Historical Perspective, Development and Applications of Next-Generation Sequencing in Plant Virology



Abstract
:1. Introduction
2. Pioneer Landmarks in DNA Sequencing
3. Major Landmarks in DNA Sequencing during the Last Three Decades
4. The Need for Fast, Inexpensive and Accurate DNA Sequencing Technologies
5. Development of NGS Platforms (2000–present)
6. Platform Selection

{kind=link}
| Sequencing platform | Amplification method | Sequencing chemistry | Read length (bp) | Sequencing Speed/h | Maximum Output Per run | Accuracy (%) | M1 I2 D3 |
|---|---|---|---|---|---|---|---|
| 454 (Roche) | Emulsion PCR | Pyrosequencing | 400–700 | 13 Mbp | 700 Mbp | 99.9 | 0.10, 0.3, 0.02 [23] |
| Illumina (Illumina) | Bridge PCR | Reversible terminators | 100–300 | 25 Mbp | 600 Gbp | 99.9 | 0.12, 0.004, 0.006 [23] |
| SOLiD (Life Technologies) | Emulsion PCR | Ligation | 75–85 | 21–28 Mbp | 80–360 Gbp | 99.9 | Error is higher than Illumina [24] |
| PacBio (Pacific Biosciences) | No amplification Single molecule real-time (or SMRT) | Fluorescently labeled nucleotides | 4, 000–5,000 | 50–115 Mbp | 200 Mb–1 Gbp | 95 | 1, 2, 12 [25] |
| Helicos (Helicos Biosciences) | No amplification Single molecule | Reversible terminators | 25–55 | 83 Mbp | 35 Gbp | 97 | Error is in the range of few percent but higher than 454 and Illumina and biased toward InDels [24] |
| Ion Torrent (Life Technologies) | Emulsion PCR | Detection of released H | 100–400 | 25 Mb–16 Gbp | 100 Mb–64 Gbp | 99 | M, 0.06, I + D 1.38 [26] |
| Nanopore (Oxford Technologies) | No amplification Single molecule | Very long reads up to 50 kbp | 150 Mbp | Tens of Gbp | 96 |
| Platform Systems | Applications |
|---|---|
| 454 GS FLX + Systems (GS FLX Titanium XL+/GS FLX Titanium XLR70) * GS Junior System (bench top) | DNA sequencing: whole genome sequencing, de novo and resequencing of large genes in a single run with read length up to 1 kbp. Amplicon sequencing; RNA sequencing: transcriptome sequencing, sequencing capture metagenomics. Run time: 10–23 h; * Fast sequencing run with read length of 400 bp. Similar applications as the above system. |
| lllumina HiSeq Systems (2500/2000/1500/1000), Genome Analyzer IIx, HiScan SQ, * MiSeq (bench top) | DNA sequencing including candidate region targeted sequencing; Epigenetic sequencing: chromatin immunoprecipitation sequencing (ChIP-Seq), methylation analysis by sequencing; RNA sequencing: transcriptome analysis, small RNA and mRNA sequencing, gene expression profiling by sequencing; Run time: 8–14 days; * Up to 15 Gbp of output with 25 M sequencing reads and 2 × 300 bp read length; Access more sequencing applications such as exome, metagenomics, human leukocyte antigen (HLA) gene typing, mRNA sequencing, targeted gene expression (proteins and non-protein coding genes) such as rRNA, tRNA, or smRNA genes; Run time: 20–35 h. |
| SOLiD 5500 W Series Genetic Analysis Systems (5500 W, 5500 xlw) | DNA sequencing: whole genome and exome; Epigenetic Sequencing; RNA Sequencing. Run time: 7–12 days. |
| PacBio PACBIO RSII | DNA sequencing using single molecule real-time (SMRT) system with the longest read lengths of any sequencing technology. Characterization of genetic variation, methylation, targeted sequencing such as SNP detection and validation, indels, structural variants, haplotypes and phasing, base modification detection to understand gene expression, host-pathogen interactions, DNA damage and DNA repair. Run time: 30 min. |
| Helicos Genetic Analyzer System | DNA sequencing and RNA sequencing. Run time: 8 days. |
| Ion Torrent Ion PGM System (bench top) *Ion Proton System (bench top) | Semiconductor sequencing with 400-bp length- Ideal for sequencing small genes and genomes. DNA sequencing for microbial: genes and genomes, amplicons, exomes (unreveal disease-causing variants), targeted sequencing, viral typing and other microbial typing. RNA sequencing. Run time: 4.5 h * Semiconductor sequencing: Sequencing microbial genomes, exomes, transcriptomes. Run time: 4.5 h |
| Nanopore GridION System (bench top) *MinION System (a miniaturized disposable device for single use) | DNA sequencing; Epigenetic sequencing; Characterization of genetic variation. RNA sequencing: the system is designed to analyze the original sample RNA directly, without undergoing conversion to cDNA. Run time possibly under 60 min. * For DNA sequencing only, i.e., blood DNA. |
| Host | Study finding/virome | Sample preparation/target | Sequencing platform | Ref. |
|---|---|---|---|---|
| Sweet potato | Detected: Sweet potato feathery mottle virus, Sweet potato chlorotic stunt virus; Discovered: two Badnavirus species (dsDNA), one Mastrevirus species (ssDNA) | siRNAs | Illumina | [27] |
| Gomphrena globosa | Plants were inoculated with unknown virus-a new Cucumovirus was identified. Proposed name: Gayfeather mild mottle virus | Total RNA + subtractive hybridization | Roche 454 GS-FLX | [28] |
| Arabidopsis thaliana | Tobacco mosaic virus siRNAs mediate virus-host interactions which may contribute to viral pathogenicity and host specificity | siRNAs | Illumina Genome Analyzer | [29] |
| Nicotiana benthamiana, Arabidopsis thaliana, Cucumis milo and tomato | Nine different viruses including Cucumber mosaic virus, Tobacco rattle virus, Pepper mild mosaic virus, Potato virus X were studied. The study extended the knowledge of distribution and composition of siRNAs in virus-infected plants and contributed to a better understanding of siRNAs biogenesis. | siRNAs of nine different viruses | Roche 454 | [30] |
| Cassava | The complete sequence of the Tanzanian strain of Cassava brown streak virus was determined and compared with that of the Ugandan strain. The virus is highly heterogeneous at both the isolate and strain levels with nucleotide identity at the isolate level of 76% | Total RNA + subtractive hybridization | Roche 454 GS− FLX | [31] |
| Nicotiana benthamiana | Profiled Cymbidium ringspot virus-derived siRNAs. These RNAs were primarily produced from the positive strand of the virus, produced with different frequency, and had 5' monophosphate and were not perfect duplexes. | siRNAs | Roche 454 and Solexa (Illumina) | [32] |
| Wild plant species from 15 families naturally infected with viruses were utilized. The families are: Acanthaceae, Bignoniaceae, Caesalpinaceae Commelinaceae, Cyperaceae, Cucurbitaceae, Euphorbiaceae, Gesneriaceae, Lamiaceae, Mimosaceae, Myrtaceae, Papilionaceae, Poaceae, Rubiaceae and Solanaceae | Identification of 11 virus families in infected plants which include: Bromoviridae, Caulimoviridae, Chrysoviridae, Closteroviridae, Endornaviridae, Luteoviridae, Narnaviridae, Partitiviridae, Potyviridae, Totiviridae, and Tymoviridae. Unclassified virus families were also identified in some samples of infected plants. Discovered several thousand novel viruses, all linked to their specific plant hosts | dsRNAs | Roche 454-GS-FLX | [33] |
| Wild cocksfoot grass | Cereal yellow dwarf virus (Luteovirus) discovered infecting this grass (not previously reported infecting this host). Cocksfoot streak virus (Potyvirus) was detected | siRNAs | Roche 454 | [34] |
| Nicotiana benthamiana, Arabidopsis thaliana | Profiling siRNAs of Bamboo mosaic virus and its interfering and non-interfering associated satellite RNAs. The overall composition of virus siRNAs and satellite RNAs in the infected plants reflect the combined action of virus, satellite RNA and different DCLs in host plants. | siRNAs (virus and satellites) | Solexa (Illumina) | [35] |
| Rice | Characterization of siRNAs of Rice stripe virus four genome RNAs in infected rice plants | siRNAs | Illumina Solexa | [36] |
| Cotton | Characterization of siRNAs of Cotton leafroll dwarf virus (genus Polerovirus, family Luteoviridae) in infected cotton plants | siRNAs | Illumina Genome Analyzer | [37] |
| Wild Passiflora caerula (Blue-crown passion flower) (a vine) | The complete nucleotide sequence of the Passion fruit woodiness virus (Potyvirus) was determined | Polyadenylated RNA | Illumina Solexa GAIIx | [38] |
| Pepper, eggplant | The complete nucleotide sequences of two new viruses Pepper yellow leaf curl virus (Polerovirus) and Eggplant mild leaf mottle virus (Ipomovirus) were determined | Purified virons viral RNA | SOLiD | [39] |
| Different hosts | A novel virus infecting watercress was identified (proposed name: Watercress white vein virus). Viruses such as Piper yellow mottle virus, Arachacha virus B, and Potato black ring virus were detected and sequenced | Partial virus and RNA purification then fragmented | Roche 454 GS−FLX | [40] |
| Tomato | siRNAs of Tomato spotted wilt virus were used to detect the virus in infected tomato before symptoms appeared at levels too low for conventional detection methods. Also, used for analysis of the virus quasi species and for identification of an unspecified Tospovirus and a squash-infecting geminivirus | siRNAs | Illumina | [41] |
| Arabidopsis thaliana | Characterization of the siRNAs and transcriptome profiles of Oilseed rape mosaic virus (Tobamovirus)-infected Arabidopsis plants. | Total RNAs and siRNAs | Illumina Genome Analyzer | [42] |
| Tomato, Nicotiana benthamiana | Characterization of the siRNAs for the monopartite begomovirus Tomato yellow leaf curl China virus and its associated betasatellite in infected tomato and Nicotiana benthamiana plants . Also, it was found that the betasatellite affected the amount of virus siRNAs detected in both plant species. | siRNAs | Solexa-Illumina | [43] |
| Tomato | Identification of Potato spindle tuber virod, Pepino mosaic virus, differentiation of two strains of the virus, and a novel Potyvirus from infected tomato plants by obtaining the complete genome sequence of the mixed infected pathogens without prior knowledge of their existence. Based on the severity of symptoms on tomato, the novel virus is provisionally named “Tomato necrotic stunt virus” | siRNAs | Illumina Genome Analyzer IIx | [44] |
| Tobacco cv Xanthi nc | Identification of gene expression changes associated with disease development in tobacco plants induced by infection with the M strain of Cucumber mosaic virus Sequencing analysis of tobacco transcriptome identified 95,916 unigenes, 34,408 of which were new transcripts by database searches. | Total RNA then treated with DNase I | Illumina Hi Seq 2000 | [45] |
| Corn (maize) | Detection and identification of Maize chlorotic mottle virus and Sugarcane mosaic virus by obtaining over 90% of both viral genomes sequencing which allowed also strain characterization. Next generation sequencing may be used for rapidly identifying potential disease causing agents, in this case viruses | Total RNA was purified from diseased tissue with virus symptoms | Roche 454 GS−FLX+ | [46] |
| Seventeen plant species, among them 14 Australian indigenous species | Detection and identification of 12 viruses described previously members of the genera Potyvirus, Nepovirus, Allexivirus and Carlavirus. Four novel viruses were identified and proposed as members of the genera Potyvirus, Sadwavirus and Trichovirus. Moreover, in 3 cases, 2–3 distinct isolates of a virus species co-infected the same plant. | Polyadenylated RNA | Illumina Genome Analyzer IIx | [47] |
| Nicotiana benthamiana, Laodelphgax striatellus (small brown leafhopper), rice | The presence Rice stripe virus (RSV) siRNAs was demonstrated in infected rice as well as in infected N. benthamiana and viruliferous L. striatellus. Also, results indicate the potential existence of RNAi-mediated immunity against RSV infection in L. striatellus, a member of Hemipteran that transmits about 55% of the known plant viruses. Moreover, demonstrated that siRNA are generated differentially in different hosts. | siRNAs | Illumina | [48] |
| Sweet potato | Detected Sweet potato feathery mottle virus strain RC, Sweet potato virus C (Potyvirus), Sweet potato chlorotic stunt virus strain WA (Crinivirus), Sweet potato leaf curl Georgia virus (Begomovirus), and Sweet potato pakakuy virus strain B (synonym: Sweet potato badnovirus B) infecting sweet potato crops in Central America. Also, 4 viruses were detected in a sweet potato sample from the Galapagos Islands. Results suggest that siRNAs deep sequencing analysis is suitable for use as a reliable method for detection of plant viruses in infected crops. | si RNA | Illumina Genome Analyzer | [49] |
| Pepper (genotype Yolo Wonder) | Developing a mathematical model that estimates genetic drift and selection intensities using next generation sequencing data of 4 variants of Potato virus Y that differ by 1–2 substitutions involved in pathogenicity. | Total RNA | Roche 454 | [50] |
| Arabidopsis thaliana | Analysis of viral siRNAs from DNA virus-infected cells showed that the entire circular genomes of Cauliflower mosaic virus (genus: Caulimovirus, family: Caulimoviridae) and cabbage leaf curl virus (genus: Begomovirus, family: Geminiviridae) are densely covered with siRNAs in both sense and antisense polarities without gaps. This would enable de novo reconstruction of the complete DNA virus genomes from siRNAs. | siRNAs | Illumina Genome Analyzer | [51,52,53] |
| Black pepper | The complete genome sequence of Piper yellow mosaic virus was determined. It was also established that the virus is a member of the genus Badnovirus and the family Caulimoviridae. Fragments of two additional novel viruses belong to Caulimoviridae were sequenced and the viruses were tentatively named Piper DNA virus 1 and 2. | Viral and plant DNA were isolated from virus-enriched fraction | Roche 454 GS−FLX Titanium | [54] |
| Host | Study finding/virome | Sample preparation/target | Sequencing platform | Ref. |
|---|---|---|---|---|
| Raspberry | A novel virus isolated from infected raspberry plants was completely sequenced and characterized. It was designated as Raspberry latent virus. The virus is a novel dicot-infecting reovirus in the family Reoviridae, subfamily Spinareovirinae. | dsRNA | Illumina | [55] |
| Citrus | In Citrus tristeza virus (CTV) infected citrus plants It was shown that the citrus homologues of Dicer-like ribonucleases mediate the genesis of the 21 and 22 nt CTV siRNAs and that the ribonucleases act not only on the genomic RNA but also on the 30 co-terminal subgenomic RNAs and, particularly, on their dsRNA forms. A novel citrus miRNAs was also indentified and how CTV influences their accumulation was determined. | CTV sRNAs, gRNA sgRNAs | Illumina | [56] |
| Citrus | Genomic organization and other molecular characterizations were determined for Citrus yellow vein clearing virus. Analyses suggested that the virus is the causal agent of yellow vein clearing disease of lemon trees and represent a new species in the genus Mandarivirus. | siRNAs | Illumina | [57] |
| Citrus | A novel DNA virus species, member of the family Geminiviridae, was identified and associated with citrus chlorotic dwarf disease. A provisional name of Citrus chlorotic dwarf-associated virus was proposed. | siRNAs and total DNA | Illumina HiSeq2000 | [58] |
| Apple, Citrus, Grapevine | Detected ASPV, ACLSV and an unknown mycovirus. Detected two variants of CTV and ASGV. Detected variants of GLRaV-3, GVA and an unknown mycovirus. | siRNAs | Illumina | [59] |
| Apple | Identified agents associated with green crinkle disease of apple trees. The disease is a complex one as the following viruses were identified associated with it: ASGV, ASPV, ACLSV, ApLV, ApPCLSV and PCMV. | siRNAs | Illumina HiSeq2000 | [60] |
| Prunus | Detected and identified known Prunus viruses such as PPV, PNRS, etc. and novel virus agents. | dsRNA | Roche 454 | [61] |
| Fig | Detected Fig mosaic virus and Fig latent virus-1 for their elimination from infected clones. It is the first application of next-generation sequencing technology to detect and identify known and new species of viruses infecting fig trees. | dsRNAs | Illumina | [62] |
| Blackberry | A novel Ampelovirus in the family Closteroviridae was identified as one of the viruses associated with blackberry yellow vein disease complex. | dsRNAs | Illumina | [63] |
| Cherry | Characterization of the genome of the divergent Little cherry virus 1 (LChV1) isolate and establishing that LChV1 isolates could be responsible for Shirofugen stunt disease syndrome. | dsRNAs | Roche 454 Pyrosequencing multiplex approach | [64] |
| Citrus | The complete nucleotide sequence and structure of a novel virus of the genus Cilevirus was determined. The novel virus causes symptoms similar to citrus leprosies and it is suggested to be called Citrus leprosis virus cytoplasmic type 2. | siRNAs | Illumina | [65] |
| Citrus | A novel virus was discovered by analysis of the contigs assembled from the virus siRNAs sequences which showed similarity with luteovirus sequence, particularly with Pea enation mosaic virus, the type member of the genus Enarnovirus. The complete genome of the virus was determined and the new virus was provisionally named Citrus vein enation virus. | siRNAs | Solexa-Illumina | [66] |
| Host | Study finding/virome | Sample preparation/target | Sequencing platform | Ref. |
|---|---|---|---|---|
| Grapevine | A novel Marafivirus (Grapevine Syrah 1virus) was identified associated with grapevine syrah decline. The virus was also identified in leafhopper vector. Also detected in plant tissue GRSPaV, GRVFV, GLRaV-9, and viroids. | Total RNA or dsRNA | Roche 454 | [67] |
| Grapevine | Grapevine virus E, not previously in South Africa. A mycovirus similar to Penicillium chrysogenum virus, two other mycoviruses, GLRaV-3, GRSPaV, GVA. | dsRNA | Illumina | [68] |
| Grapevine | Viruses of the genera Foveavirus, Maculavirus, Marafivirus, and Nepovirus were detected. siRNAs originate from both genomic and antigenomic strands with the exception of tymoviruses, the majority are derived from antigenic virus strand. | siRNAs | Illumina | [69] |
| Grapevine | A novel DNA virus was discovered associated with the grapevine vein-clearing and vine decline syndrome. The virus belongs to genus Badnavirus in the family Caulimoviridae. It is the first DNA virus discovered in grapevine. It has been provisionally named Grapevine vein clearing virus. | siRNAs | Illumina Genome Analyzer | [70] |
| Grapevine | Twenty six fungal groups were identified in a single plant source. Three of the mycoviruses were associated with Botrytis cinerea. Most of the rest were undescribed. | dsRNA | Roche 454 | [71] |
| Grapevine | A novel species of virus was discovered for which the provisional name Grapevine Pinot gris virus is proposed. Also, detected and identified GRSPaV, GRVFV, GSy 1V, and viroids. | siRNAs | Illumina | [72] |
| Grapevine | A novel circular DNA virus was identified associated with red blotch disease in grapevine in California. A provisional name of Grapevine red blotch-associated virus is proposed for the novel virus. | dsRNA extracted without DNase treatment | Illumina Genome Analyzer IIx | [73] |
| Grapevine | A novel Vitivirus was identified. The virus is provisionally named Grapevine virus F. | dsRNA | Illumina Genome Analyzer IIx | [74] |
| Grapevine | Characterization of siRNAs associated with grapevine leafroll disease. | siRNAs | Illumina | [75] |
| Grapevine | Complete sequence of a novel single-stranded DNA virus associated with grapevine red leaf disease (GRD). The virus is tentatively named Grapevine red leaf-associated virus (GRLaV). The virus represents an evolutionary distinct lineage in the family Geminiviridae. Also detected in plant tissue GRSPaV, GFV, and viroids. | Total RNA treated with DNase | Illumina Genome Analyzer IIx | [76] |
| Host | Study finding/virome | Sample preparation/target | Sequencing platform | Ref. |
|---|---|---|---|---|
| Grapevine | Different Dicer-like enzymes target RNAs of Hop stunt viroid, Grapevine yellow speckle viroid 1. Also, study suggested that the viroid RNAs may interact with host enzymes involved in the RNA-directed DNA methylation pathway | siRNAs | Solexa, Illumina | [77] |
| Grapevine | Detection and identification of Australian grapevine viroid, Hop stunt viroid and Grapevine yellow speckle viroid | Total RNA or dsRNA | Roche 454 | [67] |
| Peach | To study the genesis of Peach latent mosaic viroid siRNA and viroid pathogenesis | siRNAs | Illumina | [78] |
| Nicotiana benthamiana | RNA-dependent RNA polymerase 6 restricts accumulation and precludes meristem invasion of Potato spindle tuber viroid which replicates in nuclei | Plant and viroid siRNAs | Illumina EAS269 GAII | [79] |
| Cucumber | To study Hop stunt viroid pathway involved in the biogenesis of the viroid siRNAs | siRNAs | Illumina | [80] |
| Tomato | Detection and identification of Potato spindle tuber viroid | siRNAs | Illumina Genome Analyzer IIx | [44] |
| Grapevine | Detection and identification of Grapevine yellow speckle viroid 1 and Hop stunt viroid | siRNAs | Illumina | [72] |
| Grapevine | Detection and identification of Grapevine yellow speckle viroid 1 and Hop stunt viroid | siRNAs and dsRNAs | Illumina | [81] |
| Grapevine | Discovery of viroid-like circular RNA 375 nt long with hammerhead ribozymes. Currently, infectivity studies showed that the RNA is not infectious which may suggest that it is viral satellite | siRNAs | Illumina | [82] |
| Grapevine | Characterization of siRNAs of Hop stunt viroid, Grapevine yellow speckle viroid 1, and Grapevine yellow speckle viroid 2 | siRNAs | Illumina | [75] |
| Grapevine | Detection and identification of Grapevine yellow speckle viroid 1, Hop stunt viroid, Citrus exocortis Yucatan viroid and Citrus exocortis viroid from both symptomatic and non-symptomatic samples of grapevine read leaf disease | Total RNA treated with Dnase | Illumina Genome Analyzer IIx | [76] |
| Vector | Study finding/virome | Sample preparation/target | Sequencing platform | Ref. |
|---|---|---|---|---|
| Grapevine leafhopper | A novel Merafivirus associated with grapevine syrah decline was detected in the vector | Total nucleic acids of the viruliferous vector | Roche 454 | [67] |
| Citrus psyllid | A complete genomic sequence of the bacterium, “Candidatus Liberibacter asiaticus” was obtained. The genome is circular and its size is about 1.23 Mb, The bacterium is the causal agent of citrus Huanglongbing (greening) disease | DNA extracted from a single “Ca. L. asiaticus”-infected Asian citrus psyllid (Diaphorina citri) | Roche 454 GS−FLX | [83] |
| Bemesia tabaci | Four novel Begomovirus species were discovered in their viruliferous vectors | Purified viral DNA | Metagenomic reads 100–700 nt | [84] |
| Aodelphgax striatellus (small brown leafhopper) | The presence of Rice stripe virus siRNAs was demonstrated in the viruliferous vector L. striatellus | siRNAs | Illumina | [48] |
| Host | Study finding/pathogen | Sample preparation/target | Sequencing platform | Ref. |
|---|---|---|---|---|
| Grapevine | It was demonstrated that sequences of infected phytoplasmas belonged to 16 SrV and 16 SrXII groups, as well as to Candidatus Phytoplasma prunorum (16SrX-B) whereas some sequences could not be assigned to a single phytoplasma group. Also a high number of single nucleotide polymorphisms (SNPs) were found. Suggested NGS may be used for future phytoplasma detection in quarantine. | Total DNA from mid-vein leaf tissue | Amplicon sequencing by Roche 454 GS FLX | [85,86] |
| Grapevine | Demonstrated significant changes in the transcriptome of Aster yellows phytoplama-infected Grapevine cv. Chardonnay. The study could contribute to understanding the unknown mechanisms of phytoplasma pathogenicity. | Total RNA and DNA | Illumina HiSeq 2000 | [87] |
| Periwinkle | Genomic analysis of four phytoplasma strains of 16SrIII group and two strains of the 16SrI-B subgroup revealed the significant role of horizontal gene transfer among different “Ca. Phytoplasma” species in shaping phytoplasma genomes and promoting their diversity. | Standard DNA preparation from infected periwinkle | Illumina | [88] |
| Citrus (Mexican lime) | Identified miRNA families that are expressed differentially upon infection of Mexican lime trees with Candidatus Phytoplasma aurantifolia. The study increases our understanding of the molecular basis of witches’ broom disease which may lead to development of new strategies for its control. | miRNAs were isolated from infected and from healthy tissues | Illumina HiSeq 2000 | [89] |
| Citrus | Demonstrated that several miRNAs and siRNAs were highly induced by Ca. L. Asiaticus (Las) infection, which can be potentially developed into early diagnosis markers of huanglongbing (HLB) disease (citrus greening disease). MiR399 was induced specifically by infection of Las. MiR399 is induced by phosphorous starvation in other plant species. Applying phosphorous significantly reduced HLB symptoms in citrus. | RNA fragments 18–28 nt in length obtained after fractionation of total RNA on denaturing polyacrylamide gel electrophoresis. The total RNA was extracted from Las-infected tissue with HLB symptoms | Illumina | [90] |
7. Bioinformatics’ Software Tools for Data Analysis
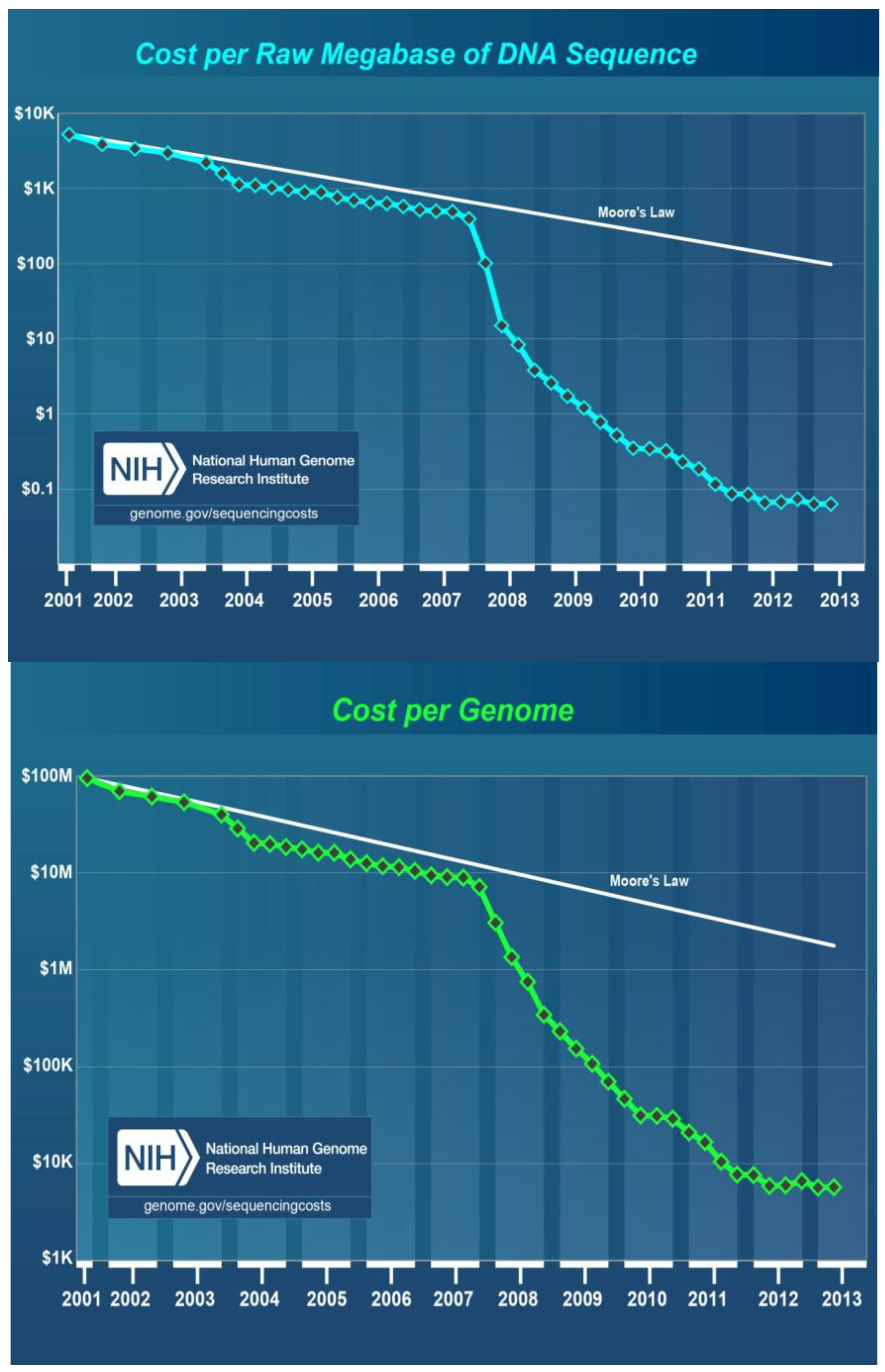
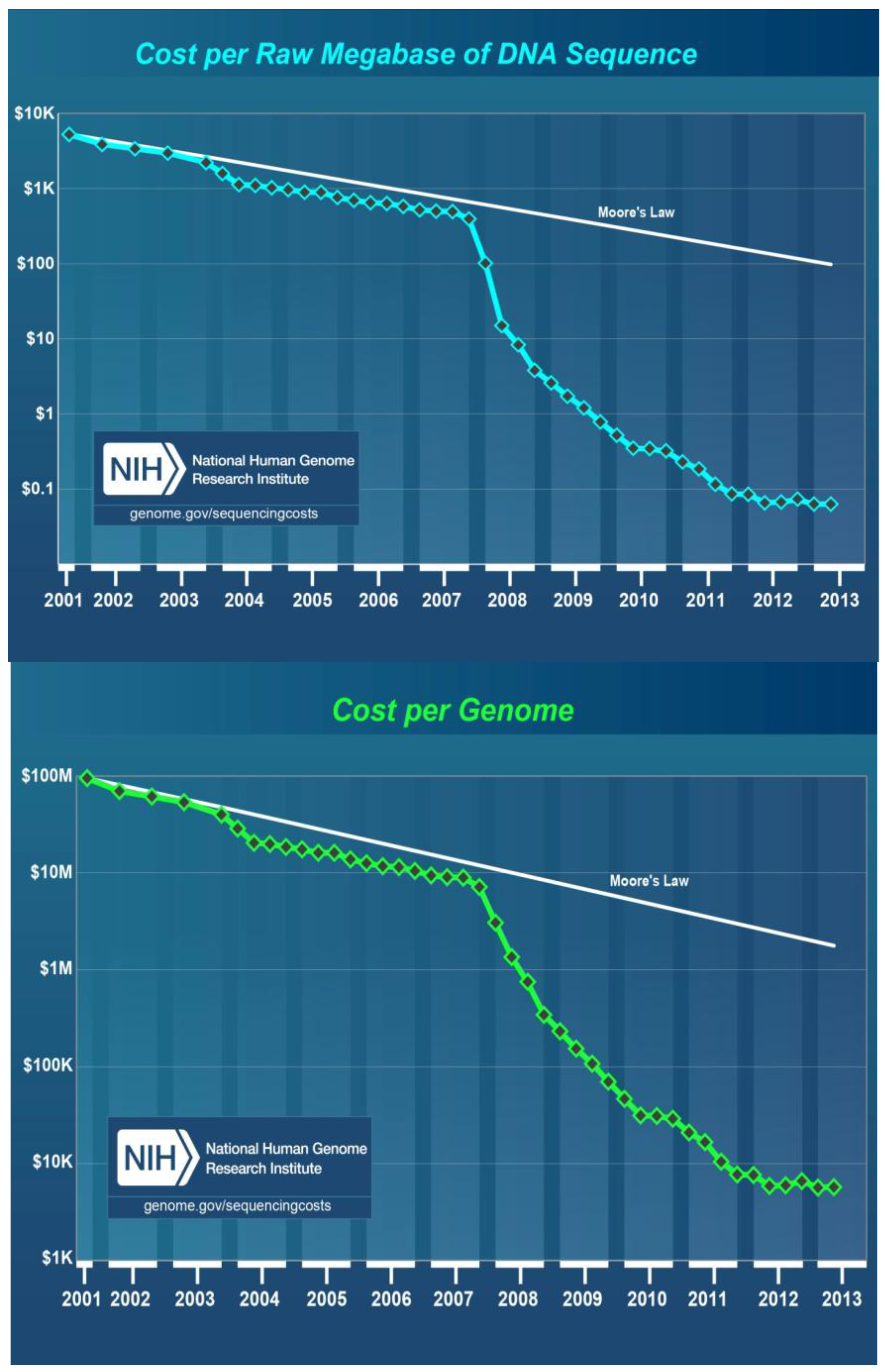
8. Cost of DNA Sequencing
9. Biological Applications of Next-Generation Sequencing
10. Applications of Next-Generation Sequencing in Medical Virology
11. Current Applications of Next-Generation Sequencing in Plant Virology
12. Next-Generation Sequencing and Revealing the Etiology of Unknown Diseases and Latent Infections
13. Prospective
14. Conclusions
Acknowledgments
Conflicts of Interest
References and Notes
- Watson, J.; Crick, F.H.C. A structure for deoxyribose nucleic acid. Nature 1953, 171, 737–738. [Google Scholar] [CrossRef]
- Holley, R.W.; Madison, J.T.; Zamir, A. A new method for sequence determination of large oligonucleotides. Biochem. Biophys. Res. Commun. 1964, 17, 389–394. [Google Scholar] [CrossRef]
- Holley, R.W.; Apgar, J.; Everett, G.A.; Madison, J.T.; Marquisee, M.; Merrill, S.H.; Penswick, J.R.; Zamir, A. Structure of a ribonucleic acid. Science 1965, 147, 1462–1465. [Google Scholar]
- Sanger, F.; Air, G.M.; Barrell, B.G.; Brown, N.L.; Coulson, A.R.; Fiddes, J.C.; Hutchison, C.A.; Smith, M. Nucleotide sequence of bacteriophage phX174DNA. Nature 1977, 265, 687–695. [Google Scholar] [CrossRef]
- Sanger, F.; Nicklen, S.; Coulsen, A.R. DNA sequencing with chain-terminator inhibitors. Proc. Natl. Acad. Sci. USA 1977, 74, 5463–5467. [Google Scholar] [CrossRef]
- Maxam, A.M.; Gilbert, W.A. A new method for sequencing DNA. Proc. Natl. Acad. Sci. USA 1977, 74, 560–564. [Google Scholar] [CrossRef]
- Jackson, D.; Symons, R.H.; Berg, P. Biochemical method for inserting new genetic information into DNA of Simian virus 40: Circular SV40 DNA molecules containing Lambda phage genes and the galactose operon of Escherichia coli. Proc. Natl. Acad. Sci. USA 1972, 69, 2904–2909. [Google Scholar] [CrossRef]
- Gilbert, W.; Maxam, A. The nucleotide sequence of the lac operator. Proc. Natl. Acad. Sci. USA 1973, 70, 3581–3584. [Google Scholar] [CrossRef]
- Baer, R.; Bankier, A.T.; Biggin, M.D.; Deininger, P.L.; Farrell, P.J.; Gibson, T.J.; Hatfull, G.; Hudson, G.S.; Satchwell, S.C.; Seguin, C.; et al. DNA sequence and expression of the B95-8 Epstein-Barr virus genome. Nature 1984, 310, 207–211. [Google Scholar] [CrossRef]
- Fleischmann, R.D.; Adams, M.D.; White, O.; Clayton, R.A.; Kirkness, E.; Kerlavage, A.R.; Bult, C.J. Whole-genome random sequencing and assembly of Haemophilus influenzae Rd. Science 1995, 269, 496–512. [Google Scholar]
- Ronaghi, M.; Karamohame’d, S.; Pettersson, B.; Uhlén, M.; Nyrén, P. Real-time DNA sequencing using detection of pyrophosphate release. Anal. Biochem. 1996, 242, 84–89. [Google Scholar] [CrossRef]
- Kawashima, E.; Farinelli, L.; Mayer, P. Method of Nucleic Acid Amplification. Int. Appl. No. PCT/GB98/00961, 8 October 1998. [Google Scholar]
- Ewing, B.; Green, P. Base-calling of automated sequencer traces using Phred. II. Error probabilities. Genomic Res. 1998, 8, 186–194. [Google Scholar]
- Life Sciences, a Roche Company. Available online: http://www.454.com/ (accessed on 22 October 2012).
- Illumina (Illumina). Available online: http://www.illumina.com/technology/sequencing_technology/ilmn/ (accessed on 22 October 2012).
- SOLiD (Life Technologies). Available online: http://www.lifetechnologies.com/us/en/home/life-science/sequencing/next-generation-sequencing/solid-next-generation-sequencing.html/ (accessed on 24 October 2012).
- Ion-Torrent (Life Technologies). Available online: http://www.lifetechnologies.com/us/en/home/life-science/sequencing/next-generation-sequencing/ion-torrent-next-generation-sequencing-orkflow/ion-torrent-next-generation-sequencing-run-sequence.html/ (accessed on 25 October 2012).
- PacBio (Pacific Bioscience). Available online: http://www.pacificbiosciences.com/ (accessed on 3 November 2012).
- Nanopore (Oxford Technologies). Available online: https://www.nanoporetech.com/ (accessed on 5 November 2012).
- Loman, N.J.; Misra, R.V.; Dallman, T.J.; Constantinidou, C.; Gharbia, S.E.; Wain, J.; Pallen, M.J. Performance comparison of benchtop high-throughput sequencing platforms. Nat. Biotechnol. 2012, 30, 434–439. [Google Scholar] [CrossRef] [Green Version]
- Radford, A.D.; Chapman, D.; Dixon, L.; Chantrey, J.; Darby, A.C.; Hall, N. Application of next-generation sequencing technologies in virology. J. Gen. Virol. 2012, 93, 1853–1868. [Google Scholar] [CrossRef]
- Liu, L.; Li, Y.; Li, S.; Hu, N.; He, Y.; Pong, R.; Lin, D.; Lu, L.; Law, M. Comparison of next-generation sequencing systems. J. Biomed. Biotechnol. 2012, 2012. [Google Scholar] [CrossRef]
- Archer, J.; Baillie, G.; Watson, S.J.; Kellam, P.; Rambaul, A.; Robertson, D.L. Analysis of high-depth sequence data for studying viral diversity: A comparison of next generation sequencing platforms using Segminator II. BMC Bioinform. 2012, 13. [Google Scholar] [CrossRef]
- Kirsher, M.; Kelso, J. High-throughput DNA sequencing-concepts and limitations. Bioassays 2010, 32, 524–536. [Google Scholar] [CrossRef]
- Carniero, M.O.; Russ, C.; Ross, M.G.; Gabriel, S.B.; Nasbaum, C.; DePristo, M.A. Pacific biosciences sequencing technology for genotyping and variation discovery in human data. BMC Genomics 2012, 13. [Google Scholar] [CrossRef]
- Bragg, L.M.; Stone, G.; Butler, M.K.; Hugenholtz, P.; Tyson, G.W. Shining a light on dark sequencing: Characterising errors in Ion Torrent PGM data. PLoS Comput. Biol. 2013, 9, e1003031. [Google Scholar] [CrossRef]
- Kreuze, J.F.; Perez, A.; Untiveros, M.; Quispe, D.; Fuentes, S.; Barker, I.; Simon, R. Complete viral genome sequence and discovery of novel viruses by deep sequencing of small RNAs: A generic method for diagnosis, discovery and sequencing of viruses. Virology 2009, 388, 1–7. [Google Scholar] [CrossRef]
- Adams, I.P.; Glover, R.H.; Monger, W.A.; Mumford, R.; Jackeviciene, E.; Navalinskiene, M.; Samuitiene, M.; Boonham, N. Next-generation sequencing and metagenomic analysis: A universal diagnostic tool in plant virology. Mol. Plant. Pathol. 2009, 10, 537–545. [Google Scholar] [CrossRef]
- Qi, X.; Bao, F.S.; Xie, Z. Small RNA deep sequencing reveals role Arabidopsis thaliana for RNA-dependent RNA polymerases in viral siRNA biogenesis. PLoS One 2009, 4, e4971. [Google Scholar]
- Donaire, L.; Wang, Y.; Gonzales-Ibeas, D.; Mayer, K.F.; Aranda, M.A.; Liave, C. Deep sequencing of plant viral small RNAs reveals effective and widespread targeting of viral genomes. Virology 2009, 392, 203–214. [Google Scholar] [CrossRef] [Green Version]
- Monger, W.A.; Alicai, T.; Ndunguru, J.; Kinyua, Z.M.; Potts, M.; Reeder, R.H.; Miano, D.W.; Adams, I.P.; Boonham, N.; Glover, R.H.; et al. The complete genome sequence of the Tanzanian strain of Cassava brown streak virus and comparison with the Ugandan strain sequence. Arch. Virol. 2010, 155, 429–433. [Google Scholar] [CrossRef]
- Szittya, G.; Moxon, S.; Pantaleo, V.; Toth, G.; Rusholme, P.R.L.; Moulton, V.; Burgyan, J.; Dalmay, T. Structural and functional analysis of viral siRNAs. PLoS Pathog. 2010, 6, e1000838. [Google Scholar] [CrossRef]
- Roossinck, M.J.; Saha, P.; Wiley, G.B.; Quan, J.; White, J.D.; Lai, H.; Chavarria, F.; Shen, G.; Roe, B.A. Ecogenomics: Using massively parallel pyrosequencing to understand virus ecology. Mol. Ecol. 2010, 19, 81–88. [Google Scholar] [CrossRef]
- Pallett, D.W.; Ho, T.; Cooper, I.; Wang, H. Detection of Cereal yellow dwarf virus using small interfering RNAs and enhanced infection rate with Cocksfoot streak virus in wild cocksfoot grass (Dactylis glomerata). J. Virol. Methods 2010, 168, 223–227. [Google Scholar] [CrossRef]
- Lin, K.-Y.; Cheng, C.-P.; Chang, B.C.-H.; Wang, W.-C.; Huang, Y.-W.; Lee, Y.-S.; Huang, H.-D.; Hsu, Y.-H.; Lin, N.-S. Global analysis of small interfering RNAs derived from Bamboo mosaic virus and its associated satellite RNAs in different plants. PLoS One 2010, 5, e11928. [Google Scholar] [CrossRef]
- Yan, F.; Zhang, H.; Adams, M.; Yang, J.; Peng, J.; Antoniw, J.; Zhou, Y.; Chen, J. Characterization of siRNAs derived from rice stripe virus in infected rice plants by deep Sequencing. Arch. Virol. 2010, 155, 935–940. [Google Scholar] [CrossRef]
- Silva, T.F.; Romanel, E.A.C.; Andrade, R.R.S.; Farinelli, L.; Osteras, M.; Deluen, C.; Correa, R.L.; Schrago, C.E.G.; Vaslin, M.F.S. Profile of small interfering RNAa from cotton plants infected with the polerovirus Cotton leafroll dwarf virus. BMC Mol. Biol. 2011, 12. [Google Scholar] [CrossRef]
- Wylie, S.J.; Jones, M.G.K. The complete genome sequence of a Passion fruit woodiness virus isolate from Australia determined using deep sequencing, and its relationship to other potyviruses. Arch. Virol. 2011, 156, 479–482. [Google Scholar] [CrossRef]
- Dombrovsky, A.; Glanz, E.; Sapkota, R.; Lachman, O.; Bronstein, M.; schnitzer, T.; Antignus, Y. Next-generation sequencing a rapid and reliable method to obtain sequence data of the genomes of undescribed plant viruses. In Proceedings of the BARD-Sponsored Workshop—Microarrays and Next-Generation Sequencing for Detection and Identification of Plant Viruses, Beltsville, MD, USA, 17–19 November 2011. Abstract No. 24.
- Mumford, R.; Adams, I.P.; Glover, R.; Hany, U.; Boonham, N. From high throughput to high output clondiag microarrays and 454 sequence for viral detection and discovery. In Proceedings of the ARD-Sponsored Workshop—Microarrays and Next-Generation Sequencing for Detection and Identification of Plant Viruses, Beltsville, MD, USA, 17–19 November 2011. Abstract No. 17.
- Hagen, C.; Frizzi, A.; Kao, J.; Jia, L.; Huang, M.; Zhang, Y.; Huang, S. Using small RNA sequences to diagnose, sequence, and investigate the infectivity characteristics of vegetable-infecting viruses. Arch. Virol. 2011, 156, 1209–1216. [Google Scholar] [CrossRef]
- Hu, Q.; Hollunder, J.; Niehl, A.; Korner, C.J.; Gereige, D.; Windels, D.; Arnold, A.; Kuiper, M.; Vasquez, F.F.; Pooggin, M.; et al. Specific impact of Tobamavirus infection on the Arabidopsis small RNA profile. PLoS One 2011, 6, e19549. [Google Scholar] [CrossRef] [Green Version]
- Yang, X.; Wang, Y.; Guo, W.; Xie, Y.; Xie, Q.; Fan, L.; Zhou, X. Characterization of small interfering RNAs derived from the geminivirus/betasatellite complex using deep sequencing. PLoS One 2011, 6, e16928. [Google Scholar]
- Li, R.; Gao, S.; Hernandez, A.G.; Wechter, W.P.; Fei, Z.; Ling, K.-S. Deep sequencing of small RNAs in tomato for virus and viroid identification and strain differentiation. PLoS One 2012, 7, e37127. [Google Scholar]
- Lu, J.; Du, Z.-X.; Kong, J.; Chen, L.-N.; Qiu, Y.-H.; Li, G.-F.; Meng, X.-H.; Zhu, S.-F. Transcriptome analysis of Nicotiana tabacum infected by Cucumber mosaic virus during systemic symptom development. PLoS One 2012, 7, e43447. [Google Scholar]
- Adams, I.P.; Miano, D.W.; Kinyua, Z.M.; Wangai, A.; Kimani, E.; Phiri, N.; Reeder, R.; Harju, V.; Glover, R.; Hany, U.; et al. Use of next generation sequencing for the identification and characterization of Maize chlorotic mottle virus and Sugarcane mosaic virus causing lethal necrosis in Kenya. Plant Pathol. 2012. [Google Scholar] [CrossRef]
- Wylie, S.J.; Luo, H.; Li, H.; Jones, M.G.K. Multiple polyadenylated RNA viruses detected in pooled cultivated and wild plant samples. Arch. Virol. 2012, 157, 271–284. [Google Scholar] [CrossRef]
- Xu, Y.; Huang, L.; Fu, S.; Wu, J.; Zhou, X. Population diversity of Rice stripe virus—Derived siRNAs in three different hosts and RNAi-based antiviral immunity in Laodelphgax striatellus. PLoS One 2012, 7, e46238. [Google Scholar]
- Kashif, M.; Pietila, S.; Artola, K.; Tugume, A.K.; Makinen, V.; Valkonen, J.P.T. Detection of viruses in sweetpotato from Honduras and Guatemala augmented by deep-sequencing of small-RNAs. Plant Dis. 2012, 96, 1430–1437. [Google Scholar] [CrossRef]
- Fabre, F.; Montarry, J.; Coville, J.; Senoussi, R.; Simon, V.; Murry, B. Modelling the evolutionary dynamics of viruses within their hosts: A case study using high-throughput sequencing. PLoS Pathog. 2012, 8, e1002654. [Google Scholar]
- Blevins, T.; Rajeswaran, R.; Aregger, M.; Borah, B.K.; Schepetilnikov, M.; Baerlocher, L.; Farinelli, L.; Meins, F., Jr.; Hohn, T.; Pooggin, M.M. Massive production of small RNAs from a non-coding region of Cauliflower mosaic virus in plant defense and viral counter-defense. Nucleic Acids Res. 2011, 39, 5003–5014. [Google Scholar] [CrossRef]
- Rajeswaran, R.; Aregger, M.; Zvereva, A.S.; Borah, B.K.; Gubaeva, E.G.; Pooggin, M.M. Sequencing of RDR6-dependent double-stranded RNAs reveals novel features of plant siRNA biogenesis. Nucleic Acids Res. 2012, 40, 6241–6254. [Google Scholar] [CrossRef]
- Aregger, M.; Borah, B.K.; Seguin, J.; Rajeswaran, R.; Gubaeva, E.G.; Zvereva, A.S.; Windels, D.; Vazquez, D.; Blevins, T.; Farinelli, L.; et al. Primary and secondary siRNAs in Geminivirus-induced gene silencing. PLoS Pathog. 2012, 8, e1002941. [Google Scholar] [CrossRef]
- Hany, U.; Adams, I.P.; Glover, R.; Bhat, A.I.; Boonham, N. The complete nucleotide sequence of Piper yellow mottle virus (PYMoV). Arch. Virol. 2013, 158. [Google Scholar] [CrossRef]
- Quito-Avila, D.F.; Jelkmann, W.; Tzanetakis, I.; Keller, K.; Martin, R.R. Complete sequence and genetic characterization of Raspberry latent virus, a novel member of the family Reoviridae. Virus Res. 2011, 155, 397–405. [Google Scholar] [CrossRef]
- Ruiz-Ruiz, S.; Navarro, B.; Gisel, A.; Pena, L.; Navarro, L.; Moreno, P.; di Serio, F.; Flores, R. Citrus tristeza virus infection induces the accumulation of viral small RNAs (21–24 nt) mapping preferentially at the terminal region 3'- of the genomic RNA and affects the host small RNA profile. Plant Mol. Biol. 2011, 75, 607–619. [Google Scholar] [CrossRef] [Green Version]
- Loconsole, G.; Onelge, N.; Potere, O.; Giampetruzzi, A.; Bozan, O.; Satar, S.; de Stradis, A.; Savino, V.; Yokomi, R.K.; Saponari, M. Identification and characterization of Citrus Yellow vein clearing virus, a putative new member of the genus Mandarivirus. Phytopathology 2012, 102, 1168–1175. [Google Scholar] [CrossRef]
- Loconsole, G.; Saldarelli, P.; Doddapaneni, H.; Savino, V.; Martelli, G.P.; Saponari, M. Identification of a single-stranded DNA virus associated with citrus chlorotic dwarf disease, a new member of the family Geminiviridae. Virology 2012, 432, 162–172. [Google Scholar] [CrossRef]
- Maree, H.J.; Nel, Y.; Visser, M.; Coetzec, B.; Manicom, B.; Burger, J.T.; Rees, D.J.G. The study of plant virus disease etiology using next-generation sequencing technologies. In Proceedings of the 22nd International Conference on Virus and Other Transmissible Diseases of Fruit Crops, Rome, Italy, 3–8 June 2012. Abstract No. 48.
- Yoshikawa, N.; Yamagishi, N.; Yaegashi, H.; Ito, T. Deep sequence analysis of viral small RNAs from a green crinkle-diseased apple tree. Petria 2012, 22, 292–297. [Google Scholar]
- Candresse, T.; Marais, A.; Faure, C.; Carriere, S.; Gentit, P. Use of 454 pyrosequencing for the fast and efficient characterization of known or novel viral agents in Prunus materials. In Proceedings of the 22nd International Conference on Virus and Other Transmissible Diseases of Fruit Crops, Rome, Italy, 3–8 June 2012. Abstract No. 47.
- Chiumenti, M.; Roberto, R.; Bottalico, G.; Campanale, A.; de Stradis, A.; Minafra, A.; Boscia, D.; Savino, V.; Martelli, G.P. Virus sanitation and deep sequence analysis of fig. In Proceedings of the 22nd International Conference on Virus and Other Transmissible Diseases of Fruit Crops, Rome, Italy, 3–8 June 2012. Abstract No. 163.
- Thekke-Veetil, T.; Sabanadzovic, S.; Keller, K.E.; Martin, R.R.; Tzanetakis, I.E. Genome organization and sequence diversity of a novel blackberry Ampelovirus. In Proceedings of the 22nd International Conference on Virus and Other Transmissible Diseases of Fruit Crops, Rome, Italy, 3–8 June 2012. Abstract No. 191.
- Candresse, T.; Marais, A.; Faure, C.; Gentit, P. Association of Little cherry virus 1 (LChV1) with the Shirofugen stunt disease and characterization of the genome of a divergent LChV1 isolate. Phytopathology 2013, 103, 293–308. [Google Scholar] [CrossRef]
- Roy, A.; Choudhary, N.; Guillermo, L.M.; Shao, J.; Govindarajulu, A.; Achor, D.; Wei, G.; Picton, D.D.; Levy, L.; Nakhla, M.K.; et al. A novel virus of the Genus Cilevirus causing symptoms similar to citrus leprosis. Phytopathology 2013, 103, 488–500. [Google Scholar] [CrossRef]
- Vives, M.C.; Velazquez, K.; Pina, J.A.; Moreno, P.; Guerri, J.; Navarro, L. Identification of a new enamovirus associated with citrus vein enation disease by deep sequencing of small RNAs. Phytopathology 2013, 103, 1077–1086. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; Golino, D.; Rowhani, A. Deep sequencing analysis of RNAs from a grapevine showing Syrah decline symptoms reveals a multiple virus infection that includes a novel virus. Virology 2009, 387, 395–401. [Google Scholar] [CrossRef]
- Coetzee, B.; Freeborough, M.J.; Maree, H.J.; Celton, J.M.; Rees, D.J.; Burger, J.T. Deep sequencing analysis of viruses infecting grapevines: Virome of a vineyard. Virology 2010, 400, 157–163. [Google Scholar] [CrossRef]
- Pantaleo, V.; Saldarelli, P.; Miozzi, L.; Giampetruzzi, A.; Gisel, A.; Moxon, S.; Dalmay, T.; Bisztray, G.; Burgyan, J. Deep sequencing analysis of viral short RNAs from an infected Pinot noir grapevine. Virology 2010, 408, 49–56. [Google Scholar] [CrossRef]
- Zhang, Y.; Singh, K.; Kaur, R.; Qiu, W. Association of a novel DNA virus with the grapevine vein-clearing and decline syndrome. Phytopathology 2011, 101, 1081–1090. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Daubert, S.; U'rbez-Torres, J.R.; Cordero, F.; Rowhani, A. Deep sequencing evidence from single grapevine plants reveals a virome dominated by mycoviruses. Arch. Virol. 2011, 156, 397–403. [Google Scholar] [CrossRef]
- Giampetruzzi, A.; Roumi, V.; Roberto, R.; Malossini, U.; Yoshikawa, N.; la Notte, P.; Terlizzi, F.; Credi, R.; Saldarelli, P. A new grapevine virus discovered by deep sequencing of virus- and viroid-derived small small RNAs in Cv Pinot gris. Virus Res. 2012, 163, 262–268. [Google Scholar] [CrossRef]
- Al Rwahnih, M.; Dave, A.; Anderson, M.; Uyemoto, J.K.; Sudarshana, M.R. Association of a circular DNA virus in grapevine affected by red blotch disease in California. In Proceedings of the 17th Congress of ICVG, Davis, CA, USA, 7–14 October 2012.
- Al Rwahnih, M.; Sudarshana, M.R.; Uyemoto, J.K.; Rowhani, A. Complete genome sequence of a novel Vitivirus isolated from grapevine. In Proceedings of the 17th Congress of ICVG, Davis, CA, USA, 7–14 October 2012.
- Alabi, O.J.; Zheng, Y.; Jagadeeswaran, G.; Sunkar, R.; Naidu, R. High-throughput sequence analysis of small RNAs in grapevine (Vitis. vinifera L.) affected by grapevine leafroll disease. In Proceedings of the 17th Congress of ICVG, Davis, CA, USA, 7–14 October 2012.
- Poojari, S.; Alabi, O.J.; Fofanov, V.Y.; Naidu, R.A. A leafhopper-transmissible DNA virus with novel evolutionary lineage in the family Geminiviridae implicated in grapevine redleaf disease by next-Generation sequencing. PLoS One 2013, 8, e64194. [Google Scholar]
- Navarro, B.; Pantaleo, V.; Gisel, A.; Moxon, S.; Dalmay, T.; Bistray, G.; di Serio, F.; Burgyan, J. Deep sequencing of viroid-derived small RNAs from grapevine provides new insight on the role of RNA silencing in plant-viroid interaction. PLoS One 2009, 4, e7686. [Google Scholar]
- Di Serio, F.; Gisel, A.; Navarro, B.; Delgado, S.; Martínez de Alba, A.-E.; Donvito, G.; Flores, R. Deep sequencing of the small RNAs derived from two symptomatic variants of a chloroplastic viroid: Implications, for their genesis and for pathogenesis. PLoS One 2009, 4, e7539. [Google Scholar]
- Di Serio, F.; Martínez de Alba, A.-E.; Navarro, B.A.; Gisel, A.; Flores, R. RNA-dependent RNA polymerase 6 delays accumulation and precludes meristem invasion of a viroid that replicates in the nucleus. J. Virol. 2010, 84, 2477–2489. [Google Scholar] [CrossRef]
- Martinez, G.; Donaire, L.; Llave, C.; Pallas, V.; Gomez, G. High-throughput sequencing of Hop stunt viroid-derived small RNAs from cucumber leaves and phloem. Mol. Plant Pathol. 2010, 11, 347–359. [Google Scholar] [CrossRef]
- Chiumenti, M.; Giampetruzzi, A.; Pirolo, C.; Morelli, M.; Saldarelli, P.; Minafra, A.; Bottalico, G.; la Notte, P.; Campanale, A.; Savino, V.; et al. Approaxhes of next generation sequencing to investigate grapevine diseases of unknown etiology. In Proceedings of the 17th Congress of ICVG, Davis, CA, USA, 7–14 October 2012.
- Wu, Q.; Wang, Y.; Cao, M.; Pantaleo, V.; Burgyan, J.; Li, W.X.; Ding, S.W. Homology-independent discovery of replicating pathogenic circular RNAs by Deep sequencing and a new computational algorithm. Proc. Natl. Acad. Sci. USA 2012, 109, 3938–3943. [Google Scholar] [CrossRef]
- Duan, Y.; Zhou, L.; Hall, D.G.; Li, W.; Doddapaneni, H.; Lin, H.; Liu, L.; Vahling, C.M.; Gabriel, D.W.; Williams, K.P.; et al. Complete genome sequence of citrus huanglongbing bacterium, “Candidatus Liberibacter asiaticus” obtaine through metagenomics. Mol. Plant MicrobeInteract. 2009, 22, 1011–1020. [Google Scholar] [CrossRef]
- Ng, T.F.F.; Duffy, S.; Polston, J.E.; Bixby, E.; Vallad, G.E.; Breitbart, M. Exploring the diversity of plant DNA viruses and their satellites using vector-enabled metagenomics on whiteflies. PLoS One 2011, 6, e19050. [Google Scholar]
- Contaldo, N.; Canel, A.; Paltrinieri, S.; Bertaccini, A.; Nicolaisen, M. Phytoplasma detection and identification in grapevine by deep amplicon sequencing. In Proceedings of the 17th Congress of ICVG, Davis, CA, USA, 7–14 October 2012.
- Nicolaisen, M.; Contaldo, N.; Makarova, O.; Paltrinieri, S.; Bertaccini, A. Deep amplicon sequencing reveals mixed phytoplasma infection within single grapevine plants. Bull. Insectol. 2011, 64, S35–S36. [Google Scholar]
- Snyman, M.C.; Solofoharivelo, M.C.; van der Walt, A.; Stephan, D.; Murray, S.; Burger, J.T. Deep sequencing analysis reveals modulated gene expression in response to aster yellow phytoplasma infection in Vitis vinifera cv. Chardonnay. In Proceedings of the 17th Congress of ICVG, Davis, CA, USA, 7–14 October 2012.
- Palmano, S.; Saccardo, F.; Martini, M.; Ermacora, P.; Scortichini, M.; Abbà, S.; Marzachì, C.; Loi, N.; Firrao, G. Insights into phytoplasma biology through next generation sequencing. J. Plant Pathol. 2012, 94, 50–51. [Google Scholar]
- Ehya, F.; Monavarfeshani, A.; Fard, E.M.; Farsad, L.K.; Nekouei, M.K.; Mardi, M.; Salekdeh, G.H. Phytoplama-responsive microRNAs modulate hormonal, nutrional, and stress signalling pathways in Mexican lime trees. PLoS One 2013, 8, e6372. [Google Scholar]
- Zhao, H.; Sun, R.; Albrecht, U.; Padmanabhan, C.; Wang, A.; Coffey, M.D.; Girke, T.; Wang, Z.; Close, T.J.; Roose, M.; et al. Small RNA profiling reveals phosphorous deficiency as a contributin factor in symptom expression for Citrus Huanglongbing disease. Mol. Plant 2013, 6, 301–310. [Google Scholar] [CrossRef]
- Paszkiewicz, K.; Studholme, D.J. De novo assembly of short sequence reads. Brief Bioinform. 2010, 11, 457–472. [Google Scholar] [CrossRef]
- Shendure, J.; Ji, H. Next-generation DNA sequencing. Nat. Biotechnol. 2008, 26, 1135–1145. [Google Scholar] [CrossRef]
- Zhang, W.; Chen, J.; Yang, Y.; Tang, Y.; Shang, J; Chen, B. A practical application of de novo genome assembly software tools for next-generation sequencing technologies. PLoS One 2011, 6, e17915. [Google Scholar]
- Horner, D.S.; Pavesi, G.; Castrignano, T.; D’Onorio de Meo, P.; Liuni, S.; Sammeth, M; Picardi, E.; Pesole, G. Bioinformatics approaches for genomics and post genomics applications of next generation sequencing. Brief Bioinform. 2009, 11, 181–197. [Google Scholar]
- Metzker, M.L. Sequencing technologies—The next generation. Nat. Rev. Genet. 2010, 11, 31–46. [Google Scholar] [CrossRef]
- Miller, J.R.; Koren, S.; Sutton, G. Assembly algorithms for next generation sequencing data. Genetics 2010, 95, 315–327. [Google Scholar]
- Pabinger, S.; Dander, A.; Fischer, M.; Snajder, R.; Sperk, M.; Efremova, M.; Krabichler, B.; Speicher, M.R.; Zschocke, J.; Trajanoski, Z. A survey of tools for variant analysis of next-generation genome sequencing data. Brief Bioinform. 2013. [Google Scholar] [CrossRef]
- Bioinformatics for Biologists; Pevzner, P.; Shamir, R. (Eds.) Cambridge University Press: Cambridge, UK, 2011; p. 392.
- Wetterstrand, K.A. DNA sequencing costs: Data from the NHGRI Genome Sequencing Program (GSP). Available online: http://www.genome.gov/sequencingcosts/ (accessed on 1 October 2013).
- Wang, Z.; Gerstein, M.; Snyder, M. RNA-Seq: A revolutionary tool for transcriptomics. Nat. Rev. Genet. 2009, 10, 57–63. [Google Scholar] [CrossRef]
- Hoffmann, C.; Minkah, N.; Leipzig, J.; Wang, G.; Arens, M.Q.; Tebas, P.; Bushman, F.D. DNA bar coding and pyrosequencing to identify rare HIV drug resistance mutations. Nucleic Acids Res. 2007, 35, e91. [Google Scholar] [CrossRef]
- Wang, C.; Mitsuya, Y.; Gharizadeh, B.; Ronaghi, M.; Shafer, R.W. Characterization of mutation spectra with ultra-deep pyrosequencing: Application to HIV-1 drug resistance. Genome Res. 2007, 17, 1195–1201. [Google Scholar] [CrossRef]
- Briese, T.; Paweska, J.T.; McMullan, L.K.; Hutchison, S.K.; Street, C.; Palacios, G.; Khristova, M.L.; Weyer, J.; Swanepoel, R.; Egholm, M.; et al. Genetic detection and characterization of Lujo virus, a new hemorrhagic fever-associated arenavirus from southern Africa. PLoS Pathog. 2009, 5, e1000455. [Google Scholar] [CrossRef]
- Barzon, L.; Lavezzo, E.; Militello, V.; Toppo, S.; Palù, G. Applications of next-generation sequencing technologies to diagnostic virology. Int. J. Mol. Sci. 2011, 12, 7861–7884. [Google Scholar] [CrossRef]
- Barba, M.; Hadidi, A. RNA silencing and viroids. J. Plant. Pathol. 2009, 91, 243–247. [Google Scholar]
- Mlotshwa, S.; Pruss, G.J.; Vance, V. Small RNAs in viral infection and host Defense. Trends Plant Sci. 2008, 13, 375–382. [Google Scholar] [CrossRef]
- Liu, S.; Vijayendran, D.; Bonning, B.C. Next generation sequencing technologies for insect virus discovery. Viruses 2011, 3, 1849–1869. [Google Scholar] [CrossRef]
- Howell, W.E.; Thompson, D.; Scott, S. Virus-like disorders of fruit trees with undetermined etiology. In Virus and Virus-Like Diseases of Pome and Stone Fruits; Hadidi, A., Barba, M., Candresse, T., Jelkmann, W., Eds.; The American Phytopathological Society Press: St. Paul, MN, USA, 2011; pp. 259–265. [Google Scholar]
- Martin, R.R. Personal communication. US Department of Agriculture, Agricultural Research Service: Corvallis, OR, USA, 2012. [Google Scholar]
- Virus Taxonomy Ninth Report of the International Committee on Taxonomy of Viruses; King, A.M.Q.; Adams, M.J.; Carstens, E.B.; Lefkowitz, E.J. (Eds.) Elsevier Academic Press: San Diego, CA, USA, 2012; p. 1327.
- Hadidi, A.; Barba, M. Next-generation sequencing: Historical perspective and current applications in plant virology. Petria 2012, 22, 262–277. [Google Scholar]
- Bar-Joseph, M.; Gera, A. On a revolutionary method for diagnosis of latent infections of viruses and the importance of its adoption to prevent the spread of citrus and other fruit tree diseases (in Hebrew). Alon Hanotea 2012, 66, 46–48. [Google Scholar]
- Martelli, G.P. Grapevine virology highlights. In Proceedings of the 17th Congress of ICVG, Davis, CA, USA, 7–14 October 2012.
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Barba, M.; Czosnek, H.; Hadidi, A. Historical Perspective, Development and Applications of Next-Generation Sequencing in Plant Virology. Viruses 2014, 6, 106-136. https://doi.org/10.3390/v6010106
Barba M, Czosnek H, Hadidi A. Historical Perspective, Development and Applications of Next-Generation Sequencing in Plant Virology. Viruses. 2014; 6(1):106-136. https://doi.org/10.3390/v6010106
Chicago/Turabian StyleBarba, Marina, Henryk Czosnek, and Ahmed Hadidi. 2014. "Historical Perspective, Development and Applications of Next-Generation Sequencing in Plant Virology" Viruses 6, no. 1: 106-136. https://doi.org/10.3390/v6010106