Bromodomain Proteins in HIV Infection

Abstract
:1. The Bromodomain Protein Family
2. HIV Infection and Reversible Acetylation
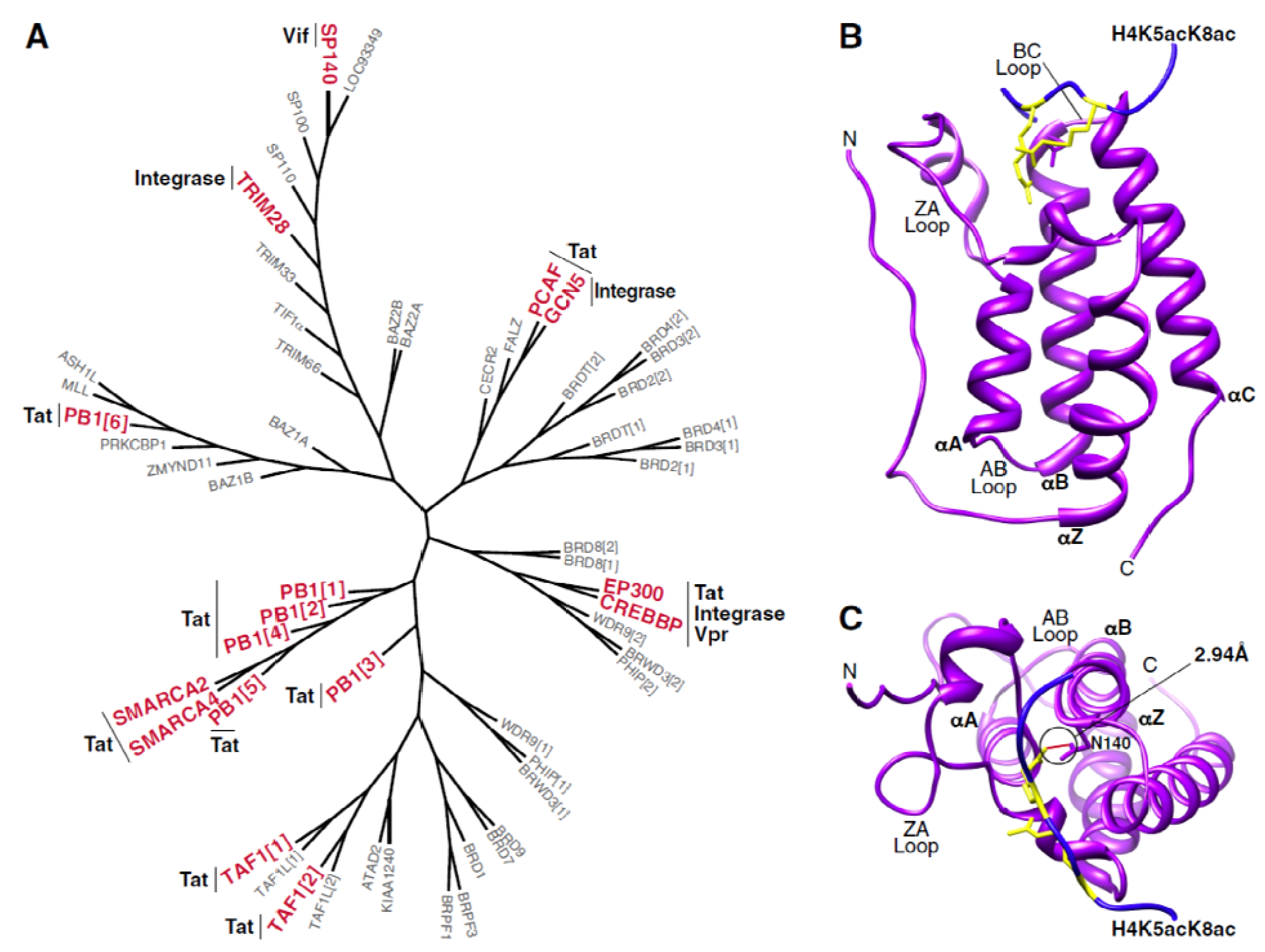
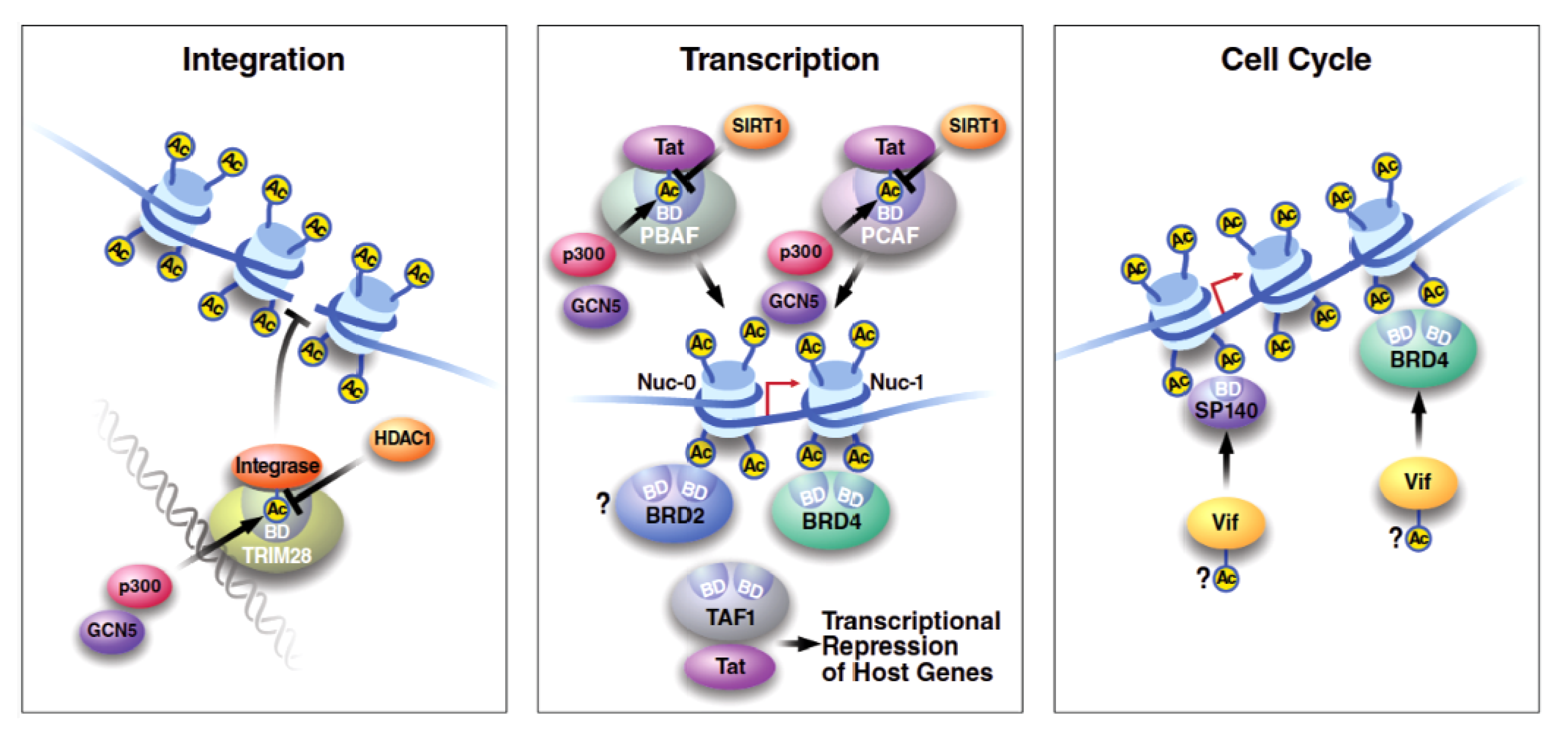
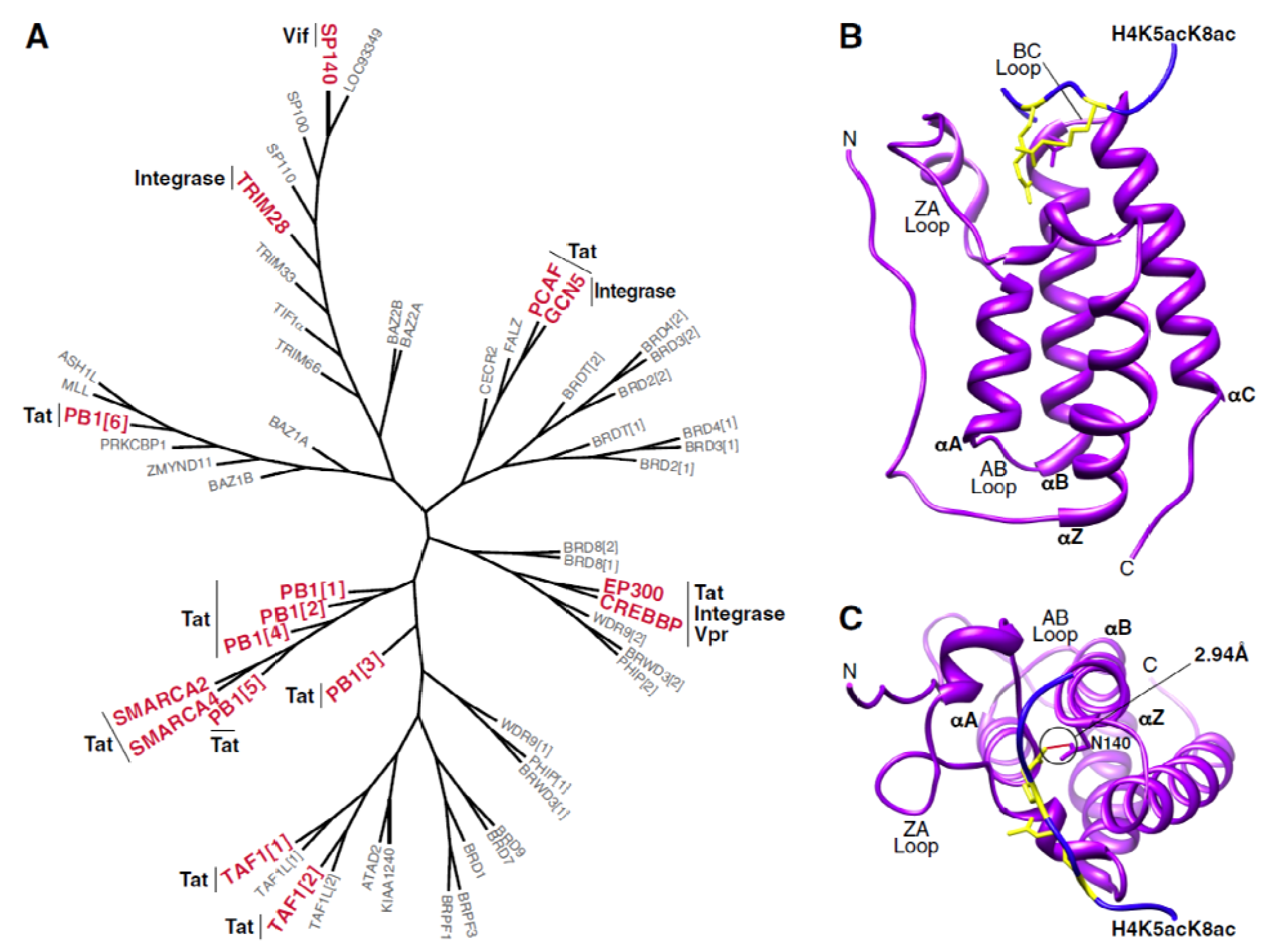
3. Interactions between Bromodomain-Containing Proteins and HIV Proteins
3.1. Acetylation-Dependent Interactions
3.1.1. p300/CBP-Associated Factor (PCAF)
3.1.2. SWItch/Sucrose Non-Fermentable (SWI/SNF)
3.1.3. Bromodomain-Containing Protein 4 (BRD4)
3.1.4. Tripartite Motif-Containing Protein 28 (TRIM28)
3.2. Other Interactions with Bromodomain-Containing Proteins
3.2.1. Transcription Initiation Factor TFIID, Subunit 1 (TAF1)
3.2.2. Nuclear Body Protein SP140
3.2.3. BRD2
3.2.4. BRD4
4. Bromodomain Inhibitors and HIV Infection

{kind=link}
{kind=link}
| Compound | Model of HIV latency | Effect | Reference |
|---|---|---|---|
| JQ1 | Ach2 | reactivation | [83] |
| JQ1 | U1 | reactivation | |
| JQ1 | J-Lat 10.6 | reactivation | |
| JQ1 | Acutely infected primary CD4+ cells | reactivation | |
| JQ1 | J∆K | reactivation | [84] |
| JQ1 | J-Lat A2 | reactivation | [85] |
| JQ1 | Jurkat 1G5 | reactivation | |
| JQ1 | HeLa NH1 and NH2 | reactivation | |
| JQ1 | HeLa-T4 | reactivation | [86] |
| JQ1 | Primary CD4+ T cells | reactivation | |
| JQ1 | Primary CD4+ T cells | inhibition | |
| JQ1 + Prostratin or PMA | J-Lat 6.3 | reactivation | |
| JQ1 + Prostratin or PMA | J-Lat 8.4 | reactivation | |
| JQ1 + Prostratin or PMA | J-Lat 9.2 | reactivation | |
| JQ1 + Prostratin or PMA | J-Lat 15.4 | reactivation | |
| JQ1 | J-Lat A2 | reactivation | [87] |
| JQ1 | J-Lat A72 | reactivation | |
| JQ1 | infected primary Bc12-transduced CD4+ T cells | reactivation | |
| JQ1 | infected primary nonpolarized T helper cells | no reactivation | |
| I-BET | infected primary Bc12-transduced CD4+ T cells | reactivation | |
| I-BET | infected primary nonpolarized T helper cells | no reactivation | |
| I-Bet151 | J-Lat A2 | reactivation | |
| I-Bet151 | J-Lat A72 | reactivation | |
| I-Bet151 | infected primary Bc12-transduced CD4+ T cells | reactivation | |
| I-Bet151 | infected primary nonpolarized T helper cells | no reactivation | |
| MS417 | J-Lat A2 | reactivation | |
| MS417 | J-Lat A72 | reactivation | |
| MS417 | infected primary Bc12-transduced CD4+ T cells | reactivation | |
| MS417 | infected primary nonpolarized T helper cells | no reactivation |
5. Concluding Remarks
Acknowledgments
Conflict of Interest
References and Notes
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar]
- Yang, X.J.; Seto, E. Lysine acetylation: Codified crosstalk with other posttranslational modifications. Mol. Cell 2008, 31, 449–461. [Google Scholar]
- Choudhary, C.; Kumar, C.; Gnad, F.; Nielsen, M.L.; Rehman, M.; Walther, T.C.; Olsen, J.V.; Mann, M. Lysine acetylation targets protein complexes and co-regulates major cellular functions. Science 2009, 325, 834–840. [Google Scholar]
- Sterner, D.E.; Berger, S.L. Acetylation of histones and transcription-related factors. Microbiol. Mol. Biol. Rev. 2000, 64, 435–459. [Google Scholar]
- Dhalluin, C.; Carlson, J.E.; Zeng, L.; He, C.; Aggarwal, A.K.; Zhou, M.M. Structure and ligand of a histone acetyltransferase bromodomain. Nature 1999, 399, 491–496. [Google Scholar]
- Filippakopoulos, P.; Knapp, S. The bromodomain interaction module. FEBS Lett. 2012, 586, 2692–2704. [Google Scholar] [CrossRef]
- Tamkun, J.W.; Deuring, R.; Scott, M.P.; Kissinger, M.; Pattatucci, A.M.; Kaufman, T.C.; Kennison, J.A. Brahma: A regulator of Drosophila homeotic genes structurally related to the yeast transcriptional activator SNF2/SWI2. Cell 1992, 68, 561–572. [Google Scholar]
- Filippakopoulos, P.; Picaud, S.; Mangos, M.; Keates, T.; Lambert, J.P.; Barsyte-Lovejoy, D.; Felletar, I.; Volkmer, R.; Muller, S.; Pawson, T.; et al. Histone recognition and large-scale structural analysis of the human bromodomain family. Cell 2012, 149, 214–231. [Google Scholar] [CrossRef]
- Moriniere, J.; Rousseaux, S.; Steuerwald, U.; Soler-Lopez, M.; Curtet, S.; Vitte, A.L.; Govin, J.; Gaucher, J.; Sadoul, K.; Hart, D.J.; et al. Cooperative binding of two acetylation marks on a histone tail by a single bromodomain. Nature 2009, 461, 664–668. [Google Scholar] [CrossRef]
- Archin, N.M.; Liberty, A.L.; Kashuba, A.D.; Choudhary, S.K.; Kuruc, J.D.; Crooks, A.M.; Parker, D.C.; Anderson, E.M.; Kearney, M.F.; Strain, M.C.; et al. Administration of vorinostat disrupts HIV-1 latency in patients on antiretroviral therapy. Nature 2012, 487, 482–485. [Google Scholar] [CrossRef]
- Kent, S.J.; Reece, J.C.; Petravic, J.; Martyushev, A.; Kramski, M.; de Rose, R.; Cooper, D.A.; Kelleher, A.D.; Emery, S.; Cameron, P.U.; et al. The search for an HIV cure: Tackling latent infection. Lancet Infect. Dis. 2013. [Google Scholar] [CrossRef]
- Contreras, X.; Schweneker, M.; Chen, C.S.; McCune, J.M.; Deeks, S.G.; Martin, J.; Peterlin, B.M. Suberoylanilide hydroxamic acid reactivates HIV from latently infected cells. J. Biol. Chem. 2009, 284, 6782–6789. [Google Scholar]
- Edelstein, L.C.; Micheva-Viteva, S.; Phelan, B.D.; Dougherty, J.P. Short communication: Activation of latent HIV type 1 gene expression by suberoylanilide hydroxamic acid (SAHA), an HDAC inhibitor approved for use to treat cutaneous T cell lymphoma. AIDS Res. Hum. Retroviruses 2009, 25, 883–887. [Google Scholar] [CrossRef]
- Kelly, W.K.; Richon, V.M.; O’Connor, O.; Curley, T.; MacGregor-Curtelli, B.; Tong, W.; Klang, M.; Schwartz, L.; Richardson, S.; Rosa, E.; et al. Phase I clinical trial of histone deacetylase inhibitor: Suberoylanilide hydroxamic acid administered intravenously. Clin. Cancer Res. 2003, 9, 3578–3588. [Google Scholar]
- Verdin, E.; Paras, P., Jr.; van Lint, C. Chromatin disruption in the promoter of human immunodeficiency virus type 1 during transcriptional activation. EMBO J. 1993, 12, 3249–3259. [Google Scholar]
- Lusic, M.; Marcello, A.; Cereseto, A.; Giacca, M. Regulation of HIV-1 gene expression by histone acetylation and factor recruitment at the LTR promoter. EMBO J. 2003, 22, 6550–6561. [Google Scholar] [CrossRef]
- Sobhian, B.; Laguette, N.; Yatim, A.; Nakamura, M.; Levy, Y.; Kiernan, R.; Benkirane, M. HIV-1 Tat assembles a multifunctional transcription elongation complex and stably associates with the 7SK snRNP. Mol. Cell 2010, 38, 439–451. [Google Scholar] [CrossRef]
- He, N.; Liu, M.; Hsu, J.; Xue, Y.; Chou, S.; Burlingame, A.; Krogan, N.J.; Alber, T.; Zhou, Q. HIV-1 Tat and host AFF4 recruit two transcription elongation factors into a bifunctional complex for coordinated activation of HIV-1 transcription. Mol. Cell 2010, 38, 428–438. [Google Scholar] [CrossRef]
- Marzio, G.; Tyagi, M.; Gutierrez, M.I.; Giacca, M. HIV-1 tat transactivator recruits p300 and CREB-binding protein histone acetyltransferases to the viral promoter. Proc. Natl. Acad. Sci. USA 1998, 95, 13519–13524. [Google Scholar] [CrossRef]
- Coull, J.J.; Romerio, F.; Sun, J.M.; Volker, J.L.; Galvin, K.M.; Davie, J.R.; Shi, Y.; Hansen, U.; Margolis, D.M. The human factors YY1 and LSF repress the human immunodeficiency virus type 1 long terminal repeat via recruitment of histone deacetylase 1. J. Virol. 2000, 74, 6790–6799. [Google Scholar] [CrossRef]
- Williams, S.A.; Chen, L.F.; Kwon, H.; Ruiz-Jarabo, C.M.; Verdin, E.; Greene, W.C. NF-kappaB p50 promotes HIV latency through HDAC recruitment and repression of transcriptional initiation. EMBO J. 2006, 25, 139–149. [Google Scholar] [CrossRef]
- Imai, K.; Okamoto, T. Transcriptional repression of human immunodeficiency virus type 1 by AP-4. J. Biol. Chem. 2006, 281, 12495–12505. [Google Scholar] [CrossRef]
- Tyagi, M.; Karn, J. CBF-1 promotes transcriptional silencing during the establishment of HIV-1 latency. EMBO J. 2007, 26, 4985–4995. [Google Scholar] [CrossRef]
- Jiang, G.; Espeseth, A.; Hazuda, D.J.; Margolis, D.M. c-Myc and Sp1 contribute to proviral latency by recruiting histone deacetylase 1 to the human immunodeficiency virus type 1 promoter. J. Virol. 2007, 81, 10914–10923. [Google Scholar] [CrossRef]
- Van Lint, C.; Emiliani, S.; Ott, M.; Verdin, E. Transcriptional activation and chromatin remodeling of the HIV-1 promoter in response to histone acetylation. EMBO J. 1996, 15, 1112–1120. [Google Scholar]
- Kiernan, R.E.; Vanhulle, C.; Schiltz, L.; Adam, E.; Xiao, H.; Maudoux, F.; Calomme, C.; Burny, A.; Nakatani, Y.; Jeang, K.T.; et al. HIV-1 tat transcriptional activity is regulated by acetylation. EMBO J. 1999, 18, 6106–6118. [Google Scholar] [CrossRef]
- Ott, M.; Schnolzer, M.; Garnica, J.; Fischle, W.; Emiliani, S.; Rackwitz, H.R.; Verdin, E. Acetylation of the HIV-1 Tat protein by p300 is important for its transcriptional activity. Curr. Biol. 1999, 9, 1489–1492. [Google Scholar] [CrossRef]
- Col, E.; Caron, C.; Seigneurin-Berny, D.; Gracia, J.; Favier, A.; Khochbin, S. The histone acetyltransferase, hGCN5, interacts with and acetylates the HIV transactivator, Tat. J. Biol. Chem. 2001, 276, 28179–28184. [Google Scholar]
- Bres, V.; Kiernan, R.; Emiliani, S.; Benkirane, M. Tat acetyl-acceptor lysines are important for human immunodeficiency virus type-1 replication. J. Biol. Chem. 2002, 277, 22215–22221. [Google Scholar]
- Kaehlcke, K.; Dorr, A.; Hetzer-Egger, C.; Kiermer, V.; Henklein, P.; Schnoelzer, M.; Loret, E.; Cole, P.A.; Verdin, E.; Ott, M. Acetylation of Tat defines a cyclinT1-independent step in HIV transactivation. Mol. Cell 2003, 12, 167–176. [Google Scholar] [CrossRef]
- Pagans, S.; Pedal, A.; North, B.J.; Kaehlcke, K.; Marshall, B.L.; Dorr, A.; Hetzer-Egger, C.; Henklein, P.; Frye, R.; McBurney, M.W.; et al. SIRT1 regulates HIV transcription via Tat deacetylation. PLoS Biol. 2005, 3, e41. [Google Scholar] [CrossRef] [Green Version]
- Huo, L.; Li, D.; Sun, X.; Shi, X.; Karna, P.; Yang, W.; Liu, M.; Qiao, W.; Aneja, R.; Zhou, J. Regulation of Tat acetylation and transactivation activity by the microtubule-associated deacetylase HDAC6. J. Biol. Chem. 2011, 286, 9280–9286. [Google Scholar]
- Cereseto, A.; Manganaro, L.; Gutierrez, M.I.; Terreni, M.; Fittipaldi, A.; Lusic, M.; Marcello, A.; Giacca, M. Acetylation of HIV-1 integrase by p300 regulates viral integration. EMBO J. 2005, 24, 3070–3081. [Google Scholar] [CrossRef]
- Terreni, M.; Valentini, P.; Liverani, V.; Gutierrez, M.I.; Di Primio, C.; Di Fenza, A.; Tozzini, V.; Allouch, A.; Albanese, A.; Giacca, M.; et al. CN5-dependent acetylation of HIV-1 integrase enhances viral integration. Retrovirology 2010, 7, 18. [Google Scholar]
- Kino, T.; Gragerov, A.; Slobodskaya, O.; Tsopanomichalou, M.; Chrousos, G.P.; Pavlakis, G.N. Human immunodeficiency virus type 1 (HIV-1) accessory protein Vpr induces transcription of the HIV-1 and glucocorticoid-responsive promoters by binding directly to p300/CBP coactivators. J. Virol. 2002, 76, 9724–9734. [Google Scholar] [CrossRef]
- Nagy, Z.; Tora, L. Distinct GCN5/PCAF-containing complexes function as co-activators and are involved in transcription factor and global histone acetylation. Oncogene 2007, 26, 5341–5357. [Google Scholar] [CrossRef]
- Wang, C.Y.; Yang, S.F.; Wang, Z.; Tan, J.M.; Xing, S.M.; Chen, D.C.; Xu, S.M.; Yuan, W. PCAF acetylates Runx2 and promotes osteoblast differentiation. J. Bone Miner. Metab. 2013. [Google Scholar] [CrossRef]
- D’Orso, I.; Frankel, A.D. Tat acetylation modulates assembly of a viral-host RNA-protein transcription complex. Proc. Natl. Acad. Sci. USA 2009, 106, 3101–3106. [Google Scholar] [CrossRef]
- Dorr, A.; Kiermer, V.; Pedal, A.; Rackwitz, H.R.; Henklein, P.; Schubert, U.; Zhou, M.M.; Verdin, E.; Ott, M. Transcriptional synergy between Tat and PCAF is dependent on the binding of acetylated Tat to the PCAF bromodomain. EMBO J. 2002, 21, 2715–2723. [Google Scholar] [CrossRef]
- Mujtaba, S.; He, Y.; Zeng, L.; Farooq, A.; Carlson, J.E.; Ott, M.; Verdin, E.; Zhou, M.M. Structural basis of lysine-acetylated HIV-1 Tat recognition by PCAF bromodomain. Mol. Cell 2002, 9, 575–586. [Google Scholar] [CrossRef]
- Zeng, L.; Li, J.; Muller, M.; Yan, S.; Mujtaba, S.; Pan, C.; Wang, Z.; Zhou, M.M. Selective small molecules blocking HIV-1 Tat and coactivator PCAF association. J. Am. Chem. Soc. 2005, 127, 2376–2377. [Google Scholar]
- Rafati, H.; Parra, M.; Hakre, S.; Moshkin, Y.; Verdin, E.; Mahmoudi, T. Repressive LTR nucleosome positioning by the BAF complex is required for HIV latency. PLoS Biol. 2011, 9, e1001206. [Google Scholar] [CrossRef]
- Mahmoudi, T. The BAF complex and HIV latency. Transcription 2012, 3, 171–176. [Google Scholar] [CrossRef]
- Van Duyne, R.; Guendel, I.; Narayanan, A.; Gregg, E.; Shafagati, N.; Tyagi, M.; Easley, R.; Klase, Z.; Nekhai, S.; Kehn-Hall, K.; et al. Varying modulation of HIV-1 LTR activity by Baf complexes. J. Mol. Biol. 2011, 411, 581–596. [Google Scholar]
- Mahmoudi, T.; Parra, M.; Vries, R.G.; Kauder, S.E.; Verrijzer, C.P.; Ott, M.; Verdin, E. The SWI/SNF chromatin-remodeling complex is a cofactor for Tat transactivation of the HIV promoter. J. Biol. Chem. 2006, 281, 19960–19968. [Google Scholar]
- Treand, C.; du Chene, I.; Bres, V.; Kiernan, R.; Benarous, R.; Benkirane, M.; Emiliani, S. Requirement for SWI/SNF chromatin-remodeling complex in Tat-mediated activation of the HIV-1 promoter. EMBO J. 2006, 25, 1690–1699. [Google Scholar] [CrossRef]
- Easley, R.; Carpio, L.; Dannenberg, L.; Choi, S.; Alani, D.; Van Duyne, R.; Guendel, I.; Klase, Z.; Agbottah, E.; Kehn-Hall, K.; et al. Transcription through the HIV-1 nucleosomes: Effects of the PBAF complex in Tat activated transcription. Virology 2010, 405, 322–333. [Google Scholar] [CrossRef]
- Dey, A.; Chitsaz, F.; Abbasi, A.; Misteli, T.; Ozato, K. The double bromodomain protein Brd4 binds to acetylated chromatin during interphase and mitosis. Proc. Natl. Acad. Sci. USA 2003, 100, 8758–8763. [Google Scholar] [CrossRef]
- Wu, S.Y.; Chiang, C.M. The double bromodomain-containing chromatin adaptor Brd4 and transcriptional regulation. J. Biol. Chem. 2007, 282, 13141–13145. [Google Scholar] [CrossRef]
- Hubner, M.R.; Eckersley-Maslin, M.A.; Spector, D.L. Chromatin organization and transcriptional regulation. Curr. Opin. Genet. Dev. 2012, 23, 89–95. [Google Scholar]
- Bisgrove, D.A.; Mahmoudi, T.; Henklein, P.; Verdin, E. Conserved P-TEFb-interacting domain of BRD4 inhibits HIV transcription. Proc. Natl. Acad. Sci. USA 2007, 104, 13690–13695. [Google Scholar]
- Schroder, S.; Cho, S.; Zeng, L.; Zhang, Q.; Kaehlcke, K.; Mak, L.; Lau, J.; Bisgrove, D.; Schnolzer, M.; Verdin, E.; et al. Two-pronged binding with bromodomain-containing protein 4 liberates positive transcription elongation factor b from inactive ribonucleoprotein complexes. J. Biol. Chem. 2012, 287, 1090–1099. [Google Scholar] [CrossRef]
- Yang, Z.; Zhu, Q.; Luo, K.; Zhou, Q. The 7SK small nuclear RNA inhibits the CDK9/cyclin T1 kinase to control transcription. Nature 2001, 414, 317–322. [Google Scholar] [CrossRef]
- Yang, Z.; Yik, J.H.; Chen, R.; He, N.; Jang, M.K.; Ozato, K.; Zhou, Q. Recruitment of P-TEFb for stimulation of transcriptional elongation by the bromodomain protein Brd4. Mol. Cell 2005, 19, 535–545. [Google Scholar] [CrossRef]
- Zhou, M.; Huang, K.; Jung, K.J.; Cho, W.K.; Klase, Z.; Kashanchi, F.; Pise-Masison, C.A.; Brady, J.N. Bromodomain protein Brd4 regulates human immunodeficiency virus transcription through phosphorylation of CDK9 at threonine 29. J. Virol. 2009, 83, 1036–1044. [Google Scholar] [CrossRef]
- Cho, S.; Schroeder, S.; Kaehlcke, K.; Kwon, H.S.; Pedal, A.; Herker, E.; Schnoelzer, M.; Ott, M. Acetylation of cyclin T1 regulates the equilibrium between active and inactive P-TEFb in cells. EMBO J. 2009, 28, 1407–1417. [Google Scholar] [CrossRef]
- Vollmuth, F.; Blankenfeldt, W.; Geyer, M. Structures of the dual bromodomains of the P-TEFb-activating protein Brd4 at atomic resolution. J. Biol. Chem. 2009, 284, 36547–36556. [Google Scholar] [CrossRef]
- Zeng, L.; Yap, K.L.; Ivanov, A.V.; Wang, X.; Mujtaba, S.; Plotnikova, O.; Rauscher, F.J., 3rd; Zhou, M.M. Structural insights into human KAP1 PHD finger-bromodomain and its role in gene silencing. Nat. Struct. Mol. Biol. 2008, 15, 626–633. [Google Scholar]
- Allouch, A.; di Primio, C.; Alpi, E.; Lusic, M.; Arosio, D.; Giacca, M.; Cereseto, A. The TRIM family protein KAP1 inhibits HIV-1 integration. Cell Host Microbe 2011, 9, 484–495. [Google Scholar] [CrossRef]
- Allouch, A.; Cereseto, A. Identification of cellular factors binding to acetylated HIV-1 integrase. Amino Acids 2011, 41, 1137–1145. [Google Scholar] [CrossRef]
- Jacobson, R.H.; Ladurner, A.G.; King, D.S.; Tjian, R. Structure and function of a human TAFII250 double bromodomain module. Science 2000, 288, 1422–1425. [Google Scholar] [CrossRef]
- Weissman, J.D.; Brown, J.A.; Howcroft, T.K.; Hwang, J.; Chawla, A.; Roche, P.A.; Schiltz, L.; Nakatani, Y.; Singer, D.S. HIV-1 tat binds TAFII250 and represses TAFII250-dependent transcription of major histocompatibility class I genes. Proc. Natl. Acad. Sci. USA 1998, 95, 11601–11606. [Google Scholar] [CrossRef]
- Granito, A.; Yang, W.H.; Muratori, L.; Lim, M.J.; Nakajima, A.; Ferri, S.; Pappas, G.; Quarneti, C.; Bianchi, F.B.; Bloch, D.B.; Muratori, P. PML nuclear body component Sp140 is a novel autoantigen in primary biliary cirrhosis. Am. J. Gastroenterol. 2010, 105, 125–131. [Google Scholar] [CrossRef]
- Bloch, D.B.; de la Monte, S.M.; Guigaouri, P.; Filippov, A.; Bloch, K.D. Identification and characterization of a leukocyte-specific component of the nuclear body. J. Biol. Chem. 1996, 271, 29198–29204. [Google Scholar]
- Dent, A.L.; Yewdell, J.; Puvion-Dutilleul, F.; Koken, M.H.; de The, H.; Staudt, L.M. LYSP100-associated nuclear domains (LANDs): Description of a new class of subnuclear structures and their relationship to PML nuclear bodies. Blood 1996, 88, 1423–1426. [Google Scholar]
- Madani, N.; Millette, R.; Platt, E.J.; Marin, M.; Kozak, S.L.; Bloch, D.B.; Kabat, D. Implication of the lymphocyte-specific nuclear body protein Sp140 in an innate response to human immunodeficiency virus type 1. J. Virol. 2002, 76, 11133–11138. [Google Scholar] [CrossRef]
- Sheehy, A.M.; Gaddis, N.C.; Malim, M.H. The antiretroviral enzyme APOBEC3G is degraded by the proteasome in response to HIV-1 Vif. Nat. Med. 2003, 9, 1404–1407. [Google Scholar] [CrossRef]
- Malovannaya, A.; Lanz, R.B.; Jung, S.Y.; Bulynko, Y.; Le, N.T.; Chan, D.W.; Ding, C.; Shi, Y.; Yucer, N.; Krenciute, G.; et al. Analysis of the human endogenous coregulator complexome. Cell 2011, 145, 787–799. [Google Scholar] [CrossRef]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar]
- Denis, G.V.; McComb, M.E.; Faller, D.V.; Sinha, A.; Romesser, P.B.; Costello, C.E. Identification of transcription complexes that contain the double bromodomain protein Brd2 and chromatin remodeling machines. J. Proteome Res. 2006, 5, 502–511. [Google Scholar] [CrossRef]
- Wang, J.; Reuschel, E.L.; Shackelford, J.M.; Jeang, L.; Shivers, D.K.; Diehl, J.A.; Yu, X.F.; Finkel, T.H. HIV-1 Vif promotes the G(1)- to S-phase cell-cycle transition. Blood 2011, 117, 1260–1269. [Google Scholar] [CrossRef]
- Yang, Z.; He, N.; Zhou, Q. Brd4 recruits P-TEFb to chromosomes at late mitosis to promote G1 gene expression and cell cycle progression. Mol. Cell. Biol. 2008, 28, 967–976. [Google Scholar] [CrossRef]
- Mochizuki, K.; Nishiyama, A.; Jang, M.K.; Dey, A.; Ghosh, A.; Tamura, T.; Natsume, H.; Yao, H.; Ozato, K. The bromodomain protein Brd4 stimulates G1 gene transcription and promotes progression to S phase. J. Biol. Chem. 2008, 283, 9040–9048. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Picaud, S.; Fedorov, O.; Keller, M.; Wrobel, M.; Morgenstern, O.; Bracher, F.; Knapp, S. Benzodiazepines and benzotriazepines as protein interaction inhibitors targeting bromodomains of the BET family. Bioorg. Med. Chem. 2012, 20, 1878–1886. [Google Scholar] [CrossRef]
- Filippakopoulos, P.; Qi, J.; Picaud, S.; Shen, Y.; Smith, W.B.; Fedorov, O.; Morse, E.M.; Keates, T.; Hickman, T.T.; Felletar, I.; et al. Selective inhibition of BET bromodomains. Nature 2010, 468, 1067–1073. [Google Scholar] [CrossRef]
- Nicodeme, E.; Jeffrey, K.L.; Schaefer, U.; Beinke, S.; Dewell, S.; Chung, C.W.; Chandwani, R.; Marazzi, I.; Wilson, P.; Coste, H.; et al. Suppression of inflammation by a synthetic histone mimic. Nature 2010, 468, 1119–1123. [Google Scholar] [CrossRef]
- Dawson, M.A.; Prinjha, R.K.; Dittmann, A.; Giotopoulos, G.; Bantscheff, M.; Chan, W.I.; Robson, S.C.; Chung, C.W.; Hopf, C.; Savitski, M.M.; et al. Inhibition of BET recruitment to chromatin as an effective treatment for MLL-fusion leukaemia. Nature 2011, 478, 529–533. [Google Scholar] [CrossRef]
- Zhang, G.; Liu, R.; Zhong, Y.; Plotnikov, A.N.; Zhang, W.; Zeng, L.; Rusinova, E.; Gerona-Nevarro, G.; Moshkina, N.; Joshua, J.; et al. Down-regulation of NF-kappaB transcriptional activity in HIV-associated kidney disease by BRD4 inhibition. J. Biol. Chem. 2012, 287, 28840–28851. [Google Scholar] [CrossRef]
- Miyoshi, S.; Ooike, S.; Iwata, K.; Hikawa, H.; Sugahara, K. International Patent PCT/JP2008/073864 (US2010/0286127 A1), 26 December 2008.
- Zuber, J.; Shi, J.; Wang, E.; Rappaport, A.R.; Herrmann, H.; Sison, E.A.; Magoon, D.; Qi, J.; Blatt, K.; Wunderlich, M.; et al. RNAi screen identifies Brd4 as a therapeutic target in acute myeloid leukaemia. Nature 2011, 478, 524–528. [Google Scholar] [CrossRef]
- Routy, J.P.; Angel, J.B.; Spaans, J.N.; Trottier, B.; Rouleau, D.; Baril, J.G.; Harris, M.; Trottier, S.; Singer, J.; Chomont, N.; et al. Design and implementation of a randomized crossover study of valproic acid and antiretroviral therapy to reduce the HIV reservoir. HIV Clin. Trials 2012, 13, 301–307. [Google Scholar] [CrossRef]
- Routy, J.P.; Tremblay, C.L.; Angel, J.B.; Trottier, B.; Rouleau, D.; Baril, J.G.; Harris, M.; Trottier, S.; Singer, J.; Chomont, N.; et al. Valproic acid in association with highly active antiretroviral therapy for reducing systemic HIV-1 reservoirs: Results from a multicentre randomized clinical study. HIV Med. 2012, 13, 291–296. [Google Scholar] [CrossRef]
- Banerjee, C.; Archin, N.; Michaels, D.; Belkina, A.C.; Denis, G.V.; Bradner, J.; Sebastiani, P.; Margolis, D.M.; Montano, M. BET bromodomain inhibition as a novel strategy for reactivation of HIV-1. J. Leukoc. Biol. 2012, 92, 1147–1154. [Google Scholar] [CrossRef]
- Bartholomeeusen, K.; Xiang, Y.; Fujinaga, K.; Peterlin, B.M. Bromodomain and extra-terminal (BET) bromodomain inhibition activate transcription via transient release of positive transcription elongation factor b (P-TEFb) from 7SK small nuclear ribonucleoprotein. J. Biol. Chem. 2012, 287, 36609–36616. [Google Scholar] [CrossRef]
- Li, Z.; Guo, J.; Wu, Y.; Zhou, Q. The BET bromodomain inhibitor JQ1 activates HIV latency through antagonizing Brd4 inhibition of Tat-transactivation. Nucleic Acids Res. 2013, 41, 277–287. [Google Scholar] [CrossRef]
- Zhu, J.; Gaiha, G.D.; John, S.P.; Pertel, T.; Chin, C.R.; Gao, G.; Qu, H.; Walker, B.D.; Elledge, S.J.; Brass, A.L. Reactivation of Latent HIV-1 by Inhibition of BRD4. Cell Rep. 2012, 2, 807–816. [Google Scholar] [CrossRef]
- Boehm, D.; Calvanese, V.; Dar, R.D.; Xing, S.; Schroeder, S.; Martins, L.; Aull, K.; Li, P.C.; Planelles, V.; Bradner, J.E.; et al. BET bromodomain-targeting compounds reactivate HIV from latency via a Tat-independent mechanism. Cell Cycle 2013, 12, 452–462. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Boehm, D.; Conrad, R.J.; Ott, M. Bromodomain Proteins in HIV Infection. Viruses 2013, 5, 1571-1586. https://doi.org/10.3390/v5061571
Boehm D, Conrad RJ, Ott M. Bromodomain Proteins in HIV Infection. Viruses. 2013; 5(6):1571-1586. https://doi.org/10.3390/v5061571
Chicago/Turabian StyleBoehm, Daniela, Ryan J. Conrad, and Melanie Ott. 2013. "Bromodomain Proteins in HIV Infection" Viruses 5, no. 6: 1571-1586. https://doi.org/10.3390/v5061571