


Viruses 2025, 17(10), 1384; https://doi.org/10.3390/v17101384 (registering DOI) - 17 Oct 2025
Abstract
Human immunodeficiency virus type 2 (HIV-2) is a lentivirus closely related to HIV-1 but exhibits distinct molecular and clinical features that influence viral infectivity and efficacy of antiretroviral therapy. The HIV capsid is a critical structural component with multifaceted roles during infection and
[...] Read more.
Human immunodeficiency virus type 2 (HIV-2) is a lentivirus closely related to HIV-1 but exhibits distinct molecular and clinical features that influence viral infectivity and efficacy of antiretroviral therapy. The HIV capsid is a critical structural component with multifaceted roles during infection and mediates some of the observed divergence between HIV-1 and HIV-2. Unlike HIV-1, study of the HIV-2 capsid is limited and standard protocols for the in vitro assembly of HIV-1 capsid protein (CA) lattice structures have not been successfully translated to the HIV-2 context. This work identifies effective approaches for the assembly of the HIV-2 CA lattice and leverages this to biochemically characterize HIV-2 CA assemblies and mutant phenotypes. Our findings elaborate on the sensitivity of HIV-2 CA to chemical conditions and reveal that it assembles into a more varied spectrum of particle morphologies compared to HIV-1. Utilizing these assemblies, we tested the hypothesis that HIV-1 and HIV-2 employ divergent mechanisms to stabilize CA oligomer forms and investigate the effects of non-conserved substitutions at the CA inter-protomer interfaces. This work advances our understanding of the key biochemical determinants of HIV-2 CA assembly that are distinct from HIV-1 and may contribute to their divergent virological properties.
Full article
(This article belongs to the Special Issue Structural and Mechanistic Advances in Retroviral Biology)
►
Show Figures

Figure 1



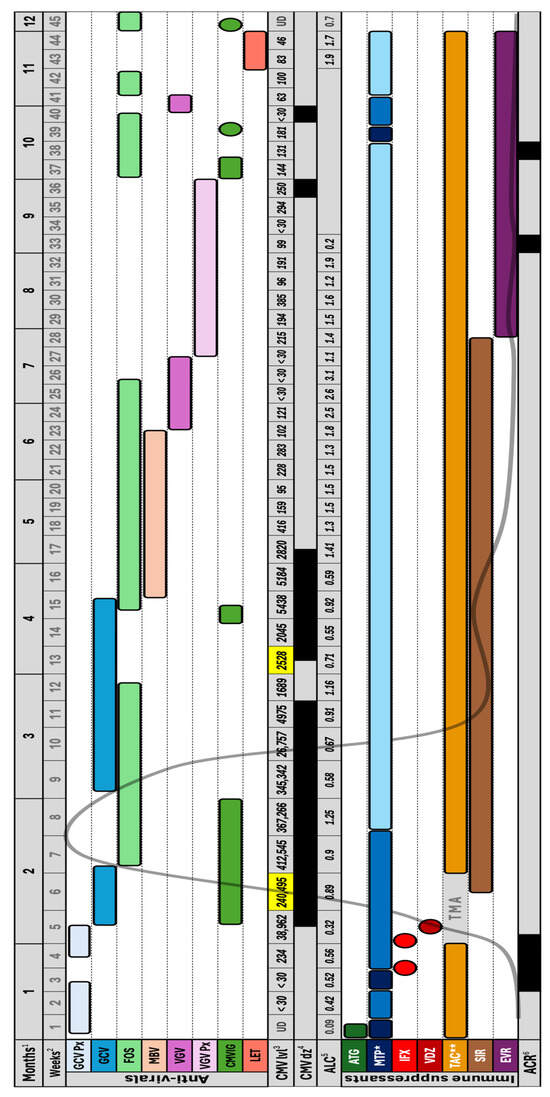

















{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}