Is There a Therapeutic Role for Selenium in Alpha-1 Antitrypsin Deficiency?



{kind=link}
Abstract
:1. Introduction
2. Selenium and the Selenoproteome
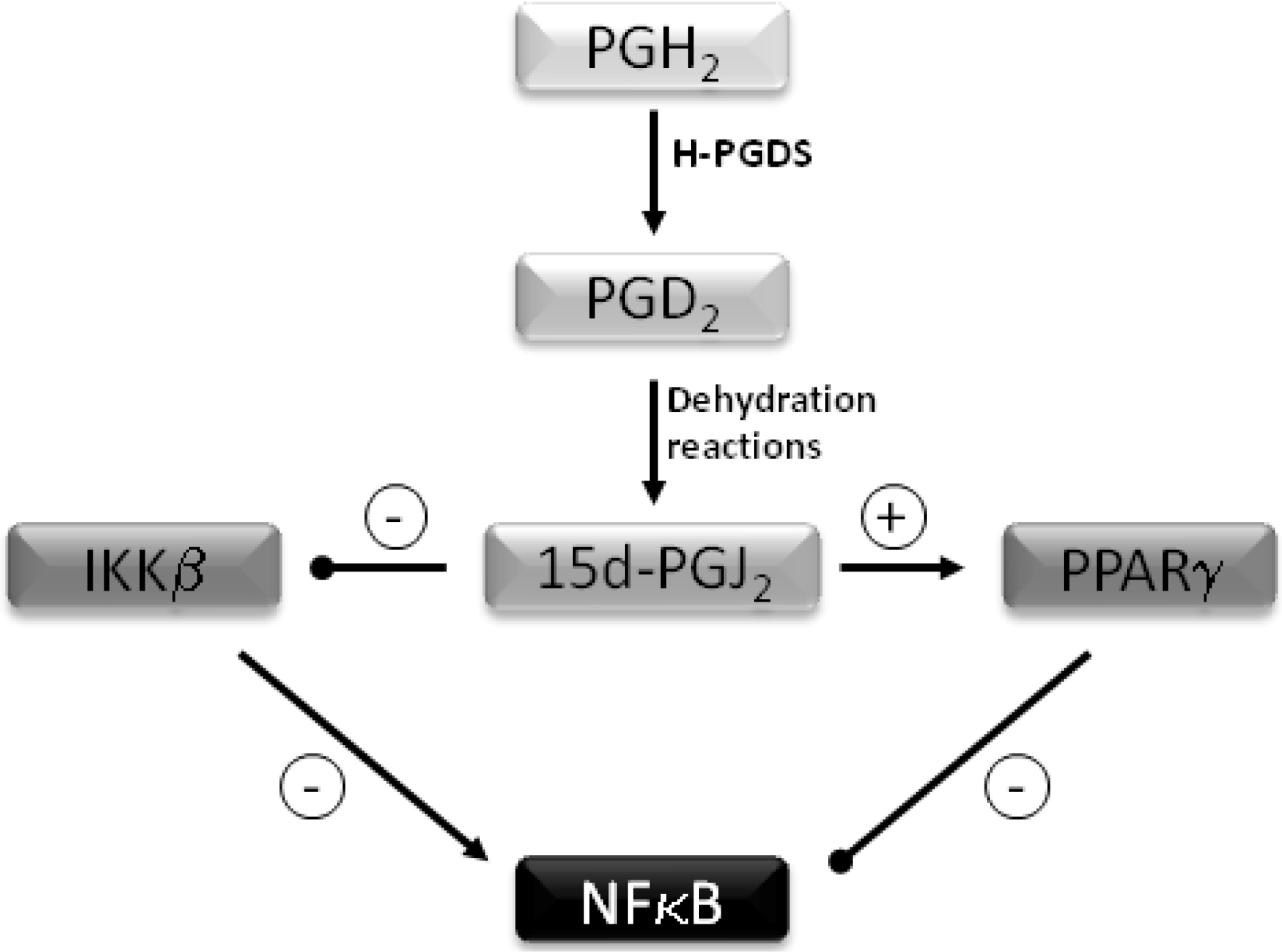
Anti-Inflammatory Properties of Selenium

3. Alpha-1 Antitrypsin Deficiency
3.1. Molecular Basis of AATD
3.2. ZAAT and Inflammation
4. Endoplasmic Reticulum Stress
5. ERAD and Selenoprotein S
6. SEPS1 and ZAAT
Selenium Status in ZAATD
7. Chronic Obstructive Pulmonary Disease
7.1. COPD and ER Stress
7.2. Selenium and COPD
8. Conclusions
Acknowledgments
Conflict of Interest
References
- Moghadaszadeh, B.; Beggs, A.H. Selenoproteins and their impact on human health through diverse physiological pathways. Physiology (Bethesda) 2006, 21, 307–315. [Google Scholar]
- Kelly, E.; Greene, C.M.; Carroll, T.P.; McElvaney, N.G.; O’Neill, S.J. Alpha-1 antitrypsin deficiency. Respir. Med. 2010, 104, 763–772. [Google Scholar] [CrossRef]
- Rayman, M.P. The importance of selenium to human health. Lancet 2000, 356, 233–241. [Google Scholar] [CrossRef]
- Combs, G.F.; Lu, J. Selenium as a cancer preventative agent. In Selenium: Its Molecular Biology and Role in Human Health, 1st; Hatfield, D.L., Berry, M.J., Gladyshev, V.N., Eds.; Springer: New York, NY, USA, 2001; pp. 205–219. [Google Scholar]
- Rayman, M.P. The argument for increasing selenium intake. Proc. Nutr. Soc. 2002, 61, 203–215. [Google Scholar] [CrossRef]
- Behne, D.; Weiler, H.; Kyriakopoulos, A. Effects of selenium deficiency on testicular morphology and function in rats. J. Reprod. Fertil. 1996, 106, 291–297. [Google Scholar] [CrossRef]
- Ge, K.; Yang, G. The epidemiology of selenium deficiency in the etiological study of endemic diseases in China. Am. J. Clin. Nutr. 1993, 57, 259S–263S. [Google Scholar]
- Beckett, G.J.; Arthur, J.R. Selenium and endocrine systems. J. Endocrinol. 2005, 184, 455–465. [Google Scholar] [CrossRef]
- Utiger, R.D. Kashin-Beck Disease—Expanding the Spectrum of Iodine-Deficiency Disorders. N. Engl. J. Med. 1998, 339, 1156–1158. [Google Scholar] [CrossRef]
- Vanderpas, J.B.; Contempré, B.; Duale, N.L.; Goossens, W.; Bebe, N.; Thorpe, R.; Ntambue, K.; Dumont, J.; Thilly, C.H.; Diplock, A.T. Iodine and selenium deficiency associated with cretinism in Northern Zaire. Am. J. Clin. Nutr. 1990, 52, 1087–1093. [Google Scholar]
- Kryukov, G.V.; Castellano, S.; Novoselov, S.V.; Lobanov, A.V.; Zehtab, O.; Guigo, R.; Gladyshev, V.N. Characterization of mammalian selenoproteomes. Science 2003, 300, 1439–1443. [Google Scholar] [CrossRef]
- Hoffmann, P.R.; Berry, M.J. Selenoprotein synthesis: A unique translational mechanism used by a diverse family of proteins. Thyroid 2005, 15, 769–775. [Google Scholar] [CrossRef]
- Kim, H.Y.; Gladyshev, V.N. Different catalytic mechanisms in mammalian selenocysteine- and cysteine-containing methionine-r-sulfoxide reductases. PLoS Biol. 2005, 3. [Google Scholar] [CrossRef]
- Vunta, H.; Davis, F.; Palempalli, U.D.; Bhat, D.; Arner, R.J.; Thompson, J.T.; Peterson, D.G.; Reddy, C.C.; Prabhu, K.S. The anti-inflammatory effects of selenium are mediated through 15-deoxy-delta12,14-prostaglandin J2 in macrophages. J. Biol. Chem. 2007, 282, 17964–17973. [Google Scholar] [CrossRef]
- Forman, B.M.; Tontonoz, P.; Chen, J.; Brun, R.P.; Spiegelman, B.M.; Evans, R.M. 15-Deoxy-Δ>12,14-Prostaglandin J2 is a ligand for the adipocyte determination factor PPARγ. Cell 1995, 83, 803–812. [Google Scholar] [CrossRef]
- Ricote, M.; Li, A.C.; Willson, T.M.; Kelly, C.J.; Glass, C.K. The peroxisome proliferator-activated receptor-γ is a negative regulator of macrophage activation. Nature 1998, 391, 79–82. [Google Scholar]
- Bailey, S.T.; Ghosh, S. 'PPAR'ting ways with inflammation. Nat. Immunol. 2005, 6, 966–967. [Google Scholar] [CrossRef]
- Pascual, G.; Fong, A.L.; Ogawa, S.; Gamliel, A.; Li, A.C.; Perissi, V.; Rose, D.W.; Willson, T.M.; Rosenfeld, M.G.; Glass, C.K. A SUMOylation-dependent pathway mediates transrepression of inflammatory response genes by PPAR-γ. Nature 2005, 437, 759–763. [Google Scholar] [CrossRef]
- Schroeder, W.T.; Miller, M.F.; Woo, S.L.; Saunders, G.F. Chromosomal localization of the human alpha 1-antitrypsin gene (Pi) to 14q31-32. Am. J. Hum. Genet. 1985, 37, 868–872. [Google Scholar]
- Hassan, T.; Smith, S.G.; Gaughan, K.; Oglesby, I.K.; O’Neill, S.; McElvaney, N.G.; Greene, C.M. Isolation and identification of cell-specific microRNAs targeting a messenger RNA using a biotinylated anti-sense oligonucleotide capture affinity technique. Nucleic Acids Res. 2013. [Google Scholar] [CrossRef]
- Brantly, M.; Nukiwa, T.; Crystal, R.G. Molecular basis of alpha-1-antitrypsin deficiency. Am. J. Med. 1988, 84, 13–31. [Google Scholar]
- McCracken, A.A.; Kruse, K.B.; Brown, J.L. Molecular basis for defective secretion of the Z variant of human alpha-1-proteinase inhibitor: Secretion of variants having altered potential for salt bridge formation between amino acids 290 and 342. Mol. Cell Biol. 1989, 9, 1406–1414. [Google Scholar]
- Lomas, D.A.; Evans, D.L.; Finch, J.T.; Carrell, R.W. The mechanism of Z alpha 1-antitrypsin accumulation in the liver. Nature 1992, 357, 605–607. [Google Scholar] [CrossRef]
- Curran, J.E.; Jowett, J.B.; Elliott, K.S.; Gao, Y.; Gluschenko, K.; Wang, J.; Abel Azim, D.M.; Cai, G.; Mahaney, M.C.; Comuzzie, A.G.; et al. Genetic variation in selenoprotein S influences inflammatory response. Nat. Genet. 2005, 37, 1234–1241. [Google Scholar] [CrossRef]
- Kim, K.H.; Gao, Y.; Walder, K.; Collier, G.R.; Skelton, J.; Kissebah, A.H. SEPS1 protects Raw264.7 cells from pharmacological ER stress agent-induced apoptosis. Biochem. Biophys. Res. Commun. 2007, 354, 127–132. [Google Scholar]
- Carroll, T.P.; Greene, C.M.; O’Connor, C.A.; Nolan, A.M.; O'Neill, S.J.; McElvaney, N.G. Evidence for unfolded protein response activation in monocytes from individuals with alpha-1 antitrypsin deficiency. J. Immunol. 2010, 184, 4538–4546. [Google Scholar] [CrossRef]
- Lawless, M.W.; Greene, C.M.; Mulgrew, A.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Activation of endoplasmic reticulum-specific stress responses associated with the conformational disease Z alpha 1-antitrypsin deficiency. J. Immunol. 2004, 172, 5722–5726. [Google Scholar]
- Hidvegi, T.; Schmidt, B.Z.; Hale, P.; Perlmutter, D.H. Accumulation of mutant alpha1-antitrypsin Z in the endoplasmic reticulum activates caspases-4 and -12, NFkappaB, and BAP31 but not the unfolded protein response. J. Biol. Chem. 2005, 280, 39002–39015. [Google Scholar]
- Rudnick, D.A.; Liao, Y.; An, J.K.; Muglia, L.J.; Perlmutter, D.H.; Teckman, J.H. Analyses of hepatocellular proliferation in a mouse model of alpha-1-antitrypsin deficiency. Hepatology 2004, 39, 1048–1055. [Google Scholar] [CrossRef]
- Miller, S.D.; Greene, C.M.; McLean, C.; Taggart, C.C.; O’Neill, S.J.; McElvaney, N.G. Tauroursodeoxycholic acid inhibits apoptosis induced by Z alpha-1 antitrypsin via inhibition of bad. Hepatology 2007, in press. [Google Scholar]
- Teckman, J.H.; An, J.K.; Blomenkamp, K.; Schmidt, B.; Perlmutter, D. Mitochondrial autophagy and injury in the liver in alpha 1-antitrypsin deficiency. Am. J. Physiol. Gastrointest. Liver Physiol. 2004, 286, G851–G862. [Google Scholar] [CrossRef]
- Palmer, E.A.; Kruse, K.B.; Fewell, S.W.; Buchanan, S.M.; Brodsky, J.L.; McCracken, A.A. Differential requirements of novel α1PiZ degradation deficient (add) genes in ER-associated protein degradation. J. Cell Sci. 2003, 116, 2361–2373. [Google Scholar] [CrossRef]
- Hosokawa, N.; Tremblay, L.O.; You, Z.; Herscovics, A.; Wada, I.; Nagata, K. Enhancement of endoplasmic reticulum (ER) degradation of misfolded null Hong Kong alpha1-antitrypsin by human ER mannosidase I. J. Biol. Chem. 2003, 278, 26287–26294. [Google Scholar]
- Greene, C.M.; McElvaney, N.G. Z alpha-1 antitrypsin deficiency and the endoplasmic reticulum stress response. World J. Gastrointest. Pharmacol. Ther. 2010, 1, 94–101. [Google Scholar] [CrossRef]
- Greene, C.M.; McElvaney, N.G. Protein misfolding and obstructive lung disease. Proc. Am. Thorac. Soc. 2010, 7, 346–355. [Google Scholar] [CrossRef]
- Greene, C.M.; Hassan, T.; Molloy, K.; McElvaney, N.G. The role of proteases, endoplasmic reticulum stress and SERPINA1 heterozygosity in lung disease and alpha-1 antitrypsin deficiency. Expert. Rev. Respir. Med. 2011, 5, 395–411. [Google Scholar] [CrossRef]
- McElvaney, N.G.; Greene, C.M. Mechanisms of protein misfolding in conformational lung diseases. Curr. Mol. Med. 2012, 12, 850–859. [Google Scholar] [CrossRef]
- Walder, K.; Kantham, L.; McMillan, J.S.; Trevaskis, J.; Kerr, L.; De Silva, A.; Sunderland, T.; Godde, N.; Gao, Y.; Bishara, N.; et al. Tanis: A link between type 2 diabetes and inflammation? Diabetes 2002, 51, 1859–1866. [Google Scholar]
- Gao, Y.; Walder, K.; Sunderland, T.; Kantham, L.; Feng, H.C.; Quick, M.; Bishara, N.; de Silva, A.; Augert, G.; Tenne-Brown, J.; et al. Elevation in TANIS expression alters glucose metabolism and insulin sensitivity in H4IIE cells. Diabetes 2003, 52, 929–934. [Google Scholar] [CrossRef]
- Gao, Y.; Feng, H.C.; Walder, K.; Bolton, K.; Sunderland, T.; Bishara, N.; Quick, M.; Kantham, L.; Collier, G.R. Regulation of the selenoprotein SELS by glucose deprivation and endoplasmic reticulum stress—SELS is a novel glucose-regulated protein. FEBS Lett. 2004, 563, 185–190. [Google Scholar] [CrossRef]
- Ye, Y.; Shibata, Y.; Yun, C.; Ron, D.; Rapoport, T.A. A membrane protein complex mediates retro-translocation from the ER lumen into the cytosol. Nature 2004, 429, 841–847. [Google Scholar] [CrossRef]
- Kelly, E.; Greene, C.M.; Carroll, T.P.; McElvaney, N.G.; O’Neill, S.J. Selenoprotein S/SEPS1 modifies endoplasmic reticulum stress in Z variant alpha1-antitrypsin deficiency. J. Biol. Chem. 2009, 284, 16891–16897. [Google Scholar]
- Rayman, M.P. Selenium in cancer prevention: A review of the evidence and mechanism of action. Proc. Nutr. Soc. 2005, 64, 527–542. [Google Scholar] [CrossRef]
- Brusselle, G.G.; Joos, G.F.; Bracke, K.R. New insights into the immunology of chronic obstructive pulmonary disease. Lancet 2011, 378, 1015–1026. [Google Scholar] [CrossRef]
- Silverman, E.K.; Sandhaus, R.A. Clinical practice. Alpha1-antitrypsin deficiency. N. Engl. J. Med. 2009, 360, 2749–2757. [Google Scholar]
- Pillai, S.G.; Ge, D.; Zhu, G.; Kong, X.; Shianna, K.V.; Need, A.C.; Feng, S.; Hersh, C.P.; Bakke, P.; Gulsvik, A.; et al. A genome-wide association study in chronic obstructive pulmonary disease (COPD): Identification of two major susceptibility loci. PLoS Genet. 2009, 5. [Google Scholar] [CrossRef]
- Kelsen, S.G. Respiratory epithelial cell responses to cigarette smoke: The unfolded protein response. Pulm. Pharmacol. Ther. 2012, 25, 447–452. [Google Scholar] [CrossRef]
- Ribeiro, C.M.; O’Neal, W.K. Endoplasmic reticulum stress in chronic obstructive lung diseases. Curr. Mol. Med. 2012, 12, 872–882. [Google Scholar] [CrossRef]
- Blumental-Perry, A. Unfolded protein response in chronic obstructive pulmonary disease: Smoking, aging and disease: A sad trifecta. Curr. Mol. Med. 2012, 12, 883–898. [Google Scholar] [CrossRef]
- Cantin, A.M.; Richter, M.V. Cigarette smoke-induced proteostasis imbalance in obstructive lung diseases. Curr. Mol. Med. 2012, 12, 836–849. [Google Scholar] [CrossRef]
- Yuan, T.; Luo, B.L.; Wei, T.H.; Zhang, L.; He, B.M.; Niu, R.C. Salubrinal protects against cigarette smoke extract-induced hbepc apoptosis likely via regulating the activity of PERK-eIF2alpha signaling pathway. Arch. Med. Res. 2012, 43, 522–529. [Google Scholar] [CrossRef]
- Geraghty, P.; Wallace, A.; D’Armiento, J.M. Induction of the unfolded protein response by cigarette smoke is primarily an activating transcription factor 4-C/EBP homologous protein mediated process. Int. J. Chron. Obstruct. Pulmon. Dis. 2011, 6, 309–319. [Google Scholar]
- Somborac-Bacura, A.; van der Toorn, M.; Franciosi, L.; Slebos, D.J.; Zanic-Grubisic, T.; Bischoff, R.; van Oosterhout, A.J. Cigarette smoke induces ER stress response and proteasomal dysfunction in human alveolar epithelial cells. Exp. Physiol. 2012, 98, 316–325. [Google Scholar]
- van Rijt, S.H.; Keller, I.E.; John, G.; Kohse, K.; Yildirim, A.O.; Eickelberg, O.; Meiners, S. Acute cigarette smoke exposure impairs proteasome function in the lung. Am. J. Physiol. Lung Cell Mol. Physiol. 2012, 303, L814–L823. [Google Scholar] [CrossRef]
- Kenche, H.; Baty, C.J.; Vedagiri, K.; Shapiro, S.D.; Blumental-Perry, A. Cigarette smoking affects oxidative protein folding in endoplasmic reticulum by modifying protein disulfide isomerase. FASEB J. 2012. [Google Scholar] [CrossRef]
- Kitaguchi, Y.; Taraseviciene-Stewart, L.; Hanaoka, M.; Natarajan, R.; Kraskauskas, D.; Voelkel, N.F. Acrolein induces endoplasmic reticulum stress and causes airspace enlargement. PLoS One 2012, 7. [Google Scholar] [CrossRef]
- Malhotra, D.; Thimmulappa, R.; Vij, N.; Navas-Acien, A.; Sussan, T.; Merali, S.; Zhang, L.; Kelsen, S.G.; Myers, A.; Wise, R.; et al. Heightened endoplasmic reticulum stress in the lungs of patients with chronic obstructive pulmonary disease: The role of Nrf2-regulated proteasomal activity. Am. J. Respir. Crit. Care Med. 2009, 180, 1196–1207. [Google Scholar] [CrossRef]
- Min, T.; Bodas, M.; Mazur, S.; Vij, N. Critical role of proteostasis-imbalance in pathogenesis of COPD and severe emphysema. J. Mol. Med. (Berl.) 2011, 89, 577–593. [Google Scholar]
- Romieu, I.; Trenga, C. Diet and obstructive lung diseases. Epidemiol. Rev. 2001, 23, 268–287. [Google Scholar] [CrossRef]
- Hirayama, F.; Lee, A.H.; Oura, A.; Mori, M.; Hiramatsu, N.; Taniguchi, H. Dietary intake of six minerals in relation to the risk of chronic obstructive pulmonary disease. Asia. Pac. J. Clin. Nutr. 2010, 19, 572–577. [Google Scholar]
- Isbaniah, F.; Wiyono, W.H.; Yunus, F.; Setiawati, A.; Totzke, U.; Verbruggen, M.A. Echinacea purpurea along with zinc, selenium and vitamin C to alleviate exacerbations of chronic obstructive pulmonary disease: Results from a randomized controlled trial. J. Clin. Pharm. Ther. 2011, 36, 568–576. [Google Scholar] [CrossRef]
- El-Attar, M.; Said, M.; El-Assal, G.; Sabry, N.A.; Omar, E.; Ashour, L. Serum trace element levels in COPD patients: The relation between trace element supplementation and period of mechanical ventilation in a randomized controlled trial. Respirology 2009, 14, 1180–1187. [Google Scholar] [CrossRef]
- McKeever, T.M.; Lewis, S.A.; Smit, H.A.; Burney, P.; Cassano, P.A.; Britton, J. A multivariate analysis of serum nutrient levels and lung function. Respir. Res. 2008, 9. [Google Scholar] [CrossRef]
- Hu, G.; Cassano, P.A. Antioxidant nutrients and pulmonary function: The third national health and nutrition examination survey (NHANES III). Am. J. Epidemiol. 2000, 151, 975–981. [Google Scholar] [CrossRef]
- Santos, M.C.; Oliveira, A.L.; Viegas-Crespo, A.M.; Vicente, L.; Barreiros, A.; Monteiro, P.; Pinheiro, T.; Bugalho De Almeida, A. Systemic markers of the redox balance in chronic obstructive pulmonary disease. Biomarkers 2004, 9, 461–469. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Greene, C.M.; Chhabra, R.; McElvaney, N.G. Is There a Therapeutic Role for Selenium in Alpha-1 Antitrypsin Deficiency? Nutrients 2013, 5, 758-770. https://doi.org/10.3390/nu5030758
Greene CM, Chhabra R, McElvaney NG. Is There a Therapeutic Role for Selenium in Alpha-1 Antitrypsin Deficiency? Nutrients. 2013; 5(3):758-770. https://doi.org/10.3390/nu5030758
Chicago/Turabian StyleGreene, Catherine M., Roohi Chhabra, and Noel G. McElvaney. 2013. "Is There a Therapeutic Role for Selenium in Alpha-1 Antitrypsin Deficiency?" Nutrients 5, no. 3: 758-770. https://doi.org/10.3390/nu5030758
APA StyleGreene, C. M., Chhabra, R., & McElvaney, N. G. (2013). Is There a Therapeutic Role for Selenium in Alpha-1 Antitrypsin Deficiency? Nutrients, 5(3), 758-770. https://doi.org/10.3390/nu5030758