakkarainen, A.; Olofsson, S.O.; Orho-Melander, M.; et al. Dual metabolic defects are required to produce hypertriglyceridemia in obese subjects. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 2144–2150. [Google Scholar] [CrossRef]
- Caron, S.; Verrijken, A.; Mertens, I.; Samanez, C.H.; Mautino, G.; Haas, J.T.; Duran-Sandoval, D.; Prawitt, J.; Francque, S.; Vallez, E.; et al. Transcriptional activation of apolipoprotein CIII expression by glucose may contribute to diabetic dyslipidemia. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 513–519. [Google Scholar] [CrossRef]
- Mamo, J.C.; Watts, G.F.; Barrett, P.H.; Smith, D.; James, A.P.; Pal, S. Postprandial dyslipidemia in men with visceral obesity: An effect of reduced LDL receptor expression? Am. J. Physiol. Endocrinol. Metab. 2001, 281, E626–E632. [Google Scholar]
- Castro Cabezas, M.; Erkelens, D.W. The direct way from gut to vessel wall: Atheroinitiation. Eur. J. Clin. Invest. 1998, 28, 504–505. [Google Scholar] [CrossRef]
- Jorgensen, A.B.; Frikke-Schmidt, R.; West, A.S.; Grande, P.; Nordestgaard, B.G.; Tybjaerg-Hansen, A. Genetically elevated non-fasting triglycerides and calculated remnant cholesterol as causal risk factors for myocardial infarction. Eur. Heart J. 2012. [Google Scholar] [CrossRef]
- Senti, M.; Nogues, X.; Pedro-Botet, J.; Rubies-Prat, J.; Vidal-Barraquer, F. Lipoprotein profile in men with peripheral vascular disease. Role of intermediate density lipoproteins and apoprotein E phenotypes. Circulation 1992, 85, 30–36. [Google Scholar] [CrossRef]
- Vogel, R.A.; Corretti, M.C.; Plotnick, G.D. Effect of a single high-fat meal on endothelial function in healthy subjects. Am. J. Cardiol. 1997, 79, 350–354. [Google Scholar] [CrossRef]
- Lundman, P.; Eriksson, M.; Schenck-Gustafsson, K.; Karpe, F.; Tornvall, P. Transient triglyceridemia decreases vascular reactivity in young, healthy men without risk factors for coronary heart disease. Circulation 1997, 96, 3266–3268. [Google Scholar] [CrossRef]
- Steinberg, H.O.; Tarshoby, M.; Monestel, R.; Hook, G.; Cronin, J.; Johnson, A.; Bayazeed, B.; Baron, A.D. Elevated circulating free fatty acid levels impair endothelium-dependent vasodilation. J. Clin. Invest. 1997, 100, 1230–1239. [Google Scholar] [CrossRef]
- Van Oostrom, A.J.; van Wijk, J.; Castro Cabezas, M. Lipaemia, inflammation and atherosclerosis: Novel opportunities in the understanding and treatment of atherosclerosis. Drugs 2004, 64, 19–41. [Google Scholar] [CrossRef]
- Alipour, A.; Elte, J.W.; van Zaanen, H.C.; Rietveld, A.P.; Castro Cabezas, M. Novel aspects of postprandial lipemia in relation to atherosclerosis. Atheroscler. Suppl. 2008, 9, 39–44. [Google Scholar]
- Couillard, C.; Bergeron, N.; Prud’homme, D.; Bergeron, J.; Tremblay, A.; Bouchard, C.; Mauriege, P.; Despres, J.P. Postprandial triglyceride response in visceral obesity in men. Diabetes 1998, 47, 953–960. [Google Scholar] [CrossRef]
- Taira, K.; Hikita, M.; Kobayashi, J.; Bujo, H.; Takahashi, K.; Murano, S.; Morisaki, N.; Saito, Y. Delayed post-prandial lipid metabolism in subjects with intra-abdominal visceral fat accumulation. Eur. J. Clin. Invest. 1999, 29, 301–308. [Google Scholar] [CrossRef]
- Su, J.W.; Nzekwu, M.M.; Cabezas, M.C.; Redgrave, T.; Proctor, S.D. Methods to assess impaired post-prandial metabolism and the impact for early detection of cardiovascular disease risk. Eur. J. Clin. Invest. 2009, 39, 741–754. [Google Scholar] [CrossRef]
- Castro Cabezas, M.; Halkes, C.J.; Meijssen, S.; van Oostrom, A.J.; Erkelens, D.W. Diurnal triglyceride profiles: A novel approach to study triglyceride changes. Atherosclerosis 2001, 155, 219–228. [Google Scholar] [CrossRef]
- Van Oostrom, A.J.; Castro Cabezas, M.; Ribalta, J.; Masana, L.; Twickler, T.B.; Remijnse, T.A.; Erkelens, D.W. Diurnal triglyceride profiles in healthy normolipidemic male subjects are associated to insulin sensitivity, body composition and diet. Eur. J. Clin. Invest. 2000, 30, 964–971. [Google Scholar] [CrossRef]
- Halkes, C.J.; Castro Cabezas, M.; van Wijk, J.P.; Erkelens, D.W. Gender differences in diurnal triglyceridemia in lean and overweight subjects. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 1767–1774. [Google Scholar] [CrossRef]
- Lean, M.E.; Han, T.S.; Morrison, C.E. Waist circumference as a measure for indicating need for weight management. Br. Med. J. 1995, 311, 158–161. [Google Scholar] [CrossRef]
- Van Gaal, L.F.; Mertens, I.L.; de Block, C.E. Mechanisms linking obesity with cardiovascular disease. Nature 2006, 444, 875–880. [Google Scholar]
- Deeb, S.S.; Zambon, A.; Carr, M.C.; Ayyobi, A.F.; Brunzell, J.D. Hepatic lipase and dyslipidemia: Interactions among genetic variants, obesity, gender, and diet. J. Lipid Res. 2003, 44, 1279–1286. [Google Scholar] [CrossRef]
- Hill, M.J.; Metcalfe, D.; McTernan, P.G. Obesity and diabetes: Lipids, “nowhere to run to”. Clin. Sci. (Lond.) 2009, 116, 113–123. [Google Scholar] [CrossRef]
- Bjorntorp, P.; Bergman, H.; Varnauskas, E. Plasma free fatty acid turnover rate in obesity. Acta Med. Scand. 1969, 185, 351–356. [Google Scholar] [CrossRef]
- Mook, S.; Halkes, C.C.; Bilecen, S.; Castro Cabezas, M. In vivo regulation of plasma free fatty acids in insulin resistance. Metabolism 2004, 53, 1197–1201. [Google Scholar] [CrossRef]
- Van Oostrom, A.J.; van Dijk, H.; Verseyden, C.; Sniderman, A.D.; Cianflone, K.; Rabelink, T.J.; Castro Cabezas, M. Addition of glucose to an oral fat load reduces postprandial free fatty acids and prevents the postprandial increase in complement component 3. Am. J. Clin. Nutr. 2004, 79, 510–515. [Google Scholar]
- Capurso, C.; Capurso, A. From excess adiposity to insulin resistance: The role of free fatty acids. Vascul. Pharmacol. 2012, 57, 91–97. [Google Scholar] [CrossRef]
- Lottenberg, A.M.; Afonso Mda, S.; Lavrador, M.S.; Machado, R.M.; Nakandakare, E.R. The role of dietary fatty acids in the pathology of metabolic syndrome. J. Nutr. Biochem. 2012, 23, 1027–1040. [Google Scholar] [CrossRef]
- Sears, B.; Ricordi, C. Role of fatty acids and polyphenols in inflammatory gene transcription and their impact on obesity, metabolic syndrome and diabetes. Eur. Rev. Med. Pharmacol. Sci. 2012, 16, 1137–1154. [Google Scholar]
- Kopp, A.; Gross, P.; Falk, W.; Bala, M.; Weigert, J.; Buechler, C.; Neumeier, M.; Scholmerich, J.; Schaffler, A. Fatty acids as metabolic mediators in innate immunity. Eur. J. Clin. Invest. 2009, 39, 924–933. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Maslowska, M.; Cianflone, K. Of mice and men (and women) and the acylation-stimulating protein pathway. Curr. Opin. Lipidol. 2000, 11, 291–296. [Google Scholar] [CrossRef]
- Cianflone, K.; Vu, H.; Walsh, M.; Baldo, A.; Sniderman, A. Metabolic response of Acylation Stimulating Protein to an oral fat load. J. Lipid Res. 1989, 30, 1727–1733. [Google Scholar]
- Cianflone, K.; Maslowska, M. Differentiation-induced production of ASP in human adipocytes. Eur. J. Clin. Invest. 1995, 25, 817–825. [Google Scholar] [CrossRef]
- Maslowska, M.; Scantlebury, T.; Germinario, R.; Cianflone, K. Acute in vitro production of acylation stimulating protein in differentiated human adipocytes. J. Lipid Res. 1997, 38, 1–11. [Google Scholar]
- Saleh, J.; Summers, L.K.; Cianflone, K.; Fielding, B.A.; Sniderman, A.D.; Frayn, K.N. Coordinated release of acylation stimulating protein (ASP) and triacylglycerol clearance by human adipose tissue in vivo in the postprandial period. J. Lipid Res. 1998, 39, 884–891. [Google Scholar]
- Skidgel, R.A. Basic carboxypeptidases: Regulators of peptide hormone activity. Trends Pharmacol. Sci. 1988, 9, 299–304. [Google Scholar] [CrossRef]
- Maslowska, M.; Sniderman, A.D.; Germinario, R.; Cianflone, K. ASP stimulates glucose transport in cultured human adipocytes. Int. J. Obes. Relat. Metab. Disord. 1997, 21, 261–266. [Google Scholar]
- Van Oostrom, A.J.; Alipour, A.; Plokker, T.W.; Sniderman, A.D.; Castro Cabezas, M. The metabolic syndrome in relation to complement component 3 and postprandial lipemia in patients from an outpatient lipid clinic and healthy volunteers. Atherosclerosis 2007, 190, 167–173. [Google Scholar] [CrossRef]
- Volp, A.C.; Barbosa, K.B.; Bressan, J. Triacylglycerols and body fat mass are possible independent predictors of C3 in apparently healthy young Brazilian adults. Nutrition 2012, 28, 544–550. [Google Scholar] [CrossRef]
- Hernandez-Mijares, A.; Banuls, C.; Bellod, L.; Jover, A.; Sola, E.; Morillas, C.; Victor, V.M.; Rocha, M. Effect of weight loss on C3 and C4 components of complement in obese patients. Eur. J. Clin. Invest. 2012, 42, 503–509. [Google Scholar] [CrossRef]
- Meijssen, S.; van Dijk, H.; Verseyden, C.; Erkelens, D.W.; Castro Cabezas, M. Delayed and exaggerated postprandial complement component 3 response in familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 811–816. [Google Scholar] [CrossRef]
- Halkes, C.J.; van Dijk, H.; de Jaegere, P.P.T.; Plokker, H.W.M.; van der Helm, Y.; Erkelens, D.W.; Castro Cabezas, M. Postprandial increase of complement component 3 in normolipidemic patients with coronary artery disease. Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1526–1530. [Google Scholar] [CrossRef]
- Kristiansson, K.; Perola, M.; Tikkanen, E.; Kettunen, J.; Surakka, I.; Havulinna, A.S.; Stancakova, A.; Barnes, C.; Widen, E.; Kajantie, E.; et al. Genome-wide screen for metabolic syndrome susceptibility Loci reveals strong lipid gene contribution but no evidence for common genetic basis for clustering of metabolic syndrome traits. Circ. Cardiovasc. Genet. 2012, 5, 242–249. [Google Scholar] [CrossRef]
- Alipour, A.; van Oostrom, A.J.; Van Wijk, J.P.; Verseyden, C.; Plokker, H.W.; Jukema, J.W.; Rabelink, A.J.; Castro Cabezas, M. Mannose binding lectin deficiency and triglyceride-rich lipoprotein metabolism in normolipidemic subjects. Atherosclerosis 2009, 206, 444–450. [Google Scholar] [CrossRef]
- Meijssen, S.; Castro Cabezas, M.; Twickler, T.B.; Jansen, H.; Erkelens, D.W. In vivo evidence of defective postprandial and postabsorptive free fatty acid metabolism in familial combined hyperlipidemia. J. Lipid Res. 2000, 41, 1096–1102. [Google Scholar]
- Halkes, C.J.; van Dijk, H.; Verseyden, C.; de Jaegere, P.P.; Plokker, H.W.; Meijssen, S.; Erkelens, D.W.; Castro Cabezas, M. Gender differences in postprandial ketone bodies in normolipidemic subjects and in untreated patients with familial combined hyperlipidemia. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1875–1880. [Google Scholar] [CrossRef]
- Castro Cabezas, M.; van Wijk, J.P.; Elte, J.W.; Klop, B. Effects of metformin on the regulation of free Fatty acids in insulin resistance: A double-blind, placebo-controlled study. J. Nutr. Metab. 2012, 2012. [Google Scholar] [CrossRef]
- Lopez-Miranda, J.; Williams, C.; Lairon, D. Dietary, physiological, genetic and pathological influences on postprandial lipid metabolism. Br. J. Nutr. 2007, 98, 458–473. [Google Scholar] [CrossRef]
- De Ruyter, J.C.; Olthof, M.R.; Seidell, J.C.; Katan, M.B. A trial of sugar-free or sugar-sweetened beverages and body weight in children. N. Engl. J. Med. 2012, 367, 1397–1406. [Google Scholar] [CrossRef]
- Patalay, M.; Lofgren, I.E.; Freake, H.C.; Koo, S.I.; Fernandez, M.L. The lowering of plasma lipids following a weight reduction program is related to increased expression of the LDL receptor and lipoprotein lipase. J. Nutr. 2005, 135, 735–739. [Google Scholar]
- Laimer, M.W.; Engl, J.; Tschoner, A.; Kaser, S.; Ritsch, A.; Tatarczyk, T.; Rauchenzauner, M.; Weiss, H.; Aigner, F.; Patsch, J.R.; et al. Effects of weight loss on lipid transfer proteins in morbidly obese women. Lipids 2009, 44, 1125–1130. [Google Scholar] [CrossRef]
- Wang, Y.; Snel, M.; Jonker, J.T.; Hammer, S.; Lamb, H.J.; de Roos, A.; Meinders, A.E.; Pijl, H.; Romijn, J.A.; Smit, J.W.; et al. Prolonged caloric restriction in obese patients with type 2 diabetes mellitus decreases plasma CETP and increases apolipoprotein AI levels without improving the cholesterol efflux properties of HDL. Diabetes Care 2011, 34, 2576–2580. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F.; Barrett, P.H.; Mamo, J.C.; Redgrave, T.G. Markers of triglyceride-rich lipoprotein remnant metabolism in visceral obesity. Clin. Chem. 2002, 48, 278–283. [Google Scholar]
- James, A.P.; Watts, G.F.; Barrett, P.H.; Smith, D.; Pal, S.; Chan, D.C.; Mamo, J.C. Effect of weight loss on postprandial lipemia and low-density lipoprotein receptor binding in overweight men. Metabolism 2003, 52, 136–141. [Google Scholar] [CrossRef]
- Roberts, C.K.; Barnard, R.J.; Liang, K.H.; Vaziri, N.D. Effect of diet on adipose tissue and skeletal muscle VLDL receptor and LPL: Implications for obesity and hyperlipidemia. Atherosclerosis 2002, 161, 133–141. [Google Scholar] [CrossRef]
- Maraki, M.I.; Aggelopoulou, N.; Christodoulou, N.; Anastasiou, C.A.; Toutouza, M.; Panagiotakos, D.B.; Kavouras, S.A.; Magkos, F.; Sidossis, L.S. Lifestyle intervention leading to moderate weight loss normalizes postprandial triacylglycerolemia despite persisting obesity. Obesity (Silver Spring) 2011, 19, 968–976. [Google Scholar] [CrossRef]
- Cruz-Teno, C.; Perez-Martinez, P.; Delgado-Lista, J.; Yubero-Serrano, E.M.; Garcia-Rios, A.; Marin, C.; Gomez, P.; Jimenez-Gomez, Y.; Camargo, A.; Rodriguez-Cantalejo, F.; et al. Detary fat modifies the postprandial inflammatory state in subjects with metabolic syndrome: The LIPGENE study. Mol. Nutr. Food Res. 2012, 56, 854–865. [Google Scholar] [CrossRef]
- Yin, R.X.; Wu, D.F.; Miao, L.; Aung, L.H.; Cao, X.L.; Yan, T.T.; Long, X.J.; Liu, W.Y.; Zhang, L.; Li, M. Several genetic polymorphisms interact with overweight/obesity to influence serum lipid levels. Cardiovasc. Diabetol. 2012, 11. [Google Scholar] [CrossRef]
- Corella, D.; Peloso, G.; Arnett, D.K.; Demissie, S.; Cupples, L.A.; Tucker, K.; Lai, C.Q.; Parnell, L.D.; Coltell, O.; Lee, Y.C.; et al. APOA2, dietary fat, and body mass index: replication of a gene-diet interaction in 3 independent populations. Arch. Intern. Med. 2009, 169, 1897–1906. [Google Scholar] [CrossRef]
- Lai, C.Q.; Corella, D.; Demissie, S.; Cupples, L.A.; Adiconis, X.; Zhu, Y.; Parnell, L.D.; Tucker, K.L.; Ordovas, J.M. Dietary intake of n-6 fatty acids modulates effect of apolipoprotein A5 gene on plasma fasting triglycerides, remnant lipoprotein concentrations, and lipoprotein particle size: The Framingham Heart Study. Circulation 2006, 113, 2062–2070. [Google Scholar] [CrossRef]
- Sanchez-Moreno, C.; Ordovas, J.M.; Smith, C.E.; Baraza, J.C.; Lee, Y.C.; Garaulet, M. APOA5 gene variation interacts with dietary fat intake to modulate obesity and circulating triglycerides in a Mediterranean population. J. Nutr. 2012, 141, 380–385. [Google Scholar]
- Thomas, T.R.; Horner, K.E.; Langdon, M.M.; Zhang, J.Q.; Krul, E.S.; Sun, G.Y.; Cox, R.H. Effect of exercise and medium-chain fatty acids on postprandial lipemia. J. Appl. Physiol. 2001, 90, 1239–1246. [Google Scholar]
- Ferguson, M.A.; Alderson, N.L.; Trost, S.G.; Essig, D.A.; Burke, J.R.; Durstine, J.L. Effects of four different single exercise sessions on lipids, lipoproteins, and lipoprotein lipase. J. Appl. Physiol. 1998, 85, 1169–1174. [Google Scholar]
- Harrison, M.; Moyna, N.M.; Zderic, T.W.; O’Gorman, D.J.; McCaffrey, N.; Carson, B.P.; Hamilton, M.T. Lipoprotein particle distribution and skeletal muscle lipoprotein lipase activity after acute exercise. Lipids. Health Dis. 2012, 11. [Google Scholar] [CrossRef]
- Slivkoff-Clark, K.M.; James, A.P.; Mamo, J.C. The chronic effects of fish oil with exercise on postprandial lipaemia and chylomicron homeostasis in insulin resistant viscerally obese men. Nutr. Metab. (Lond.) 2012, 9, 9. [Google Scholar] [CrossRef]
- Sullivan, S.; Kirk, E.P.; Mittendorfer, B.; Patterson, B.W.; Klein, S. Randomized trial of exercise effect on intrahepatic triglyceride content and lipid kinetics in nonalcoholic fatty liver disease. Hepatology 2012, 55, 1738–1745. [Google Scholar] [CrossRef]
- Magkos, F. Exercise and fat accumulation in the human liver. Curr. Opin. Lipidol. 2010, 21, 507–517. [Google Scholar] [CrossRef]
- van Herpen, N.A.; Schrauwen-Hinderling, V.B.; Schaart, G.; Mensink, R.P.; Schrauwen, P. Three weeks on a high-fat diet increases intrahepatic lipid accumulation and decreases metabolic flexibility in healthy overweight men. J. Clin. Endocrinol. Metab. 2012, 96, E691–E695. [Google Scholar]
- Mestek, M.L. Physical activity, blood lipids, and lipoproteins. Am. J. Lifestyle Med. 2009, 3, 279–283. [Google Scholar] [CrossRef]
- Thompson, P.D.; Rader, D.J. Does exercise increase HDL cholesterol in those who need it the most? Arterioscler. Thromb. Vasc. Biol. 2001, 21, 1097–1098. [Google Scholar] [CrossRef]
- Maki, K.C.; Pelkman, C.L.; Finocchiaro, E.T.; Kelley, K.M.; Lawless, A.L.; Schild, A.L.; Rains, T.M. Resistant starch from high-amylose maize increases insulin sensitivity in overweight and obese men. J. Nutr. 2012, 142, 717–723. [Google Scholar] [CrossRef]
- Robertson, M.D.; Wright, J.W.; Loizon, E.; Debard, C.; Vidal, H.; Shojaee-Moradie, F.; Russell-Jones, D.; Umpleby, A.M. Insulin-sensitizing effects on muscle and adipose tissue after dietary fiber intake in men and women with metabolic syndrome. J. Clin. Endocrinol. Metab. 2012, 97, 3326–3332. [Google Scholar] [CrossRef]
- Zhou, Y.H.; Ma, X.Q.; Wu, C.; Lu, J.; Zhang, S.S.; Guo, J.; Wu, S.Q.; Ye, X.F.; Xu, J.F.; He, J. Effect of anti-obesity drug on cardiovascular risk factors: A systematic review and meta-analysis of randomized controlled trials. PLoS One 2012, 7, e39062. [Google Scholar] [CrossRef]
- Aron-Wisnewsky, J.; Julia, Z.; Poitou, C.; Bouillot, J.L.; Basdevant, A.; Chapman, M.J.; Clement, K.; Guerin, M. Effect of bariatric surgery-induced weight loss on SR-BI-, ABCG1-, and ABCA1-mediated cellular cholesterol efflux in obese women. J. Clin. Endocrinol. Metab. 2011, 96, 1151–1159. [Google Scholar] [CrossRef]
- Catapano, A.L.; Reiner, Z.; de Backer, G.; Graham, I.; Taskinen, M.R.; Wiklund, O.; Agewall, S.; Alegria, E.; Chapman, M.J.; Durrington, P.; et al. ESC/EAS Guidelines for the management of dyslipidaemias: The Task Force for the management of dyslipidaemias of the European Society of Cardiology (ESC) and the European Atherosclerosis Society (EAS). Atherosclerosis 2011, 217, 1–44. [Google Scholar]
- Kushner, R.F. Clinincal assessment and management of adult obesity. Circulation 2012, 126, 2870–2877. [Google Scholar] [CrossRef]
- Berglund, L.; Brunzell, J.D.; Goldberg, A.C.; Goldberg, I.J.; Sacks, F.; Murad, M.H.; Stalenhoef, A.F. Evaluation and treatment of hypertriglyceridemia: An endocrine society clinical practice guideline. J. Clin. Endocrinol. Metab. 2012, 97, 2969–2989. [Google Scholar] [CrossRef]
- Sniderman, A.D.; Williams, K.; Contois, J.H.; Monroe, H.M.; McQueen, M.J.; de Graaf, J.; Furberg, C.D. A meta-analysis of low-density lipoprotein cholesterol, non-high-density lipoprotein cholesterol, and apolipoprotein B as markers of cardiovascular risk. Circ. Cardiovasc. Qual. Outcomes 2011, 4, 337–345. [Google Scholar] [CrossRef]
- Mora, S.; Glynn, R.J.; Boekholdt, S.M.; Nordestgaard, B.G.; Kastelein, J.J.; Ridker, P.M. On-treatment non-high-density lipoprotein cholesterol, apolipoprotein B, triglycerides, and lipid ratios in relation to residual vascular risk after treatment with potent statin therapy: JUPITER (justification for the use of statins in prevention: An intervention trial evaluating rosuvastatin). J. Am. Coll. Cardiol. 2012, 59, 1521–1528. [Google Scholar]
- Boekholdt, S.M.; Arsenault, B.J.; Mora, S.; Pedersen, T.R.; LaRosa, J.C.; Nestel, P.J.; Simes, R.J.; Durrington, P.; Hitman, G.A.; Welch, K.M.; et al. Association of LDL cholesterol, non-HDL cholesterol, and apolipoprotein B levels with risk of cardiovascular events among patients treated with statins: A meta-analysis. JAMA 2012, 307, 1302–1309. [Google Scholar] [CrossRef]
- Robinson, J.G.; Wang, S.; Jacobson, T.A. Meta-analysis of comparison of effectiveness of lowering apolipoprotein B versus low-density lipoprotein cholesterol and nonhigh-density lipoprotein cholesterol for cardiovascular risk reduction in randomized trials. Am. J. Cardiol 2012, 110, 1468–1476. [Google Scholar] [CrossRef]
- Klop, B.; Cohn, J.S.; van Oostrom, A.J.; van Wijk, J.P.; Birnie, E.; Castro Cabezas, M. Daytime triglyceride variability in men and women with different levels of triglyceridemia. Clin. Chim. Acta 2011, 412, 2183–2189. [Google Scholar] [CrossRef]
- Brunzell, J.D. Clinical practice. Hypertriglyceridemia. N. Engl. J. Med. 2007, 357, 1009–1017. [Google Scholar] [CrossRef]
- Watts, G.F.; Karpe, F. Triglycerides and atherogenic dyslipidaemia: Extending treatment beyond statins in the high-risk cardiovascular patient. Heart 2011, 97, 350–356. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F. Dyslipidaemia in the metabolic syndrome and type 2 diabetes: Pathogenesis, priorities, pharmacotherapies. Expert Opin. Pharmacother. 2011, 12, 13–30. [Google Scholar] [CrossRef]
- Watts, G.F.; Karpe, F. Why, when and how should hypertriglyceridemia be treated in the high-risk cardiovascular patient? Expert. Rev. Cardiovasc. Ther. 2011, 9, 987–997. [Google Scholar] [CrossRef]
- Dujovne, C.A.; Williams, C.D.; Ito, M.K. What combination therapy with a statin, if any, would you recommend? Cur. Atheroscler. Rep. 2011, 13, 12–22. [Google Scholar]
- Rubenfire, M.; Brook, R.D.; Rosenson, R.S. Treating mixed hyperlipidemia and the atherogenic lipid phenotype for prevention of cardiovascular events. Am. J. Med. 2010, 123, 892–898. [Google Scholar] [CrossRef]
- Toth, P.P. Drug treatment of hyperlipidaemia: A guide to the rational use of lipid-lowering drugs. Drugs 2010, 70, 1363–1379. [Google Scholar] [CrossRef]
- Tenenbaum, A.; Motro, M.; Fisman, E.Z.; Tanne, D.; Boyko, V.; Behar, S. Bezafibrate for the secondary prevention of myocardial infarction in patients with metabolic syndrome. Arch. Intern. Me.d 2005, 165, 1154–1160. [Google Scholar]
- Tenkanen, L.; Manttari, M.; Manninen, V. Some coronary risk factors related to the insulin resistance syndrome and treatment with gemfibrozil. Experience from the Helsinki Heart Study. Circulation 1995, 92, 1779–1785. [Google Scholar] [CrossRef]
- Tenkanen, L.; Manttari, M.; Kovanen, P.T.; Virkkunen, H.; Manninen, V. Gemfibrozil in the treatment of dyslipidemia: An 18-year mortality follow-up of the Helsinki Heart Study. Arch. Intern. Med. 2006, 166, 743–748. [Google Scholar] [CrossRef]
- Rubins, H.B.; Robins, S.J.; Collins, D.; Nelson, D.B.; Elam, M.B.; Schaefer, E.J.; Faas, F.H.; Anderson, J.W. Diabetes, plasma insulin, and cardiovascular disease: subgroup analysis from the Department of Veterans Affairs high-density lipoprotein intervention trial (VA-HIT). Arch. Intern. Med. 2002, 162, 2597–2604. [Google Scholar] [CrossRef]
- Scott, R.; O’Brien, R.; Fulcher, G.; Pardy, C.; D’Emden, M.; Tse, D.; Taskinen, M.R.; Ehnholm, C.; Keech, A. Effects of fenofibrate treatment on cardiovascular disease risk in 9795 individuals with type 2 diabetes and various components of the metabolic syndrome: The Fenofibrate Intervention and Event Lowering in Diabetes (FIELD) study. Diabetes Care 2009, 32, 493–498. [Google Scholar] [CrossRef]
- Ginsberg, H.N.; Elam, M.B.; Lovato, L.C.; Crouse, J.R., III; Leiter, L.A.; Linz, P.; Friedewald, W.T.; Buse, J.B.; Gerstein, H.C.; Probstfield, J.; et al. Effects of combination lipid therapy in type 2 diabetes mellitus. N. Engl. J. Med. 2010, 362, 1563–1574. [Google Scholar] [CrossRef]
- Boden, W.E.; Probstfield, J.L.; Anderson, T.; Chaitman, B.R.; Desvignes-Nickens, P.; Koprowicz, K.; McBride, R.; Teo, K.; Weintraub, W. Niacin in patients with low HDL cholesterol levels receiving intensive statin therapy. N. Engl. J. Med. 2011, 365, 2255–2267. [Google Scholar] [CrossRef]
- Watts, G.F.; Chan, D.C.; Ooi, E.M.; Nestel, P.J.; Beilin, L.J.; Barrett, P.H. Fish oils, phytosterols and weight loss in the regulation of lipoprotein transport in the metabolic syndrome: Lessons from stable isotope tracer studies. Clin. Exp. Pharmacol. Physiol. 2006, 33, 877–882. [Google Scholar] [CrossRef]
- Chan, D.C.; Watts, G.F.; Barrett, P.H.; Beilin, L.J.; Redgrave, T.G.; Mori, T.A. Regulatory effects of HMG CoA reductase inhibitor and fish oils on apolipoprotein B-100 kinetics in insulin-resistant obese male subjects with dyslipidemia. Diabetes 2002, 51, 2377–2386. [Google Scholar] [CrossRef]
- Van Wijk, J.P.; de Koning, E.J.; Martens, E.P.; Rabelink, T.J. Thiazolidinediones and blood lipids in type 2 diabetes. Arterioscler. Thromb. Vasc. Biol. 2003, 23, 1744–1749. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Klop, B.; Elte, J.W.F.; Cabezas, M.C. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients 2013, 5, 1218-1240. https://doi.org/10.3390/nu5041218
Klop B, Elte JWF, Cabezas MC. Dyslipidemia in Obesity: Mechanisms and Potential Targets. Nutrients. 2013; 5(4):1218-1240. https://doi.org/10.3390/nu5041218
Chicago/Turabian StyleKlop, Boudewijn, Jan Willem F. Elte, and Manuel Castro Cabezas. 2013. "Dyslipidemia in Obesity: Mechanisms and Potential Targets" Nutrients 5, no. 4: 1218-1240. https://doi.org/10.3390/nu5041218