Histone Deacetylases in Herpesvirus Replication and Virus-Stimulated Host Defense

Abstract
:1. Introduction
2. Viral Control of HDAC Complexes

{kind=link}
{kind=link}
{kind=link}
| Enzyme | Virus | Interaction | Functional consequence | Ref. |
|---|---|---|---|---|
| HDAC1 | HSV-1 | ICP8 | Redistribution of HDAC1/CoREST and LSD1 to cytoplasm | [28,29,30,31] |
| HSV-1 | ICP0 | Disrupts CoREST association; localizes HDAC1/ICP0 to ND10 bodies | [28,32,33,34,35] | |
| HSV-1 | US3 | Upstream effector of HDAC1 phosphorylation | [32,36,37] | |
| HCMV | pUL29/28 | Associates with HDAC1/HDAC2 and NuRD to promote expression of viral genes | [38,39] | |
| HCMV | pUL38 | Associates with HDAC1/HDAC2 and NuRD complex members via pUL29/28 bridge | [38,39] | |
| HCMV | IE86 | Co-expression promotes MIEP repression | [40] | |
| EBV | ENBA3C | Represses Cp promoter via association with co-repressor complexes (e.g., mSin3A and NCoR) | [41,42] | |
| EBV | TRF2 | Deacetylation of OriP; promotes stability of latent genome | [43] | |
| KSHV | ORF50 promoter | Proposed to modulate promoter acetylation status and LANA acetylation | [2,44] | |
| HDAC2 | HSV-1 | US3 | Upstream effector of HDAC2 phosphorylation | [32,36,37] |
| HCMV | pUL29/28 | Associates with HDAC1/HDAC2 and NuRD to promote expression of viral genes | [38,39] | |
| HCMV | IE2 | De-represses pUL54 promoter; promotes localization of HDAC2 to replications sites | [45] | |
| HCMV | pUL38 | Associates with HDAC1/HDAC2 and NuRD complex members via pUL29/28 bridge | [38,39] | |
| EBV | ENBA3C | Represses Cp promoter via association with co-repressor complexes (e.g., mSin3A and NCoR) | [41,42] | |
| EBV | TRF2 | Deacetylation of OriP; promotes stability of latent genome | [43] | |
| HDAC3 | HCMV | IE1 | Increased acetylation at viral promoter | [46,47] |
| HCMV | IE2 | Increased acetylation at viral promoter | [46,47] | |
| HDAC4 | HSV-1 | ICP0 | Relieves MEF2-binding domain-mediated repression | [48] |
| HDAC5 | HSV-1 | ICP0 | Relieves MEF2-binding domain-mediated repression | [48] |
| KSHV | ORF50 promoter | Proposed to modulate promoter acetylation status and LANA acetylation | [2,44] | |
| HDAC7 | HSV-1 | ICP0 | Relieves MEF2-binding domain-mediated repression | [48] |
| KSHV | ORF50 promoter | Proposed to modulate promoter acetylation status and LANA acetylation | [2,44] |
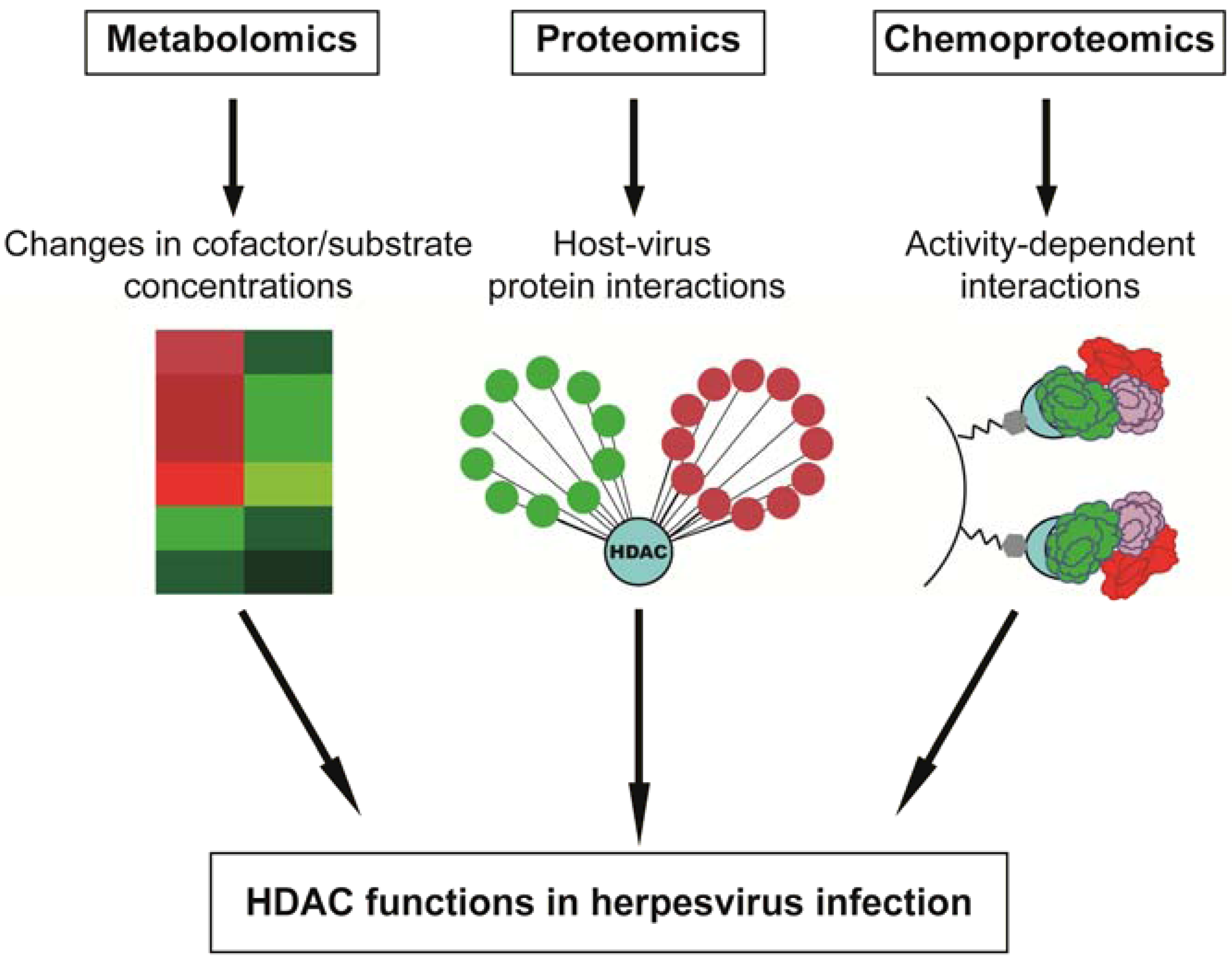
2.1. Infection with the Alpha-Herpesvirus HSV-1 Promotes Misregulation of HDAC1/HDAC2-Containing Complexes
2.2. Beta-Herpesvirus HCMV Proteins Target Class I HDACs to Modulate Viral Gene Transcription
2.3. The Gamma-Herpesviruses EBV and KSHV Regulate HDAC-Containing Co-Repressor Complexes through Protein Interactions and Phosphorylation-Dependent Signal Cascades
3. Host Employment of HDACs and Acetylation in Defense against Herpesviruses
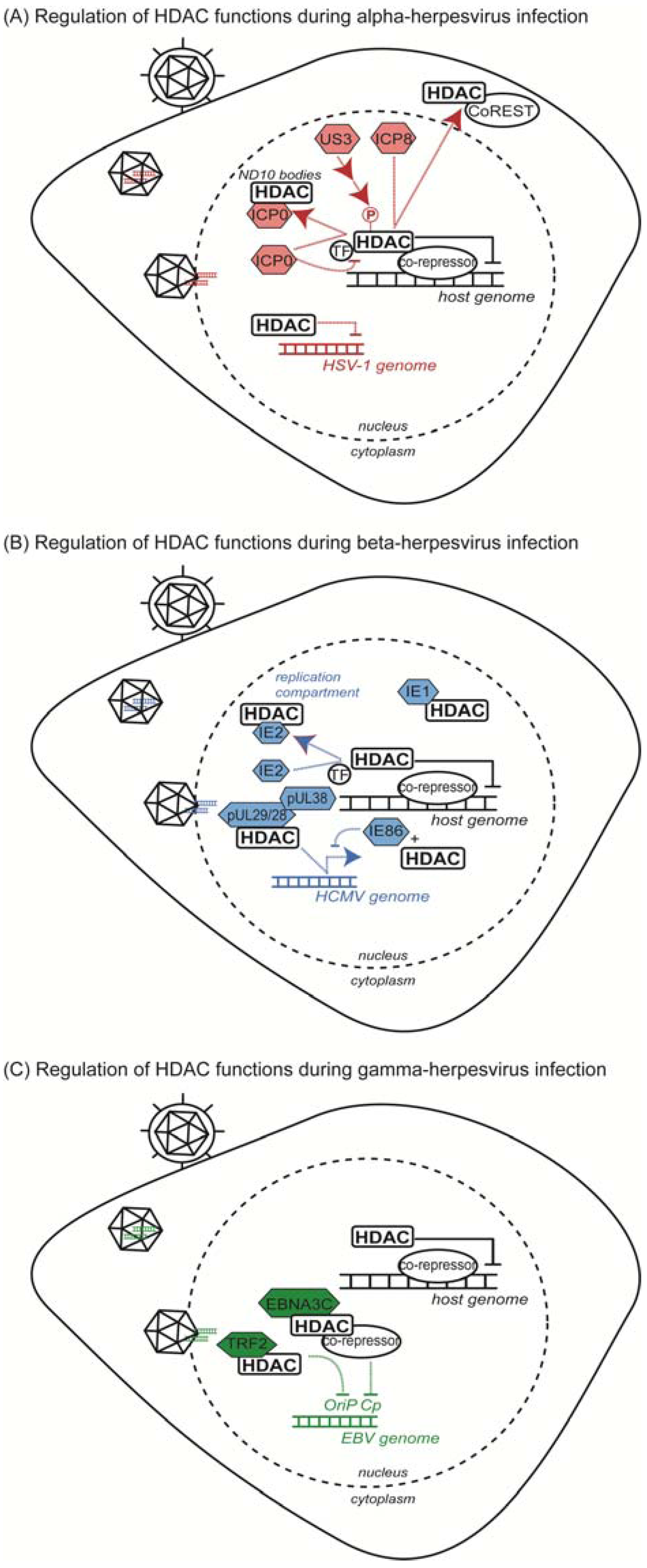
3.1. IFI16 Acetylation and Host Detection of Viral DNA
3.2. HDAC Association with PML/ND10 Bodies during Herpesvirus Infection
3.3. HDAC Non-Histone Substrates in Host Defense
3.4. Roles for Additional Acetylation-Modulating Enzymes during Infection: SIRTs and HATs
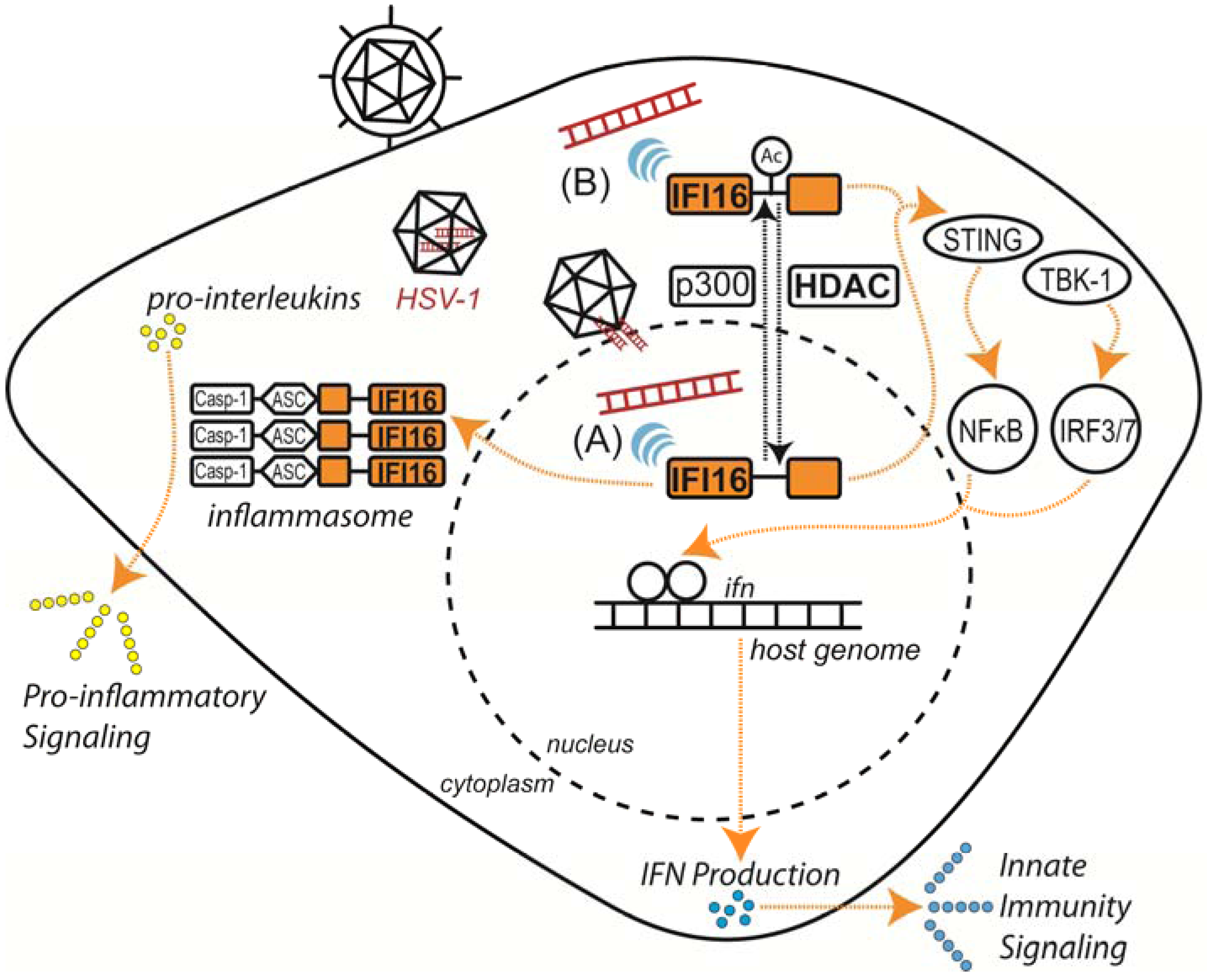
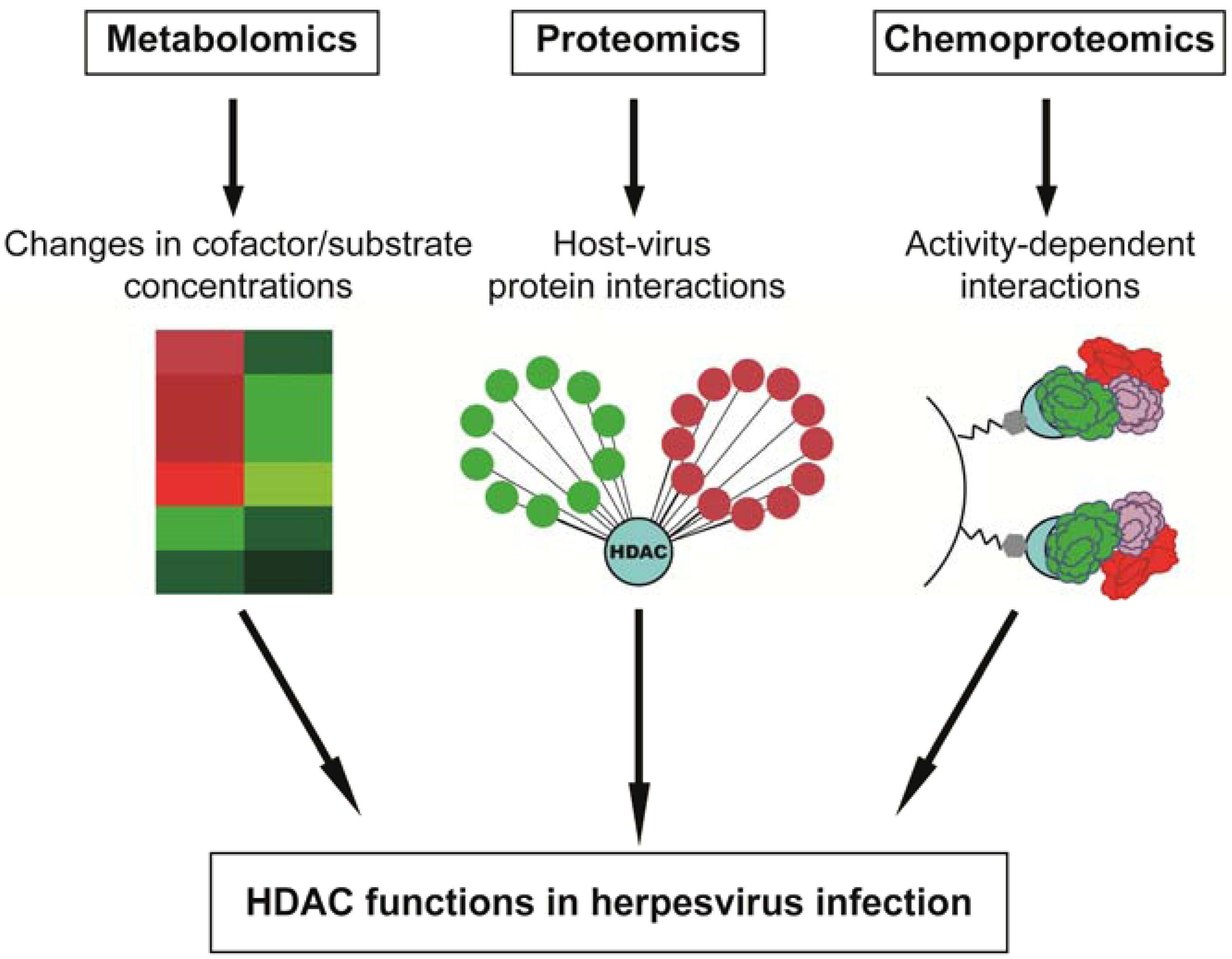
4. Perspective: “Omic” Approaches in Characterizing HDAC Functions in Virus Infection
Acknowledgments
Conflict of Interest
References and Notes
- Meier, J.L. Reactivation of the human cytomegalovirus major immediate-early regulatory region and viral replication in embryonal NTera2 cells: Role of trichostatin a, retinoic acid, and deletion of the 21-base-pair repeats and modulator. J. Virol. 2001, 75, 1581–1593. [Google Scholar] [CrossRef]
- Lu, F.; Zhou, J.; Wiedmer, A.; Madden, K.; Yuan, Y.; Lieberman, P.M. Chromatin remodeling of the Kaposi’s sarcoma-associated herpesvirus ORF50 promoter correlates with reactivation from latency. J. Virol. 2003, 77, 11425–11435. [Google Scholar] [CrossRef]
- Danaher, R.J.; Jacob, R.J.; Steiner, M.R.; Allen, W.R.; Hill, J.M.; Miller, C.S. Histone deacetylase inhibitors induce reactivation of herpes simplex virus type 1 in a latency-associated transcript-independent manner in neuronal cells. J. Neurovirol. 2005, 11, 306–317. [Google Scholar]
- Sinclair, J.; Sissons, P. Latency and reactivation of human cytomegalovirus. J. Gen. Virol. 2006, 87, 1763–1779. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Fraser, N.W. During latency, herpes-simplex virus type-1 DNA is associated with nucleosomes in a chromatin structure. J. Virol. 1989, 63, 943–947. [Google Scholar]
- Kent, J.R.; Zeng, P.Y.; Atanasiu, D.; Gardner, J.; Fraser, N.W.; Berger, S.L. During lytic infection herpes simplex virus type 1 is associated with histones bearing modifications that correlate with active transcription. J. Virol. 2004, 78, 10178–10186. [Google Scholar] [CrossRef]
- Jenkins, P.J.; Binne, U.K.; Farrell, P.J. Histone acetylation and reactivation of Epstein-Barr virus from latency. J. Virol. 2000, 74, 710–720. [Google Scholar] [CrossRef]
- Murphy, J.C.; Fischle, W.; Verdin, E.; Sinclair, J.H. Control of cytomegalovirus lytic gene expression by histone acetylation. EMBO J. 2002, 21, 1112–1120. [Google Scholar] [CrossRef]
- Kubat, N.J.; Tran, R.K.; McAnany, P.; Bloom, D.C. Specific histone tail modification and not DNA methylation is a determinant of herpes simplex virus type 1 latent gene expression. J. Virol. 2004, 78, 1139–1149. [Google Scholar] [CrossRef]
- Cuevas-Bennett, C.; Shenk, T. Dynamic histone H3 acetylation and methylation at human cytomegalovirus promoters during replication in fibroblasts. J. Virol. 2008, 82, 9525–9536. [Google Scholar] [CrossRef]
- Toth, Z.; Maglinte, D.T.; Lee, S.H.; Lee, H.-R.; Wong, L.-Y.; Brulois, K.F.; Lee, S.; Buckley, J.D.; Laird, P.W.; Marquez, V.E.; et al. Epigenetic analysis of KSHV latent and lytic genomes. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Kim, S.C.; Sprung, R.; Chen, Y.; Xu, Y.; Ball, H.; Pei, J.; Cheng, T.; Kho, Y.; Xiao, H.; Xiao, L.; et al. Substrate and functional diversity of lysine acetylation revealed by a proteomics survey. Mol. Cell 2006, 23, 607–618. [Google Scholar] [CrossRef]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Inoue, A.; Fujimoto, D. Enzymatic deacetylation of histone. Biochem. Biophys. Res. Commun. 1969, 36, 146–150. [Google Scholar] [CrossRef]
- Berger, S.L. The complex language of chromatin regulation during transcription. Nature 2007, 447, 407–412. [Google Scholar] [CrossRef]
- Yang, X.-J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar] [CrossRef]
- Gregoretti, I.V.; Lee, Y.M.; Goodson, H.V. Molecular evolution of the histone deacetylase family: Functional implications of phylogenetic analysis. J. Mol. Biol. 2004, 338, 17–31. [Google Scholar] [CrossRef]
- Herbein, G.; Wendling, D. Histone deacetylases in viral infections. Clin. Epigenetics 2010, 1, 13–24. [Google Scholar] [CrossRef]
- Hassig, C.A.; Fleischer, T.C.; Billin, A.N.; Schreiber, S.L.; Ayer, D.E. Histone deacetylase activity is required for full transcriptional repression by mSin3A. Cell 1997, 89, 341–347. [Google Scholar] [CrossRef]
- Wade, P.A.; Jones, P.L.; Vermaak, D.; Wolffe, A.P. A multiple subunit Mi-2 histone deacetylase from Xenopus laevis cofractionates with an associated Snf2 superfamily ATPase. Curr. Biol. 1998, 8, 843–846. [Google Scholar] [CrossRef]
- Zhang, Y.; LeRoy, G.; Seelig, H.P.; Lane, W.S.; Reinberg, D. The dermatomyositis-specific autoantigen Mi2 is a component of a complex containing histone deacetylase and nucleosome remodeling activities. Cell 1998, 95, 279–289. [Google Scholar] [CrossRef]
- Tong, J.K.; Hassig, C.A.; Schnitzler, G.R.; Kingston, R.E.; Schreiber, S.L. Chromatin deacetylation by an ATP-dependent nucleosome remodelling complex. Nature 1998, 395, 917–921. [Google Scholar] [CrossRef]
- Guenther, M.G.; Lane, W.S.; Fischle, W.; Verdin, E.; Lazar, M.A.; Shiekhattar, R. A core SMRT corepressor complex containing HDAC3 and TBL1, a WD40-repeat protein linked to deafness. Genes Dev. 2000, 14, 1048–1057. [Google Scholar]
- Li, J.W.; Wang, J.; Wang, J.X.; Nawaz, Z.; Liu, J.M.; Qin, J.; Wong, J.M. Both corepressor proteins SMRT and N-CoR exist in large protein complexes containing HDAC3. EMBO J. 2000, 19, 4342–4350. [Google Scholar]
- You, A.; Tong, J.K.; Grozinger, C.M.; Schreiber, S.L. CoREST is an integral component of the CoREST-human histone deacetylase complex. Proc. Natl. Acad. Sci. USA 2001, 98, 1454–1458. [Google Scholar]
- Bantscheff, M.; Hopf, C.; Savitski, M.M.; Dittmann, A.; Grandi, P.; Michon, A.-M.; Schlegl, J.; Abraham, Y.; Becher, I.; Bergamini, G.; et al. Chemoproteomics profiling of HDAC inhibitors reveals selective targeting of HDAC complexes. Nat. Biotechnol. 2011, 29, 255–265. [Google Scholar]
- Joshi, P.; Greco, T.M.; Guise, A.J.; Luo, Y.; Yu, F.; Nesvizhskii, A.I.; Cristea, I.M. The functional interactome landscape of the human histone deacetylase family. Mol. Syst. Biol. 2013, 9. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. Engagement of the lysine-specific demethylase/HDAC1/CoREST/REST complex by herpes simplex virus 1. J. Virol. 2009, 83, 4376–4385. [Google Scholar] [CrossRef]
- Shi, Y.; Lan, F.; Matson, C.; Mulligan, P.; Whetstine, J.R.; Cole, P.A.; Casero, R.A. Histone demethylation mediated by the nuclear amine oxidase homolog LSD1. Cell 2004, 119, 941–953. [Google Scholar] [CrossRef]
- Shi, Y.J.; Matson, C.; Lan, F.; Iwase, S.; Baba, T.; Shi, Y. Regulation of LSD1 histone demethylase activity by its associated factors. Mol. Cell 2005, 19, 857–864. [Google Scholar] [CrossRef]
- Gu, H.D.; Liang, Y.; Mandel, G.; Roizman, B. Components of the REST/CoREST/histone deacetylase repressor complex are disrupted, modified, and translocated in HSV-1-infected cells. Proc. Natl. Acad. Sci. USA 2005, 102, 7571–7576. [Google Scholar] [CrossRef]
- Poon, A.P.W.; Gu, H.; Roizman, B. ICP0 and the U(S)3 protein kinase of herpes simplex virus 1 independently block histone deacetylation to enable gene expression. Proc. Natl. Acad. Sci. USA 2006, 103, 9993–9998. [Google Scholar] [CrossRef]
- Zhang, Y.G.; Jones, C. The bovine herpesvirus 1 immediate-early protein (bICP0) associates with histone deacetylase 1 to activate transcription. J. Virol. 2001, 75, 9571–9578. [Google Scholar] [CrossRef]
- Gu, H.; Roizman, B. Herpes simplex virus-infected cell protein 0 blocks the silencing of viral DNA by dissociating histone deacetylases from the CoREST-REST complex. Proc. Natl. Acad. Sci. USA 2007, 104, 17134–17139. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. Nuclear retention of ICP0 in cells exposed to HDAC inhibitor or transfected with DNA before infection with herpes simplex virus 1. Proc.Natl.Acad.Sci.USA 2008, 105, 20488–20493. [Google Scholar] [CrossRef]
- Poon, A.P.W.; Liang, Y.; Roizman, B. Herpes simplex virus 1 gene expression is accelerated by inhibitors of histone deacetylases in rabbit skin cells infected with a mutant carrying a cDNA copy of the infected-cell protein No. 0. J. Virol. 2003, 77, 12671–12678. [Google Scholar] [CrossRef]
- Walters, M.S.; Kinchington, P.R.; Banfield, B.W.; Silverstein, S. Hyperphosphorylation of histone deacetylase 2 by alphaherpesvirus US3 kinases. J. Virol. 2010, 84, 9666–9676. [Google Scholar] [CrossRef]
- Moorman, N.J.; Cristea, I.M.; Terhune, S.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe 2008, 3, 253–262. [Google Scholar] [CrossRef]
- Terhune, S.S.; Moorman, N.J.; Cristea, I.M.; Savaryn, J.P.; Cuevas-Bennett, C.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus UL29/28 protein interacts with components of the NuRD complex which promote accumulation of immediate-early RNA. PLoS Pathog. 2010, 6. [Google Scholar] [CrossRef]
- Reeves, M.; Murphy, J.; Greaves, R.; Fairley, J.; Brehm, A.; Sinclair, J. Autorepression of the human cytomegalovirus major immediate-early promoter/enhancer at late times of infection is mediated by the recruitment of chromatin remodelina enzymes by IE86. J. Virol. 2006, 80, 9998–10009. [Google Scholar] [CrossRef]
- Radkov, S.A.; Touitou, R.; Brehm, A.; Rowe, M.; West, M.; Kouzarides, T.; Allday, M.J. Epstein-Barr virus nuclear antigen 3C interacts with histone deacetylase to repress transcription. J. Virol. 1999, 73, 5688–5697. [Google Scholar]
- Knight, J.S.; Lan, K.; Subramanian, C.; Robertson, E.S. Epstein-Barr virus nuclear antigen 3C recruits histone deacetylase activity and associates with the corepressors mSin3A and NCoR in human B-cell lines. J. Virol. 2003, 77, 4261–4272. [Google Scholar] [CrossRef]
- Zhou, J.; Snyder, A.R.; Lieberman, P.M. Epstein-barr virus episome stability is coupled to a delay in replication timing. J. Virol. 2009, 83, 2154–2162. [Google Scholar] [CrossRef]
- Lu, F.; Day, L.; Gao, S.J.; Lieberman, P.M. Acetylation of the latency-associated nuclear antigen regulates repression of Kaposi’s sarcoma-associated herpesvirus lytic transcription. J. Virol. 2006, 80, 5273–5282. [Google Scholar] [CrossRef]
- Park, J.J.; Kim, Y.E.; Pham, H.T.; Kim, E.T.; Chung, Y.H.; Ahn, J.H. Functional interaction of the human cytomegalovirus IE2 protein with histone deacetylase 2 in infected human fibroblasts. J. Gen. Virol. 2007, 88, 3214–3223. [Google Scholar] [CrossRef]
- Nevels, M.; Paulus, C.; Shenk, T. Human cytomegalovirus immediate-early 1 protein facilitates viral replication by antagonizing histone deacetylation. Proc. Natl. Acad. Sci. USA 2004, 101, 17234–17239. [Google Scholar] [CrossRef]
- Saffert, R.T.; Kalejta, R.F. Inactivating a cellular intrinsic immune defense mediated by Daxx is the mechanism through which the human cytomegalovirus pp71 protein stimulates viral immediate-early gene expression. J. Virol. 2006, 80, 3863–3871. [Google Scholar] [CrossRef]
- Lomonte, P.; Thomas, J.; Texier, P.; Caron, C.; Khochbin, S.; Epstein, A.L. Functional interaction between class II histone deacetylases and ICPO of herpes simplex virus type 1. J. Virol. 2004, 78, 6744–6757. [Google Scholar]
- Pflum, M.K.H.; Tong, J.K.; Lane, W.S.; Schreiber, S.L. Histone deacetylase I phosphorylation promotes enzymatic activity and complex formation. J. Biol. Chem. 2001, 276, 47733–47741. [Google Scholar]
- Tsai, S.C.; Seto, E. Regulation of histone deacetylase 2 by protein kinase CK2. J. Biol. Chem. 2002, 277, 31826–31833. [Google Scholar] [CrossRef]
- Grozinger, C.M.; Schreiber, S.L. Regulation of histone deacetylase 4 and 5 and transcriptional activity by 14-3-3-dependent cellular localization. Proc. Natl. Acad. Sci. USA 2000, 97, 7835–7840. [Google Scholar] [CrossRef]
- Wang, A.H.; Kruhlak, M.J.; Wu, J.; Bertos, N.R.; Vezmar, M.; Posner, B.I.; Bazett-Jones, D.P.; Yang, X.J. Regulation of histone deacetylase 4 by binding of 14-3-3 proteins. Mol. Cell. Biol. 2000, 20, 6904–6912. [Google Scholar] [CrossRef]
- Kao, H.Y.; Verdel, A.; Tsai, C.C.; Simon, C.; Juguilon, H.; Khochbin, S. Mechanism for nucleocytoplasmic shuttling of histone deacetylase 7. J. Biol. Chem. 2001, 276, 47496–47507. [Google Scholar]
- Greco, T.M.; Yu, F.; Guise, A.J.; Cristea, I.M. Nuclear import of histone deacetylase 5 by requisite nuclear localization signal phosphorylation. Mol. Cell. Proteomics 2011, 10. [Google Scholar] [CrossRef]
- Guise, A.J.; Greco, T.M.; Zhang, I.Y.; Yu, F.; Cristea, I.M. Aurora B-dependent regulation of Class IIa histone deacetylases by mitotic nuclear localization signal phosphorylation. Mol. Cell. Proteomics 2012, 11, 1220–1229. [Google Scholar] [CrossRef]
- Ahn, J.H.; Brignole, E.J.; Hayward, G.S. Disruption of PML subnuclear domains by the acidic IE1 protein of human cytomegalovirus is mediated through interaction with PML and may modulate a RING finger-dependent cryptic transactivator function of PML. Mol. Cell. Biol. 1998, 18, 4899–4913. [Google Scholar]
- Wu, W.S.; Vallian, S.; Seto, E.; Yang, W.M.; Edmondson, D.; Roth, S.; Chang, K.S. The growth suppressor PML represses transcription by functionally and physically interacting with histone deacetylases. Mol. Cell. Biol. 2001, 21, 2259–2268. [Google Scholar] [CrossRef]
- Kao, H.Y.; Ordentlich, P.; Koyano-Nakagawa, N.; Tang, Z.; Downes, M.; Kintner, C.R.; Evans, R.M.; Kadesch, T. A histone deacetylase corepressor complex regulates the Notch signal transduction pathway. Genes Dev. 1998, 12, 2269–2277. [Google Scholar] [CrossRef]
- Fischle, W.; Dequiedt, F.; Hendzel, M.J.; Guenther, M.G.; Lazar, M.A.; Voelter, W.; Verdin, E. Enzymatic activity associated with class IIHDACs is dependent on a multiprotein complex containing HDAC3 and SMRT/N-CoR. Mol. Cell 2002, 9, 45–57. [Google Scholar] [CrossRef]
- Gruffat, H.; Manet, E.; Sergeant, A. MEF2-mediated recruitment of class 11 HDAC at the EBV immediate early gene BZLF1 links latency and chromatin remodeling. EMBO Rep. 2002, 3, 141–146. [Google Scholar] [CrossRef]
- Bryant, H.; Farrell, P.J. Signal transduction and transcription factor modification during reactivation of Epstein-Barr virus from latency. J. Virol. 2002, 76, 10290–10298. [Google Scholar] [CrossRef]
- Bottero, V.; Sharma-Walia, N.; Kerur, N.; Paul, A.G.; Sadagopan, S.; Cannon, M.; Chandran, B. Kaposi Sarcoma-associated herpes virus (KSHV) G protein-coupled receptor (vGPCR) activates the ORF50 lytic switch promoter: A potential positive feedback loop for sustained ORF50 gene expression. Virology 2009, 392, 34–51. [Google Scholar] [CrossRef]
- Lukac, D.M.; Renne, R.; Kirshner, J.R.; Ganem, D. Reactivation of Kaposi’s sarcoma-associated herpesvirus infection from latency by expression of the ORF 50 transactivator, a homolog of the EBV R protein. Virology 1998, 252, 304–312. [Google Scholar] [CrossRef]
- Vega, R.B.; Harrison, B.C.; Meadows, E.; Roberts, C.R.; Papst, P.J.; Olson, E.N.; McKinsey, T.A. Protein kinases C and D mediate agonist-dependent cardiac hypertrophy through nuclear export of histone deacetylase 5. Mol. Cell. Biol. 2004, 24, 8374–8385. [Google Scholar] [CrossRef]
- Krithivas, A.; Young, D.B.; Liao, G.L.; Greene, D.; Hayward, S.D. Human herpesvirus 8 LANA interacts with proteins of the mSin3 corepressor complex and negatively regulates Epstein-Barr virus gene expression in dually infected PEL cells. J. Virol. 2000, 74, 9637–9645. [Google Scholar] [CrossRef]
- Gwack, Y.; Byun, H.; Hwang, S.; Lim, C.; Choe, J. CREB-binding protein and histone deacetylase regulate the transcriptional activity of Kaposi's sarcoma-associated herpesvirus open reading frame 50. J. Virol. 2001, 75, 1909–1917. [Google Scholar] [CrossRef]
- Gwack, Y.; Hwang, S.; Byun, H.; Lim, C.; Kim, J.W.; Choi, E.J.; Choe, J. Kaposi’s sarcoma-associated herpesvirus open reading frame 50 represses p53-induced transcriptional activity and apoptosis. J. Virol. 2001, 75, 6245–6248. [Google Scholar] [CrossRef]
- Gu, W.; Roeder, R.G. Activation of p53 sequence-specific DNA binding by acetylation of the p53 C-terminal domain. Cell 1997, 90, 595–606. [Google Scholar] [CrossRef]
- Luo, J.Y.; Li, M.Y.; Tang, Y.; Laszkowska, M.; Roeder, R.G.; Gu, W. Acetylation of p53 augments its site-specific DNA binding both in vitro and in vivo. Proc. Nat. Acad. Sci. USA 2004, 101, 2259–2264. [Google Scholar]
- Li, T.; Diner, B.A.; Chen, J.; Cristea, I.M. Acetylation modulates cellular distribution and DNA sensing ability of interferon-inducible protein IFI16. Proc. Natl. Acad. Sci. USA 2012, 109, 10558–10563. [Google Scholar] [CrossRef]
- Nencioni, A.; Beck, J.; Werth, D.; Gruenebach, F.; Patrone, F.; Ballestrero, A.; Brossart, P. Histone deacetylase inhibitors affect dendritic cell differentiation and immunogenicity. Clin. Cancer Res. 2007, 13, 3933–3941. [Google Scholar] [CrossRef]
- Halili, M.A.; Andrews, M.R.; Labzin, L.I.; Schroder, K.; Matthias, G.; Cao, C.; Lovelace, E.; Reid, R.C.; Le, G.T.; Hume, D.A.; et al. Differential effects of selective HDAC inhibitors on macrophage inflammatory responses to the Toll-like receptor 4 agonist LPS. J. Leukocyte Biol. 2010, 87, 1103–1114. [Google Scholar] [CrossRef]
- Roger, T.; Lugrin, J.; le Roy, D.; Goy, G.; Mombelli, M.; Koessler, T.; Ding, X.C.; Chanson, A.L.; Reymond, M.K.; Miconnet, I.; et al. Histone deacetylase inhibitors impair innate immune responses to Toll-like receptor agonists and to infection. Blood 2011, 117, 1205–1217. [Google Scholar] [CrossRef]
- Mombelli, M.; Lugrin, J.; Rubino, I.; Chanson, A.L.; Giddey, M.; Calandra, T.; Roger, T. Histone deacetylase inhibitors impair antibacterial defenses of macrophages. J. Infect. Dis. 2011, 204, 1367–1374. [Google Scholar] [CrossRef]
- Unterholzner, L.; Keating, S.E.; Baran, M.; Horan, K.A.; Jensen, S.B.; Sharma, S.; Sirois, C.M.; Jin, T.; Latz, E.; et al. IFI16 is an innate immune sensor for intracellular DNA. Nat. Immunol. 2010, 11, 997–1004. [Google Scholar] [CrossRef]
- Kerur, N.; Veettil, M.V.; Sharma-Walia, N.; Bottero, V.; Sadagopan, S.; Otageri, P.; Chandran, B. IFI16 acts as a nuclear pathogen sensor to induce the inflammasome in response to Kaposi Sarcoma-associated herpesvirus infection. Cell Host Microbe 2011, 9, 363–375. [Google Scholar] [CrossRef]
- Orzalli, M.H.; DeLuca, N.A.; Knipe, D.M. Nuclear IFI16 induction of IRF-3 signaling during herpesviral infection and degradation of IFI16 by the viral ICP0 protein. Proc. Natl. Acad. Sci. USA 2012, 109, E3008–E3017. [Google Scholar] [CrossRef]
- Singh, V.V.; Kerur, N.; Bottero, V.; Dutta, S.; Chakraborty, S.; Ansari, M.A.; Paudel, N.; Chikoti, L.; Chandran, B. Kaposi’s sarcoma-associated herpesvirus latency in endothelial and B cells activates interferon gamma-inducible protein 16 (IFI16) mediated inflammasomes. J. Virol. 2013, 87, 4417–4431. [Google Scholar] [CrossRef]
- Johnson, K.E.; Chikoti, L.; Chandran, B. HSV-1 infection induces activation and subsequent inhibition of the IFI16 and NLRP3 inflammasomes. J. Virol. 2013, 87, 5005–5018. [Google Scholar] [CrossRef]
- Horan, K.A.; Hansen, K.; Jakobsen, M.R.; Holm, C.K.; Soby, S.; Unterholzner, L.; Thompson, M.; West, J.A.; Iversen, M.B.; Rasmussen, S.B.; et al. Proteasomal degradation of herpes simplex virus capsids in macrophages releases DNA to the cytosol for recognition by DNA sensors. J. Immunol. 2013, 190, 2311–2319. [Google Scholar] [CrossRef]
- Fernandes-Alnemri, T.; Yu, J.W.; Datta, P.; Wu, J.; Alnemri, E.S. AIM2 activates the inflammasome and cell death in response to cytoplasmic DNA. Nature 2009, 458, 509–513. [Google Scholar] [CrossRef]
- Rathinam, V.A.; Jiang, Z.; Waggoner, S.N.; Sharma, S.; Cole, L.E.; Waggoner, L.; Vanaja, S.K.; Monks, B.G.; Ganesan, S.; Latz, E.; et al. The AIM2 inflammasome is essential for host defense against cytosolic bacteria and DNA viruses. Nat. Immunol. 2009, 11, 395–402. [Google Scholar]
- Yoneyama, M.; Kikuchi, M.; Natsukawa, T.; Shinobu, N.; Imaizumi, T.; Miyagishi, M.; Taira, K.; Akira, S.; Fujita, T. The RNA helicase RIG-I has an essential function in double-stranded RNA-induced innate antiviral responses. Nat. Immunol. 2004, 5, 730–737. [Google Scholar] [CrossRef]
- Maul, G.G.; Ishov, A.M.; Everett, R.D. Nuclear domain 10 as preexisting potential replication start sites of herpes simplex virus type-1. Virology 1996, 217, 67–75. [Google Scholar] [CrossRef]
- Tavalai, N.; Papior, P.; Rechter, S.; Leis, M.; Stamminger, T. Evidence for a role of the cellular ND10 protein PML in mediating intrinsic immunity against human cytomegalovirus infections. J. Virol. 2006, 80, 8006–8018. [Google Scholar] [CrossRef]
- Tang, Q.Y.; Maul, G.G. Mouse cytomegalovirus immediate-early protein 1 binds with host cell repressors to relieve suppressive effects on viral transcription and replication during lytic infection. J. Virol. 2003, 77, 1357–1367. [Google Scholar] [CrossRef]
- Groves, I.J.; Reeves, M.B.; Sinclair, J.H. Lytic infection of permissive cells with human cytomegalovirus is regulated by an intrinsic “pre-immediate-early” repression of viral gene expression mediated by histone post-translational modification. J. Gen. Virol. 2009, 90, 2364–2374. [Google Scholar] [CrossRef]
- Burkham, J.; Coen, D.M.; Hwang, C.B.C.; Weller, S.K. Interactions of herpes simplex virus type 1 with ND10 and recruitment of PML to replication compartments. J. Virol. 2001, 75, 2353–2367. [Google Scholar] [CrossRef]
- Tang, X.; Gao, J.S.; Guan, Y.J.; McLane, K.E.; Yuan, Z.L.; Ramratnam, B.; Chin, Y.E. Acetylation-dependent signal transduction for type I interferon receptor. Cell 2007, 131, 93–105. [Google Scholar] [CrossRef]
- Yuan, Z.L.; Guan, Y.J.; Chatterjee, D.; Chin, Y.E. Stat3 dimerization regulated by reversible acetylation of a single lysine residue. Science 2005, 307, 269–273. [Google Scholar] [CrossRef]
- Kramer, O.H.; Knauer, S.K.; Greiner, G.; Jandt, E.; Reichardt, S.; Guhrs, K.H.; Stauber, R.H.; Bohmer, F.D.; Heinzel, T. A phosphorylation-acetylation switch regulates STAT1 signaling. Genes Dev. 2009, 23, 223–235. [Google Scholar] [CrossRef]
- Chen, L.F.; Greene, W.C. Regulation of distinct biological activities of the NF-kappaB transcription factor complex by acetylation. J. Mol. Med. 2003, 81, 549–557. [Google Scholar] [CrossRef]
- Quivy, V.; van Lint, C. Regulation at multiple levels of NF-kappaB-mediated transactivation by protein acetylation. Biochem. Pharmacol. 2004, 68, 1221–1229. [Google Scholar] [CrossRef]
- Kiernan, R.; Bres, V.; Ng, R.W.; Coudart, M.P.; el Messaoudi, S.; Sardet, C.; Jin, D.Y.; Emiliani, S.; Benkirane, M. Post-activation turn-off of NF-kappa B-dependent transcription is regulated by acetylation of p65. J. Biol. Chem. 2003, 278, 2758–2766. [Google Scholar] [CrossRef]
- Chen, L.; Fischle, W.; Verdin, E.; Greene, W.C. Duration of nuclear NF-kappaB action regulated by reversible acetylation. Science 2001, 293, 1653–1657. [Google Scholar] [CrossRef]
- Yeung, F.; Hoberg, J.E.; Ramsey, C.S.; Keller, M.D.; Jones, D.R.; Frye, R.A.; Mayo, M.W. Modulation of NF-kappaB-dependent transcription and cell survival by the SIRT1 deacetylase. EMBOJ. 2004, 23, 2369–2380. [Google Scholar]
- Rothgiesser, K.M.; Erener, S.; Waibel, S.; Luscher, B.; Hottiger, M.O. SIRT2 regulates NF-kappaB dependent gene expression through deacetylation of p65 Lys310. J. Cell Sci. 2010, 123, 4251–4258. [Google Scholar] [CrossRef] [Green Version]
- Yang, H.; Zhang, W.; Pan, H.; Feldser, H.G.; Lainez, E.; Miller, C.; Leung, S.; Zhong, Z.; Zhao, H.; Sweitzer, S.; et al. SIRT1 activators suppress inflammatory responses through promotion of p65 deacetylation and inhibition of NF-kappaB activity. PLoS One 2012, 7, e46364. [Google Scholar]
- Amici, C.; Rossi, A.; Costanzo, A.; Ciafre, S.; Marinari, B.; Balsamo, M.; Levrero, M.; Santoro, M.G. Herpes simplex virus disrupts NF-kappaB regulation by blocking its recruitment on the IkappaBalpha promoter and directing the factor on viral genes. J. Biol. Chem. 2006, 281, 7110–7117. [Google Scholar] [CrossRef]
- El Mjiyad, N.; Bontems, S.; Gloire, G.; Horion, J.; Vandevenne, P.; Dejardin, E.; Piette, J.; Sadzot-Delvaux, C. Varicella-zoster virus modulates NF-kappaB recruitment on selected cellular promoters. J. Virol. 2007, 81, 13092–13104. [Google Scholar] [CrossRef]
- Katsura, T.; Iwai, S.; Ota, Y.; Shimizu, H.; Ikuta, K.; Yura, Y. The effects of trichostatin A on the oncolytic ability of herpes simplex virus for oral squamous cell carcinoma cells. Canc. Gene Ther. 2009, 16, 237–245. [Google Scholar]
- Kwon, H.S.; Brent, M.M.; Getachew, R.; Jayakumar, P.; Chen, L.F.; Schnolzer, M.; McBurney, M.W.; Marmorstein, R.; Greene, W.C.; Ott, M. Human immunodeficiency virus type 1 Tat protein inhibits the SIRT1 deacetylase and induces T cell hyperactivation. Cell Host Microbe 2008, 3, 158–167. [Google Scholar] [CrossRef]
- Munoz-Fontela, C.; Gonzalez, D.; Marcos-Villar, L.; Campagna, M.; Gallego, P.; Gonzalez-Santamaria, J.; Herranz, D.; Gu, W.; Serrano, M.; Aaronson, S.A.; et al. Acetylation is indispensable for p53 antiviral activity. Cell Cycle 2011, 10, 3701–3705. [Google Scholar] [CrossRef]
- Hsu, C.H.; Chang, M.D.; Tai, K.Y.; Yang, Y.T.; Wang, P.S.; Chen, C.J.; Wang, Y.H.; Lee, S.C.; Wu, C.W.; Juan, L.J. HCMV IE2-mediated inhibition of HAT activity downregulates p53 function. EMBO J. 2004, 23, 2269–2280. [Google Scholar] [CrossRef]
- Melroe, G.T.; Silva, L.; Schaffer, P.A.; Knipe, D.M. Recruitment of activated IRF-3 and CBP/p300 to herpes simplex virus ICP0 nuclear foci: Potential role in blocking IFN-beta induction. Virology 2007, 360, 305–321. [Google Scholar] [CrossRef]
- Juan, L.J.; Shia, W.J.; Chen, M.H.; Yang, W.M.; Seto, E.; Lin, Y.S.; Wu, C.W. Histone deacetylases specifically down-regulate p53-dependent gene activation. J. Biol. Chem. 2000, 275, 20436–20443. [Google Scholar]
- Pagans, S.; Pedal, A.; North, B.J.; Kaehlcke, K.; Marshall, B.L.; Dorr, A.; Hetzer-Egger, C.; Henklein, P.; Frye, R.; McBurney, M.W.; et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005, 3, 210–220. [Google Scholar] [CrossRef] [Green Version]
- Zhang, H.-S.; Zhou, Y.; Wu, M.-R.; Zhou, H.-S.; Xu, F. Resveratrol inhibited Tat-induced HIV-1 LTR transactivation via NAD(+)-dependent SIRT1 activity. Life Sci. 2009, 85, 484–489. [Google Scholar] [CrossRef]
- Thakur, B.K.; Chandra, A.; Dittrich, T.; Welte, K.; Chandra, P. Inhibition of SIRT1 by HIV-1 viral protein Tat results in activation of p53 pathway. Biochem. Biophys. Res. Commun. 2012, 424, 245–250. [Google Scholar]
- Allison, S.J.; Jiang, M.; Milner, J. Oncogenic viral protein HPV E7 up-regulates the SIRT1 longevity protein in human cervical cancer cells. Aging 2009, 1, 316–327. [Google Scholar]
- Campagna, M.; Herranz, D.; Garcia, M.A.; Marcos-Villar, L.; Gonzalez-Santamaria, J.; Gallego, P.; Gutierrez, S.; Collado, M.; Serrano, M.; Esteban, M.; et al. SIRT1 stabilizes PML promoting its sumoylation. Cell Death Differ. 2011, 18, 72–79. [Google Scholar] [CrossRef]
- Picchione, K.E.; Bhattacharjee, A. Viral genome silencing by neuronal sirtuin 1. J. Neurovirol. 2011, 17, 184–188. [Google Scholar] [CrossRef]
- Marfe, G.; Tafani, M.; Fiorito, F.; Pagnini, U.; Iovane, G.; De Martino, L. Involvement of FOXO transcription factors, TRAIL-FasL/Fas, and sirtuin proteins family in canine coronavirus type II-Induced apoptosis. PLoS One 2011, 6, e27313. [Google Scholar]
- Kalamvoki, M.; Roizman, B. Circadian CLOCK histone acetyl transferase localizes at ND10 nuclear bodies and enables herpes simplex virus gene expression. Proc. Natl. Acad. Sci. USA 2010, 107, 17721–17726. [Google Scholar] [CrossRef]
- Kalamvoki, M.; Roizman, B. The histone acetyltransferase CLOCK is an essential component of the herpes simplex virus 1 transcriptome that includes TFIID, ICP4, ICP27, and ICP22. J. Virol. 2011, 85, 9472–9477. [Google Scholar] [CrossRef]
- Li, R.; Zhu, J.; Xie, Z.; Liao, G.; Liu, J.; Chen, M.-R.; Hu, S.; Woodard, C.; Lin, J.; Taverna, S.D.; et al. Conserved herpesvirus kinases target the DNA damage response pathway and TIP60 histone acetyltransferase to promote virus replication. Cell Host Microbe 2011, 10, 390–400. [Google Scholar]
- Zhang, Y.G.; Jiang, Y.Q.; Geiser, V.; Zhou, J.; Jones, C. Bovine herpesvirus 1 immediate-early protein (bICP0) interacts with the histone acetyltransferase p300, which stimulates productive infection and gC promoter activity. J. Gen. Virol. 2006, 87, 1843–1851. [Google Scholar] [CrossRef]
- Skiba, M.; Mettenleiter, T.C.; Karger, A. Quantitative whole-cell proteome analysis of pseudorabies virus-infected cells. J. Virol. 2008, 82, 9689–9699. [Google Scholar] [CrossRef]
- Cristea, I.M.; Carroll, J.-W.N.; Rout, M.P.; Rice, C.M.; Chait, B.T.; MacDonald, M.R. Tracking and elucidating Alphavirus-host protein interactions. J. Biol. Chem. 2006, 281, 30269–30278. [Google Scholar]
- Calderwood, M.A.; Venkatesan, K.; Xing, L.; Chase, M.R.; Vazquez, A.; Holthaus, A.M.; Ewence, A.E.; Li, N.; Hirozane-Kishikawa, T.; Hill, D.E.; et al. Epstein-Barr virus and virus human protein interaction maps. Proc. Natl. Acad. Sci. USA 2007, 104, 7606–7611. [Google Scholar] [CrossRef]
- Moorman, N.J.; Sharon-Friling, R.; Shenk, T.; Cristea, I.M. A targeted spatial-temporal proteomics approach implicates multiple cellular trafficking pathways in human cytomegalovirus virion maturation. Mol. Cell. Proteomics 2010, 9, 851–860. [Google Scholar] [CrossRef]
- Cristea, I.M.; Rozjabek, H.; Molloy, K.R.; Karki, S.; White, L.L.; Rice, C.M.; Rout, M.P.; Chait, B.T.; MacDonald, M.R. Host factors associated with the sindbis virus RNA-dependent RNA polymerase: Role for G3BP1 and G3BP2 in virus replication. J. Virol. 2010, 84, 6720–6732. [Google Scholar] [CrossRef]
- Cristea, I.M.; Moorman, N.J.; Terhune, S.S.; Cuevas, C.D.; O’Keefe, E.S.; Rout, M.P.; Chait, B.T.; Shenk, T. Human cytomegalovirus pUL83 stimulates activity of the viral immediate-early promoter through its interaction with the cellular IFI16 protein. J. Virol. 2010, 84, 7803–7814. [Google Scholar] [CrossRef]
- Doolittle, J.M.; Gomez, S.M. Mapping protein interactions between Dengue virus and its human and insect hosts. PLoS Negl. Trop. Dis. 2011, 5, e954. [Google Scholar] [CrossRef]
- Jager, S.; Cimermancic, P.; Gulbahce, N.; Johnson, J.R.; McGovern, K.E.; Clarke, S.C.; Shales, M.; Mercenne, G.; Pache, L.; Li, K.; et al. Global landscape of HIV-human protein complexes. Nature 2012, 481, 365–370. [Google Scholar]
- Kramer, T.; Greco, T.M.; Taylor, M.P.; Ambrosini, A.E.; Cristea, I.M.; Enquist, L.W. Kinesin-3 mediates axonal sorting and directional transport of alphaherpesvirus particles in neurons. Cell Host Microbe 2012, 12, 806–814. [Google Scholar] [CrossRef]
- Miteva, Y.V.; Budayeva, H.G.; Cristea, I.M. Proteomics-based methods for discovery, quantification, and validation of protein-protein interactions. Anal. Chem. 2013, 85, 749–768. [Google Scholar] [CrossRef]
- Cristea, I.M.; Williams, R.; Chait, B.T.; Rout, M.P. Fluorescent proteins as proteomic probes. Mol. Cell. Proteomics 2005, 4, 1933–1941. [Google Scholar] [CrossRef]
- Kaczkowski, B.; Rossing, M.; Andersen, D.K.; Dreher, A.; Morevati, M.; Visser, M.A.; Winther, O.; Nielsen, F.C.; Norrild, B. Integrative analyses reveal novel strategies in HPV11,-16 and -45 early infection. Sci. Rep. 2012, 2, 515. [Google Scholar]
- Choi, H.; Larsen, B.; Lin, Z.-Y.; Breitkreutz, A.; Mellacheruvu, D.; Fermin, D.; Qin, Z.S.; Tyers, M.; Gingras, A.-C.; Nesvizhskii, A.I. SAINT: Probabilistic scoring of affinity purification-mass spectrometry data. Nat. Methods 2011, 8, 70–73. [Google Scholar] [CrossRef]
- Ptak, R.G.; Fu, W.; Sanders-Beer, B.E.; Dickerson, J.E.; Pinney, J.W.; Robertson, D.L.; Rozanov, M.N.; Katz, K.S.; Maglott, D.R.; Pruitt, K.D.; et al. Cataloguing the HIV type 1 human protein interaction network. AIDS Res. Hum. Retroviruses 2008, 24, 1497–1502. [Google Scholar]
- Fu, W.; Sanders-Beer, B.E.; Katz, K.S.; Maglott, D.R.; Pruitt, K.D.; Ptak, R.G. Human immunodeficiency virus type 1, human protein interaction database at NCBI. Nucleic Acids Res. 2009, 37, D417–D422. [Google Scholar] [CrossRef]
- Chatr-aryamontri, A.; Ceol, A.; Peluso, D.; Nardozza, A.; Panni, S.; Sacco, F.; Tinti, M.; Smolyar, A.; Castagnoli, L.; Vidal, M.; et al. VirusMINT: A viral protein interaction database. Nucleic Acids Res. 2009, 37, D669–D673. [Google Scholar]
- Pinney, J.W.; Dickerson, J.E.; Fu, W.; Sanders-Beer, B.E.; Ptak, R.G.; Robertson, D.L. HIV-host interactions: A map of viral perturbation of the host system. AIDS 2009, 23, 549–554. [Google Scholar]
- Bell, C.; Desjardins, M.; Thibault, P.; Radtke, K. Proteomics analysis of herpes simplex virus type 1-infected cells reveals dynamic changes of viral protein expression, ubiquitylation, and phosphorylation. J. Proteome Res. 2013, 12, 1820–1829. [Google Scholar] [CrossRef]
- Gredmark, S.; Schlieker, C.; Quesada, V.; Spooner, E.; Ploegh, H.L. A functional ubiquitin-specific protease embedded in the large tegument protein (ORF64) of murine gammaherpesvirus 68 is active during the course of infection. J. Virol. 2007, 81, 10300–10309. [Google Scholar]
- Salisbury, C.M.; Cravatt, B.F. Activity-based probes for proteomic profiling of histone deacetylase complexes. Proc. Natl. Acad. Sci. USA 2007, 104, 1171–1176. [Google Scholar] [CrossRef] [Green Version]
- Salisbury, C.M.; Cravatt, B.F. Optimization of activity-based probes for proteomic profiling of histone deacetylase complexes. J. Am. Chem. Soc. 2008, 130, 2184–2194. [Google Scholar] [CrossRef]
- Munger, J.; Bajad, S.U.; Coller, H.A.; Shenk, T.; Rabinowitz, J.D. Dynamics of the cellular metabolome during human cytomegalovirus infection. PLoS Pathog. 2006, 2, e132. [Google Scholar] [CrossRef]
- Milne, S.B.; Mathews, T.P.; Myers, D.S.; Ivanova, P.T.; Brown, H.A. Sum of the parts: Mass spectrometry-based metabolomics. Biochemistry 2013, 52, 3829–3840. [Google Scholar]
- Munger, J.; Bennett, B.D.; Parikh, A.; Feng, X.J.; McArdle, J.; Rabitz, H.A.; Shenk, T.; Rabinowitz, J.D. Systems-level metabolic flux profiling identifies fatty acid synthesis as a target for antiviral therapy. Nat. Biotechnol. 2008, 26, 1179–1186. [Google Scholar]
- Chambers, J.W.; Maguire, T.G.; Alwine, J.C. Glutamine metabolism is essential for human cytomegalovirus infection. J. Virol. 2010, 84, 1867–1873. [Google Scholar] [CrossRef]
- Yu, Y.; Clippinger, A.J.; Alwine, J.C. Viral effects on metabolism: Changes in glucose and glutamine utilization during human cytomegalovirus infection. Trends Microbiol. 2011, 19, 360–367. [Google Scholar] [CrossRef]
- Vastag, L.; Koyuncu, E.; Grady, S.L.; Shenk, T.E.; Rabinowitz, J.D. Divergent effects of human cytomegalovirus and herpes simplex virus-1 on cellular metabolism. PLoS Pathog. 2011, 7, e1002124. [Google Scholar] [CrossRef]
- Ivanova, P.T.; Milne, S.B.; Myers, D.S.; Brown, H.A. Lipidomics: A mass spectrometry based systems level analysis of cellular lipids. Curr. Opin. Chem. Biol. 2009, 13, 526–531. [Google Scholar] [CrossRef]
- Brugger, B.; Glass, B.; Haberkant, P.; Leibrecht, I.; Wieland, F.T.; Krausslich, H.G. The HIV lipidome: A raft with an unusual composition. Proc. Natl. Acad. Sci. USA 2006, 103, 2641–2646. [Google Scholar]
- Bruegger, B.; Krautkraemer, E.; Tibroni, N.; Munte, C.E.; Rauch, S.; Leibrecht, I.; Glass, B.; Breuer, S.; Geyer, M.; Kraeusslich, H.-G.; et al. Human immunodeficiency virus type 1 nef protein modulates the lipid composition of virions and host cell membrane microdomains. Retrovirology 2007, 4. [Google Scholar] [CrossRef]
- Diamond, D.L.; Syder, A.J.; Jacobs, J.M.; Sorensen, C.M.; Walters, K.-A.; Proll, S.C.; McDermott, J.E.; Gritsenko, M.A.; Zhang, Q.; Zhao, R.; et al. Temporal proteome and lipidome profiles reveal hepatitis C virus-associated reprogramming of hepatocellular metabolism and bioenergetics. PLoS Pathog. 2010, 6, e1000719. [Google Scholar] [CrossRef]
- Liu, S.T.H.; Sharon-Friling, R.; Ivanova, P.; Milne, S.B.; Myers, D.S.; Rabinowitz, J.D.; Brown, H.A.; Shenk, T. Synaptic vesicle-like lipidome of human cytomegalovirus virions reveals a role for SNARE machinery in virion egress. Proc. Natl. Acad. Sci. USA 2011, 108, 12869–12874. [Google Scholar] [CrossRef]
- Gudleski-O'Regan, N.; Greco, T.M.; Cristea, I.M.; Shenk, T. Increased expression of LDL receptor-related protein 1 during human cytomegalovirus infection reduces virion cholesterol and infectivity. Cell Host Microbe 2012, 12, 86–96. [Google Scholar] [CrossRef]
- Rabinowitz, J.D.; Purdy, J.G.; Vastag, L.; Shenk, T.; Koyuncu, E. Metabolomics in drug target discovery. Cold Spring Harb. Symp. Quant. Biol. 2011, 76, 235–246. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Guise, A.J.; Budayeva, H.G.; Diner, B.A.; Cristea, I.M. Histone Deacetylases in Herpesvirus Replication and Virus-Stimulated Host Defense. Viruses 2013, 5, 1607-1632. https://doi.org/10.3390/v5071607
Guise AJ, Budayeva HG, Diner BA, Cristea IM. Histone Deacetylases in Herpesvirus Replication and Virus-Stimulated Host Defense. Viruses. 2013; 5(7):1607-1632. https://doi.org/10.3390/v5071607
Chicago/Turabian StyleGuise, Amanda J., Hanna G. Budayeva, Benjamin A. Diner, and Ileana M. Cristea. 2013. "Histone Deacetylases in Herpesvirus Replication and Virus-Stimulated Host Defense" Viruses 5, no. 7: 1607-1632. https://doi.org/10.3390/v5071607