1. Introduction
Labeling of biomolecules with enzymes has been widely applied in various bioassays due to the advantages of the inherent “biocatalytic” signal amplification. Each enzyme molecule, tethered to an antibody, a nucleic acid probe or other recognition element, can convert many molecules of a suitable substrate into detectable indicator product. This renders the enzyme-linked assays considerably sensitive. The immunochemical technique well known as ELISA [
1-
3] belongs to the core methodology of analysis of proteins and other biologically important species. Colorimetric or chemiluminescence-based techniques employing enzyme-labeled antibodies have successfully been applied, for example, in studies of proteins posttranslation modifications [
4], DNA-protein interactions [
3,
5], gene expression [
1], etc. Electrochemical enzyme-linked immunoassay techniques have also been reported [
6-
10].
Electrochemical DNA hybridization techniques involving enzyme-labeled probes have recently been proposed as well [
7,
8,
11-
18]. For electrochemical detection, the marker enzyme is required to catalyze conversion of an inactive substrate into electrochemically active or surface-active indicator which can subsequently be detected by voltammetry [
13,
14], amperometry [
12], impedance spectroscopy [
16] or other technique. For instance, alkaline phosphatase (ALP) has been applied [
8,
13-
15] in connection with phosphor-esters of phenols such as 1-naphthol or p-aminophenol. The phenol phosphates are electrochemically inactive while the parent phenols, released from the esters by the ALP, are electrochemically oxidizable. Hence, the DNA hybridization events can be detected via measurements of the released phenol signals which appear only in the presence of the enzyme tag. Besides ALP, other enzymes such as peroxidases [
11,
12] or β-galactosidase [
17], in connection with suitable substrates, have been applied in electrochemical DNA hybridization assays.
Enzyme-linked DNA sensing techniques have been applied in various experimental arrangements. The ELISA microwells [
11] or different types of magnetic beads [
8,
14,
15,
17] have been utilized as solid substrates for immobilization of capture probes and/or the target DNAs (tDNA) and performing hybridization of the tDNAs with the enzyme-labeled reporter probes. In other approaches, the capture probes or tDNAs were attached to the detection (transducer) electrode and both DNA hybridization and electrochemical detection were conducted at the same surface [
12,
13]. Using thermostable soybean peroxidase, a sensor for mismatch-sensitive enzyme-amplified detection of DNA hybridization was proposed [
12]. Another approach, based on hybridization between tDNAs adsorbed at carbon electrodes and ALP-labeled signaling probes, has recently been proposed [
13] for the determination of trinucleotide repeat lengths in polymerase chain reaction (PCR)-amplified genomic DNA fragments.
In the DNA hybridization experiments, the enzymes have often been coupled to the probe via biotin-(strept)avidin linkage, i.e., commercially available (strept)avidin-enzyme conjugates were attached to a biotinylated nucleic acid. Employment of the biotin-(strept)avidin technology offers utilization of an alternative approach, based on incorporation of the biotin-labeled nucleotides into DNA using DNA polymerases (instead of hybridization between tDNA with a biotinylated probe). Primer extension (PEX)-based assays involving labeled deoxynucleotide triphosphates (dNTPs) are routinely used in modern DNA sequencing techniques [
19,
20] and have been applied also in connection with the electrochemical detection platform [
16,
21-
25]. Besides ferrocene-dNTP conjugates most frequently used for these purposes [
21-
24], other electrochemically active moieties such as nitro- or amino phenyl derivatives have recently been coupled to dNTPs and incorporated into DNA [
25]. PEX incorporation of biotinylated dNTPs, followed by attachment of enzymes producing electrochemically detectable indicators, has also been reported [
16].
In this paper, applications of simple enzyme-linked electrochemical techniques, in connection with disposable screen-printed carbon electrodes (SPCE), for the detection of non-repetitive genomic DNA sequences amplified by PCR are proposed. The detection system involves biotin DNA labels, ALP-streptavidin conjugate (SALP) and 1-naphthyl phosphate as a substrate to be converted into the 1-naphthol indicator. We show an excellent differentiation between complementary and non-complementary DNA sequences via hybridization of tDNA adsorbed at SPCE with biotinylated probes, as well as highly specific detection of PCR-amplified genomic DNA fragments by means of a newly introduced PEX-based assay. In connection with a reverse transcription-PCR (RT-PCR) technique, application of the enzyme-linked electrochemical assay in gene expression monitoring is demonstrated.
2. Results and Discussion
It has been reported previously that some types of carbon electrodes (such as carbon paste [
26,
27], graphite-composite [
18], pyrolytic graphite or screen-printed electrodes [
13]) can be used as substrates for DNA hybridization without any special surface modifications, interfacing and/or covalent immobilization of capture probes (reviewed in [
28,
29]). Physisorbed single-stranded (ss) DNA (or peptide nucleic acid [
26]) was shown to be able of forming duplex with complementary DNA strands in solution to which the ssDNA-modified electrode was exposed. Sensors based on probe or tDNA adsorption at carbon electrodes have been combined with various detection principles, including label-free detection employing intrinsic DNA electroactivity [
30], application of non-covalent redox indicators (such as [Co(phen)
3]
3+/2+, methylene blue [
27], Meldola's blue [
31] or others) or probes labeled with enzymes [
13,
18].
Here we applied an enzyme-linked voltammetric technique, proposed previously for hybridization analysis of repetitive DNA sequences [
13], to detect hybridization between non-repetitive, random-sequence tDNAs and complementary biotinylated probes at the SPCE surface.
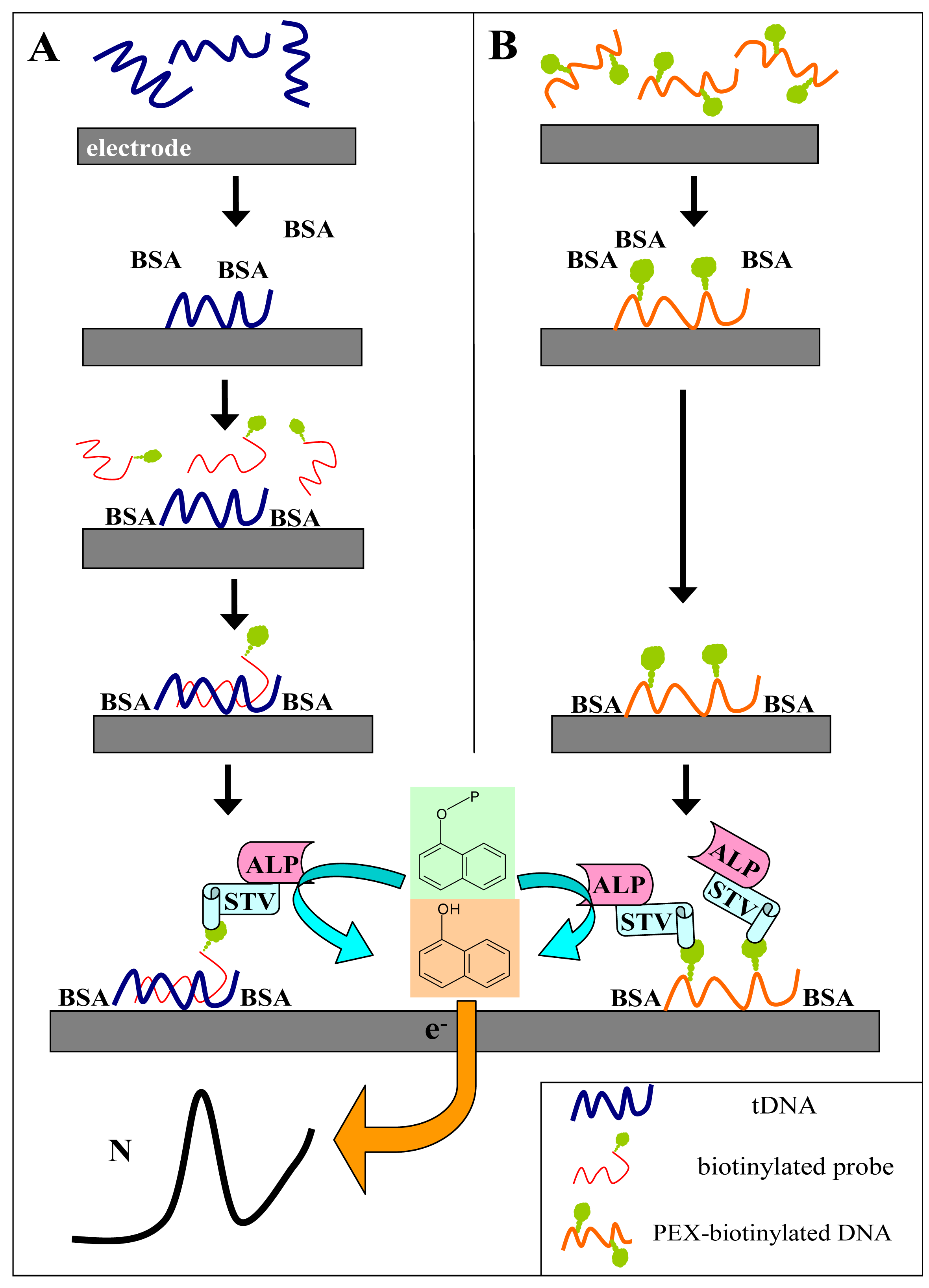
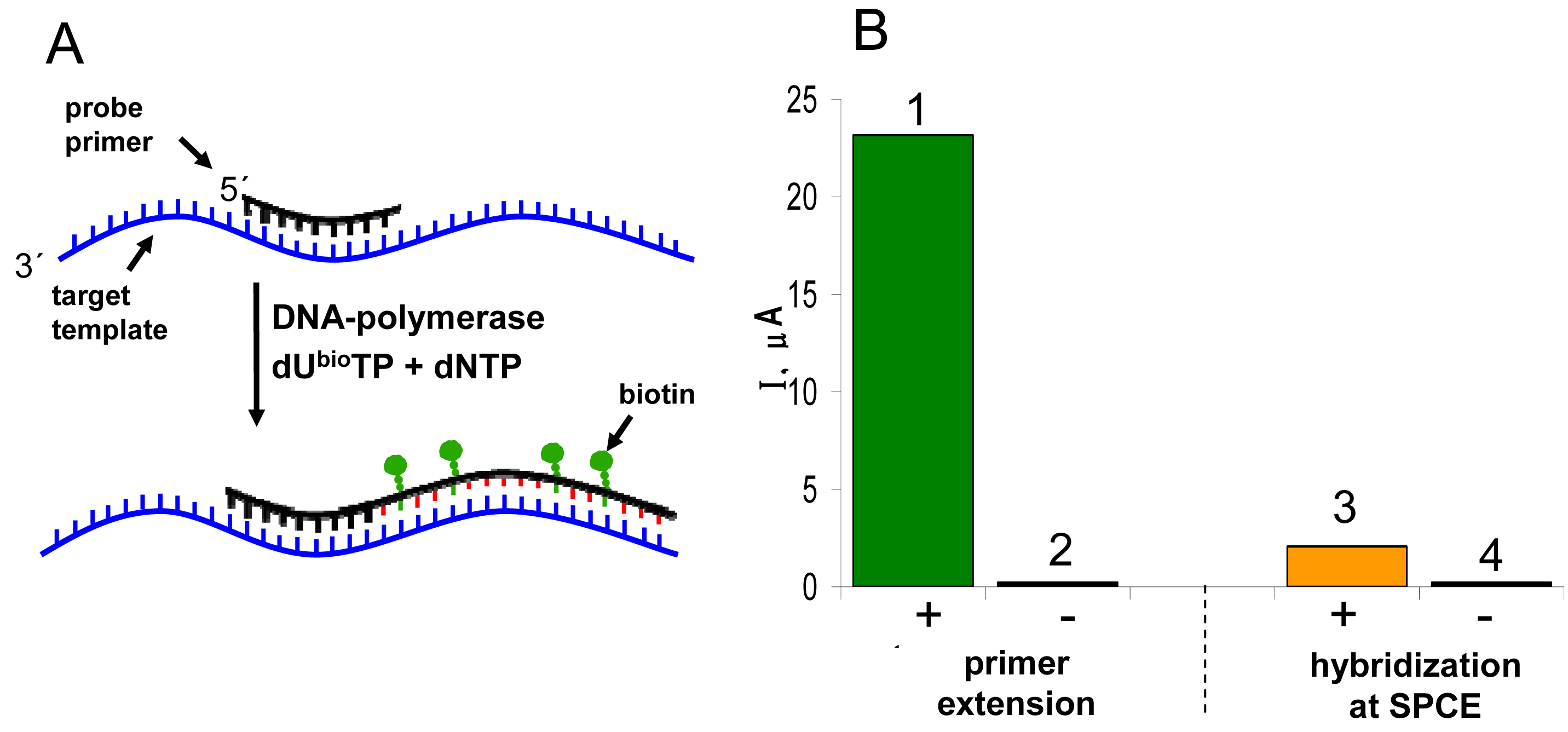
Figure 1A shows scheme of the experiment. The tDNA was adsorbed at the SPCE at open current circuit from a small (6-μl) aliquot of the sample. When the Gwent C 10903P14 ink
† was used for the SPCE preparation, no pretreatment of the electrode prior to DNA adsorption was necessary to obtain well defined and reasonably reproducible responses. After adsorption of the tDNA, the unoccupied electrode surface was blocked by bovine serum albumin, followed by subsequent application of the biotinylated probe solution and the SALP conjugate solution. Blocking of the electrode prior to the hybridization step was critical for specificity of the sensor responses: when it was omitted or performed after incubation with the biotinylated probe, false positive responses were obtained due to unspecific adsorption of the probe at the electrode surface [
13]. Finally, the electrode was dipped into solution of the substrate (1-naphthyl phosphate) in background electrolyte, and after a short incubation time during which the substrate was enzymatically converted into the electroactive indicator (1-naphthol), signal of the latter was measured using linear sweep voltammetry (LSV).
2.1 Hybridization with synthetic oligonucleotides
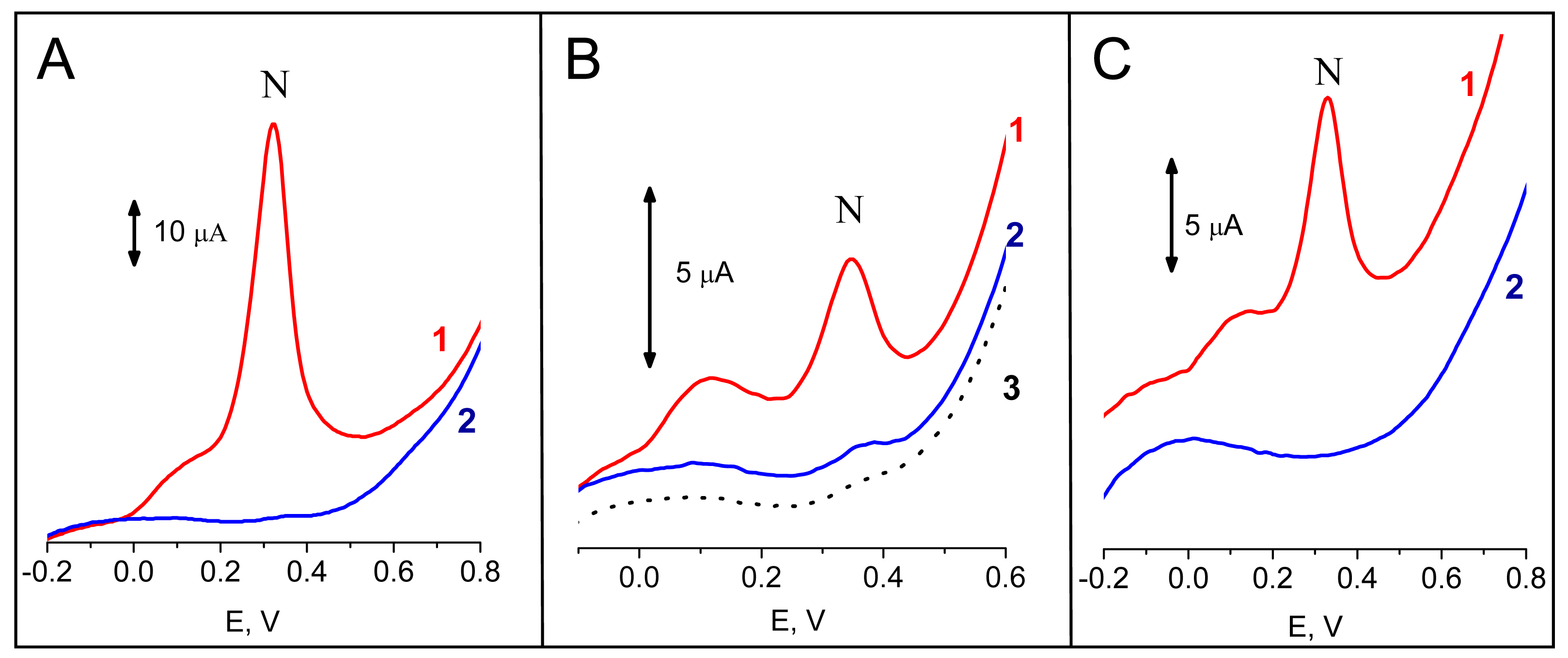
Typical hybridization response obtained for a model tDNA
Tp53 (
Table I), a synthetic 40-mer oligonucleotide (ODN) involving nucleotide sequence derived from coding part of human tumor suppressor gene
p53 [
3-
5], is shown in
Figure 2A. The target ODN was adsorbed at the SPCE from 5 μg mL
-1 solution in 0.3 M NaCl, 10 mM Tris-HCl, pH 7.6 (buffer H) and hybridized with the complementary
Pp53 probe (a 20-mer ODN bearing biotin tag at its 3′-end; 5 μg mL
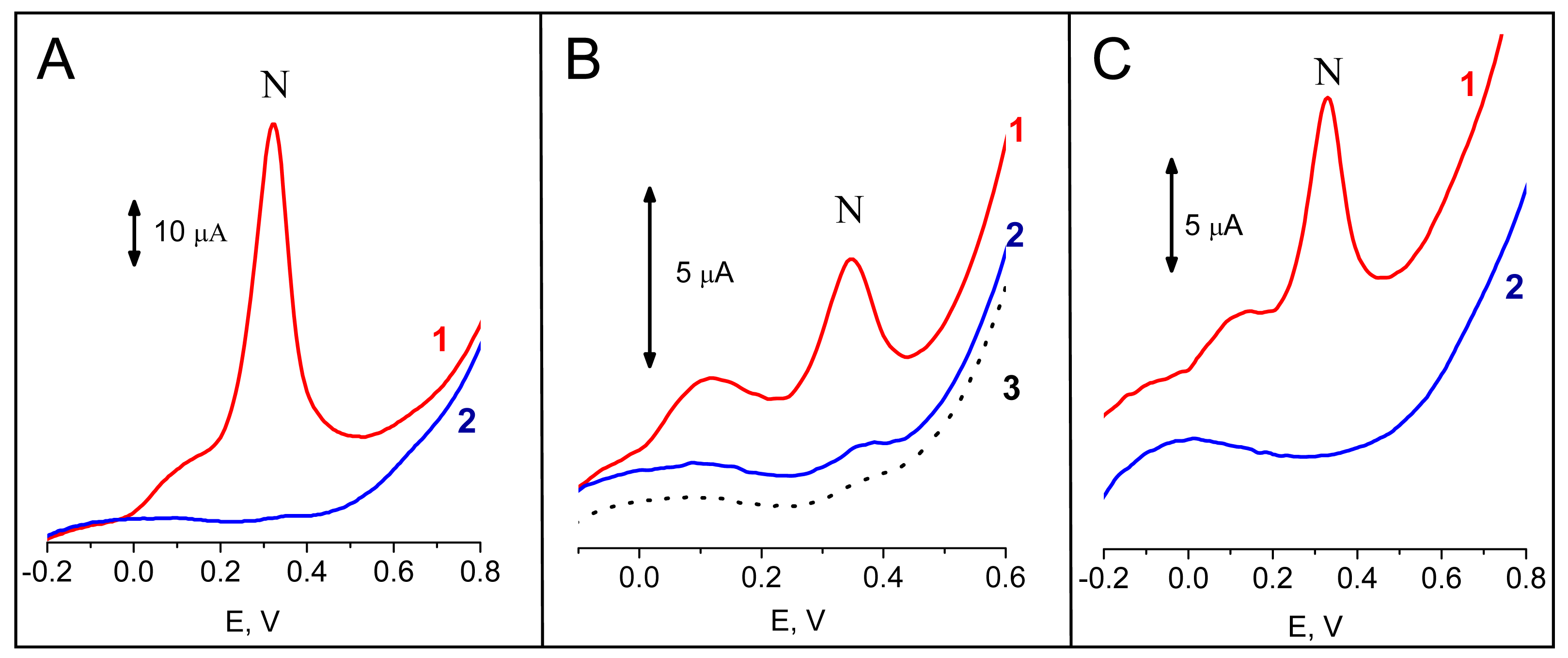
-1 in the buffer H) for 120 s. Incubation of the sensor in the substrate solution prior to the voltammetric measurement took 60 s. Under these conditions, a large peak N due to electrochemical oxidation of 1-naphthol at about +0.32 V was detected (
Fig, 2A). When
PrbcL probe was used instead of
Pp53, only negligible peak N was observed, in agreement with a lack of complementarity between the
Tp53 target and the
PrbcL probe (
Fig. 2A). Similarly, when
Tp53 was replaced by an ODN
NTgtt7, no significant signal was observed after incubation with the
Pp53 (
Fig. 3) or
PrbcL (not shown) probes. Hence, an excellent discrimination between ODNs complementary and non-complementary to the biotinylated probe was attained using the enzyme-linked electrochemical DNA hybridization assay.
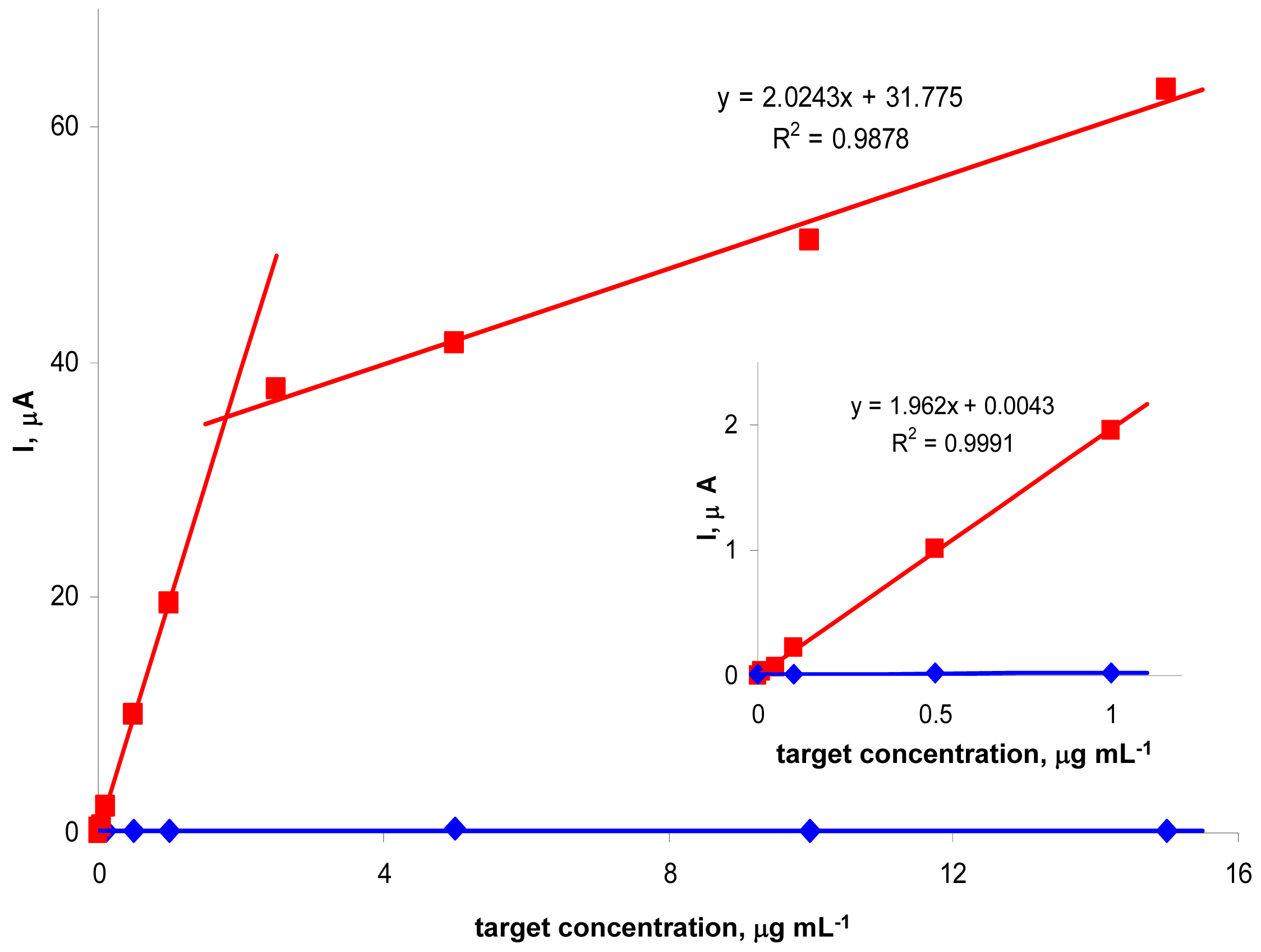
We further studied effects of tDNA concentration on the peak N intensity (
Fig. 3). For the
Tp53 target (applied at concentrations varying between 0 and 15 μg mL
-1) and
Pp53 (applied always at a concentration of 5 μg mL
-1), a biphasic dependence of the signal intensity on tDNA concentration, consisting of two linear segments, was observed. Between 0 and ∼5 μg mL
-1 the peak N height increased steeply with increasing
Tp53 concentration, followed by a region of less steeply increasing signal. Such shape of the dependence suggested saturation of the electrode surface by the tDNA molecules. In addition, other phenomena such as sterical clashes affecting the hybridization process (and/or SALP binding) at high surface concentrations of the tDNA [
28,
29], may contribute to the observed break on the tDNA concentration-signal intensity dependence. Under the given conditions, the
Tp53 target ODN was detectable (reliably distinguishable from the non-specific ODN
NTgtt7) down to 50 ng mL
-1 (corresponding to 300 pg in a 6-μl sample) i.e., about 3.8 nM (22 fmol in 6 μl). With the non-complementary
NTgtt7, only the negligible background signal was observed for any tDNA concentration between 0 and 16 μg mL
-1.
2.2 Hybridization with PCR-amplified genomic fragments
In contrast to the model synthetic ODNs, PCR-amplified DNA fragments are inherently double-stranded. Hence, denaturation of the duplex PCR product prior to adsorption at the electrode surface is necessary to make hybridization between the biotinylated probe and complementary stretch within one of the amplicon strands possible. Previously we showed [
13] that tDNA can be adsorbed at the carbon electrode surface from denaturing medium (20 mM NaOH) without loss of its hybridization capacity. The efficacy of tDNA hybridization with the signaling probe (performed in neutral medium) was under such conditions even higher, when compared to results obtained with thermally denatured tDNAs adsorbed at the electrode from physiological media, due to prevention of undesired renaturation of the amplicon duplexes. Here we used an analogous procedure and adsorbed the PCR products at the SPCE from solution containing 20 mM NaOH and 180 mM NaCl; other steps of the analytical protocol were performed as with the model target ODNs.
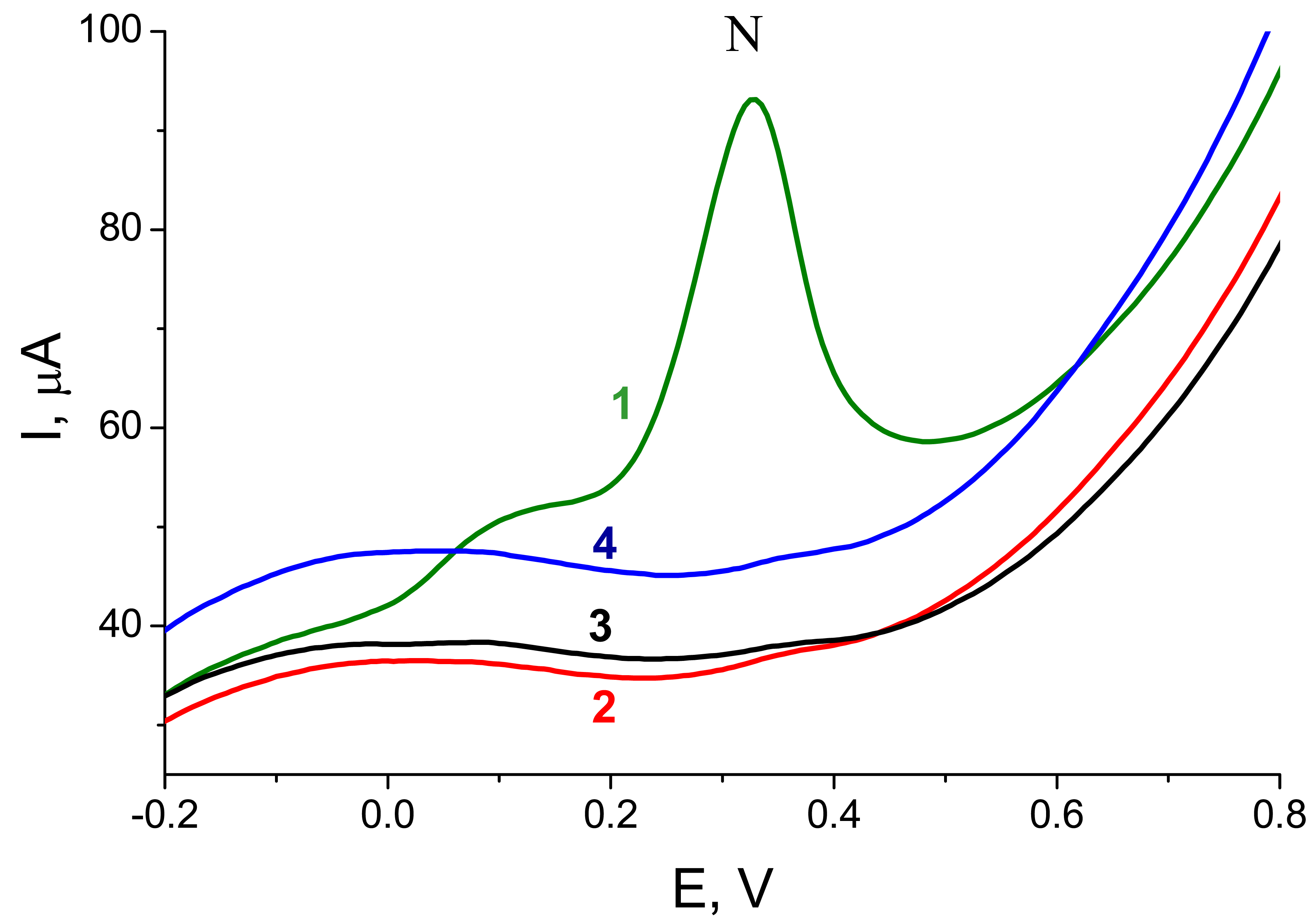
Figure 2B shows responses resulting from hybridization of a PCR-amplified 347-bp fragment of
p53 cDNA,
frp53, with probes
Pp53 (complementary) or
PrbcL (non-complementary). For the
Pp53 probe, a well defined peak N was detected, while for the
PrbcL probe only the small background signal was observed. When the
PrbcL probe was hybridized with
frrbcL, a 264-bp amplicon of the
rbcL cDNA possessing a stretch complementary to the
PrbcL, the resulting response was similar to that observed for the
frp53 -
Pp53 pair (see
Fig. 4). The presented electrochemical enzyme-linked technique thus provided a reliable discrimination between specific and non-specific (complementary or non-complementary to the probe used) PCR-amplified genomic DNA sequences. Intensities of signals obtained with the complementary PCR products (applied at the electrode at concentrations between 15 to 20 μg mL
-1 in 6-μl aliquots) were 20 to 30-times lower, compared to signals obtained for similar mass amounts of the target ODN (
Fig. 3). This could be expected, considering relative contents of the specific DNA stretches forming duplexes with the probes in the tDNAs. While in the single-stranded 40-mer ODN, 50 % of the total DNA corresponded to the 20-nucleotide sequence recognized by the probe, proportions of the recognized stretches in the PCR amplicons were 3-4 %. Molar amounts of the specific sequences in the PCR amplicons per sample were, for the given DNA amounts, around 0.75 pmol. Such molar amounts corresponded to about 1 μg mL
-1 of the 40-mer
Tp53 ODN which gave a signal of comparable intensity (
Fig. 3). Hence, these results suggest an excellent performance of the technique, providing well-defined and reliable responses not only for the model synthetic ODNs, but also for the “real” samples of the PCR-amplified genomic DNA elements.
2.2.1 Monitoring of gene expression using DNA hybridization
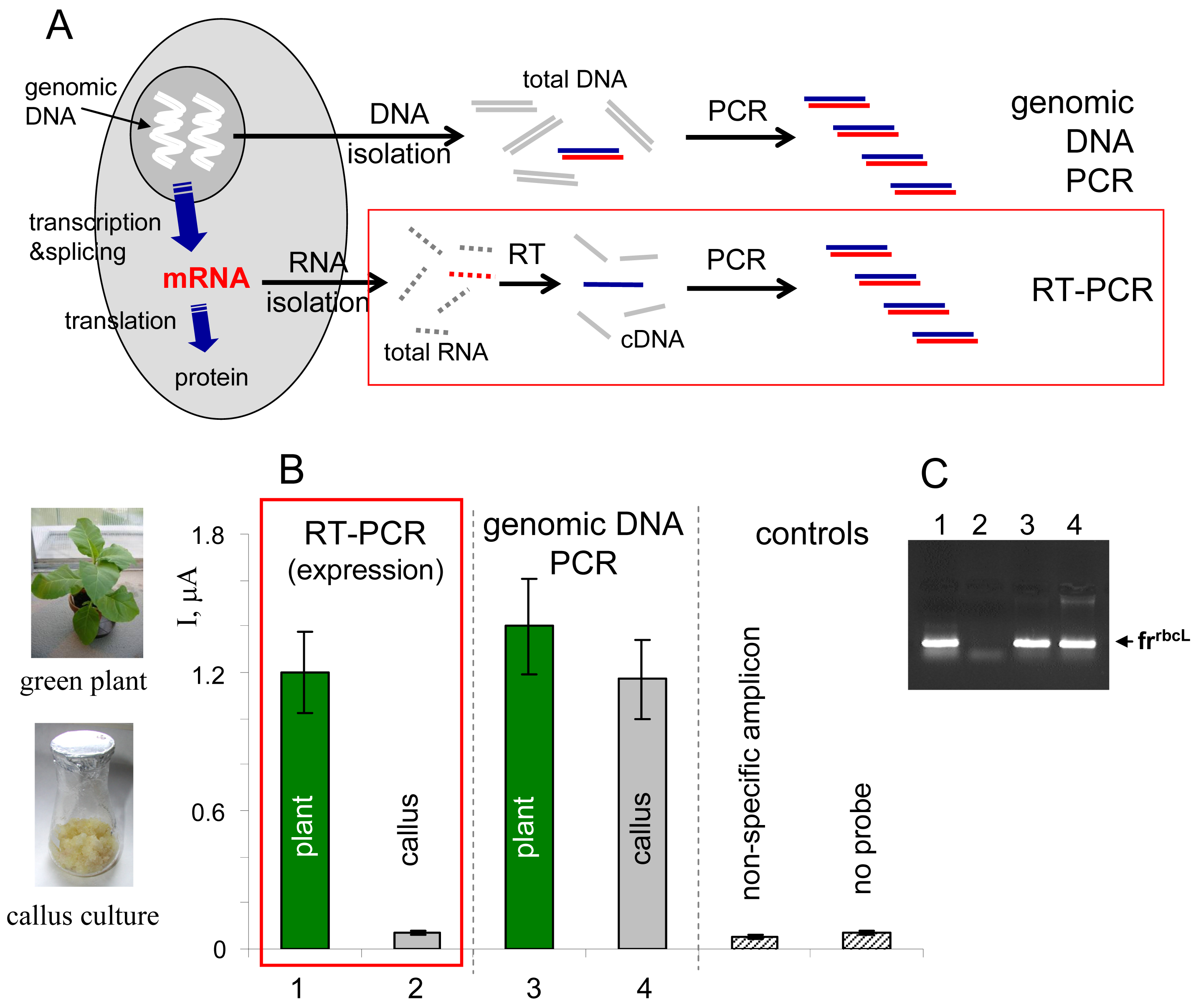
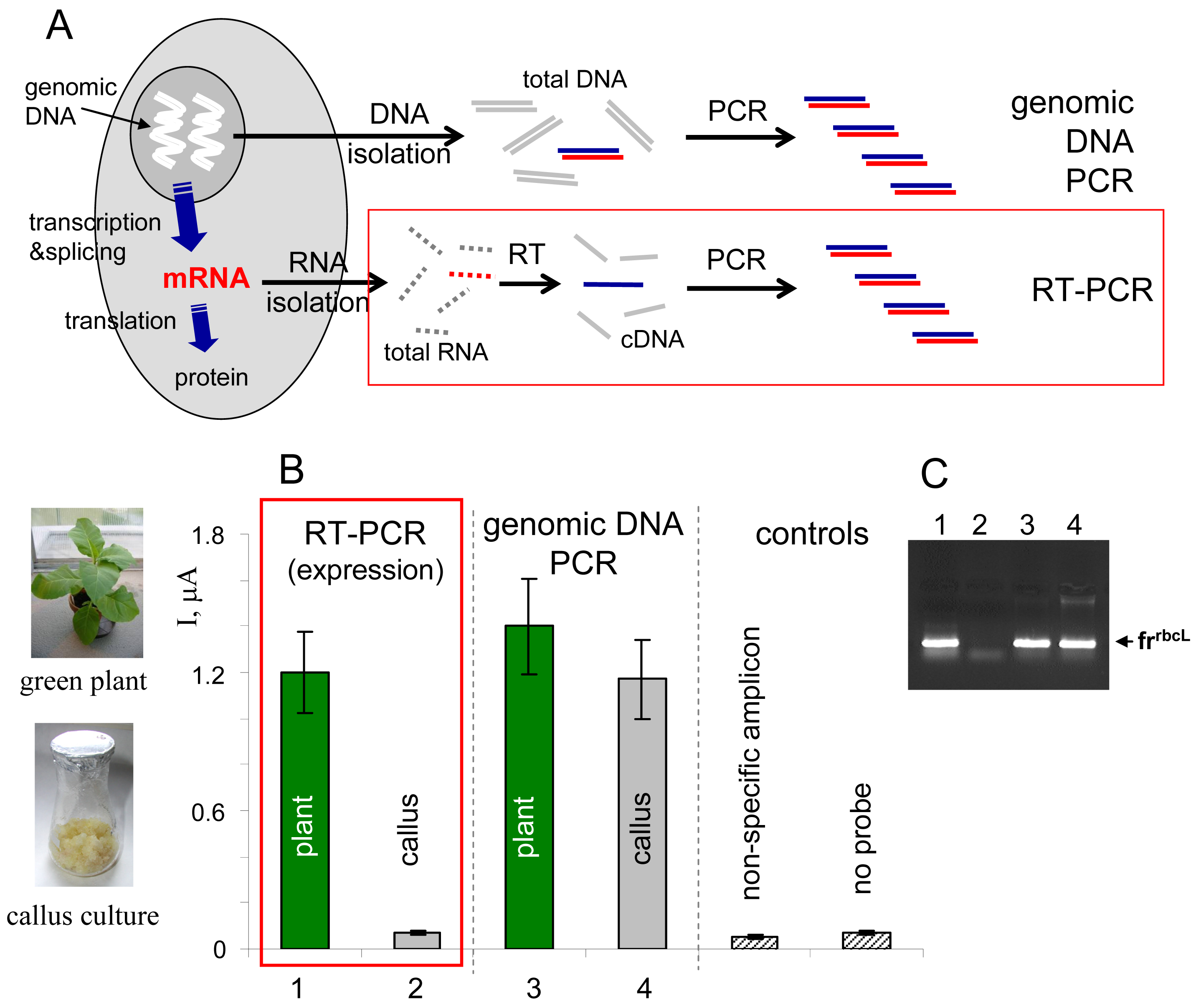
Gene expression is a process involving transcription of DNA sequence into RNA sequence, processing of the primary transcript into mRNA and translation of the mRNA sequence into amino acid sequence during proteosynthesis (
Fig. 4A). The gene expression can in principle be monitored at the RNA or protein levels [
1]. Specific mRNA can be detected, for example, using northern hybridization (total RNA isolated from biological material is separated by gel electrophoresis, blotted onto a membrane and hybridized with a gene-specific probe), or using an approach involving reverse transcription of RNA into cDNA, followed by PCR amplification of the cDNA using gene-specific primers (RT-PCR,
Fig. 4A) [
1,
32,
33]. RT-PCR has been widely applied in connection with various detection platforms, including simple agarose gel electrophoresis of the amplified cDNA fragments (
Fig. 4C), gene arrays employing cDNA hybridization with surface-confined capture probes, as well as real-time PCR techniques allowing precise qualitative analyses [
34,
35].
Here we used the electrochemical enzyme-linked DNA hybridization technique in connection with RT-PCR for monitoring of expression of a gene
rbcL [
36] in two types of plant tissues, green leaves and non-green callus cultures of tobacco (
Nicotiana tabacum). The
rbcL gene, encoding one of subunits of the enzyme Rubisco involved in photosynthesis, is known to be expressed in green parts of plants upon illumination, but not in the non-green plant cells [
36]. Total RNA was isolated from the tobacco tissues and reversely transcribed into cDNA using random nonamer primers, and the
frrbcL fragment was amplified using
rbcL-specific primers (
Table I). In parallel to this RT-PCR experiment, total genomic DNA was isolated from the plant material and used as a template for PCR amplification of the same
frrbcL fragment. All PCR products were applied as tDNAs in the electrochemical enzyme-linked DNA hybridization assay. Positive signal observed in the RT-PCR experiment for green tobacco leaves was in agreement with the expected active
rbcL expression in this tissue (
Fig. 4B). On the contrary, response obtained for the
rbcL non-expressing callus culture was at the level of negative controls. When the
frrbcL fragment was amplified from the total genomic DNA (“genomic DNA PCR” in
Fig. 4), positive signals were detected for both tobacco tissues, in agreement with presence of the gene in the plant genome regardless of the cell or tissue type (the sense of information gained from the latter experiment is “the gene is present”, while that obtained from the RT-PCR experiment means “the gene is active/inactive”). Control agarose gel electrophoresis (
Fig. 4C) confirmed presence of the
frrbcL amplicon in lanes corresponding to samples that gave positive responses in the electrochemical assay (
Fig. 4B).
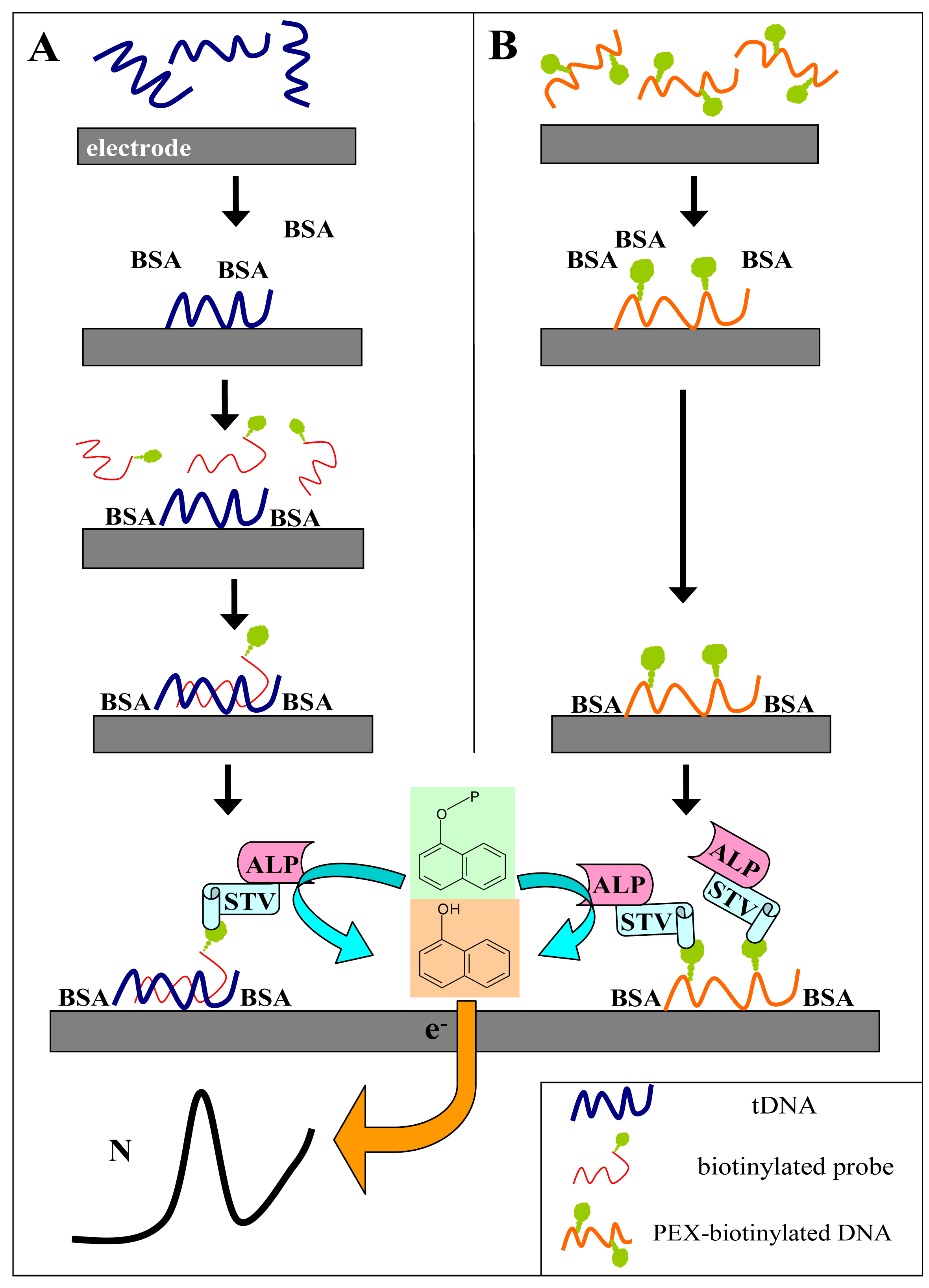
2.3 Primer extension-based DNA sensing
Sequence-specific DNA detection using various DNA labels need not necessarily involve DNA hybridization with labeled probes at surfaces. The markers, when available in the form of labeled dNTPs, can be introduced in specific DNA regions by DNA polymerases [
16,
21-
23,
25]. In this case, the sequence specificity is achieved through using a (unlabeled) probe which hybridizes with the tDNA of interest and serves as a primer for the labeled DNA synthesis (
Fig. 5A). When the primer extension (PEX) reaction mixture contains a biotin-labeled dNTP (such as dU
bioTP), the biotin tags are introduced into the synthesized DNA stretch. Incorporation of multiple tags per PEX product (i.e., per a primer molecule) offers a possibility of signal enhancement [
21]. The biotinylated PEX product can be detected using the electrochemical enzyme-linked assay depicted in
Figure 1B. The procedure is analogous to that used above for the DNA hybridization assays (
Fig. 1A) but incubation with the biotinylated probe is omitted.
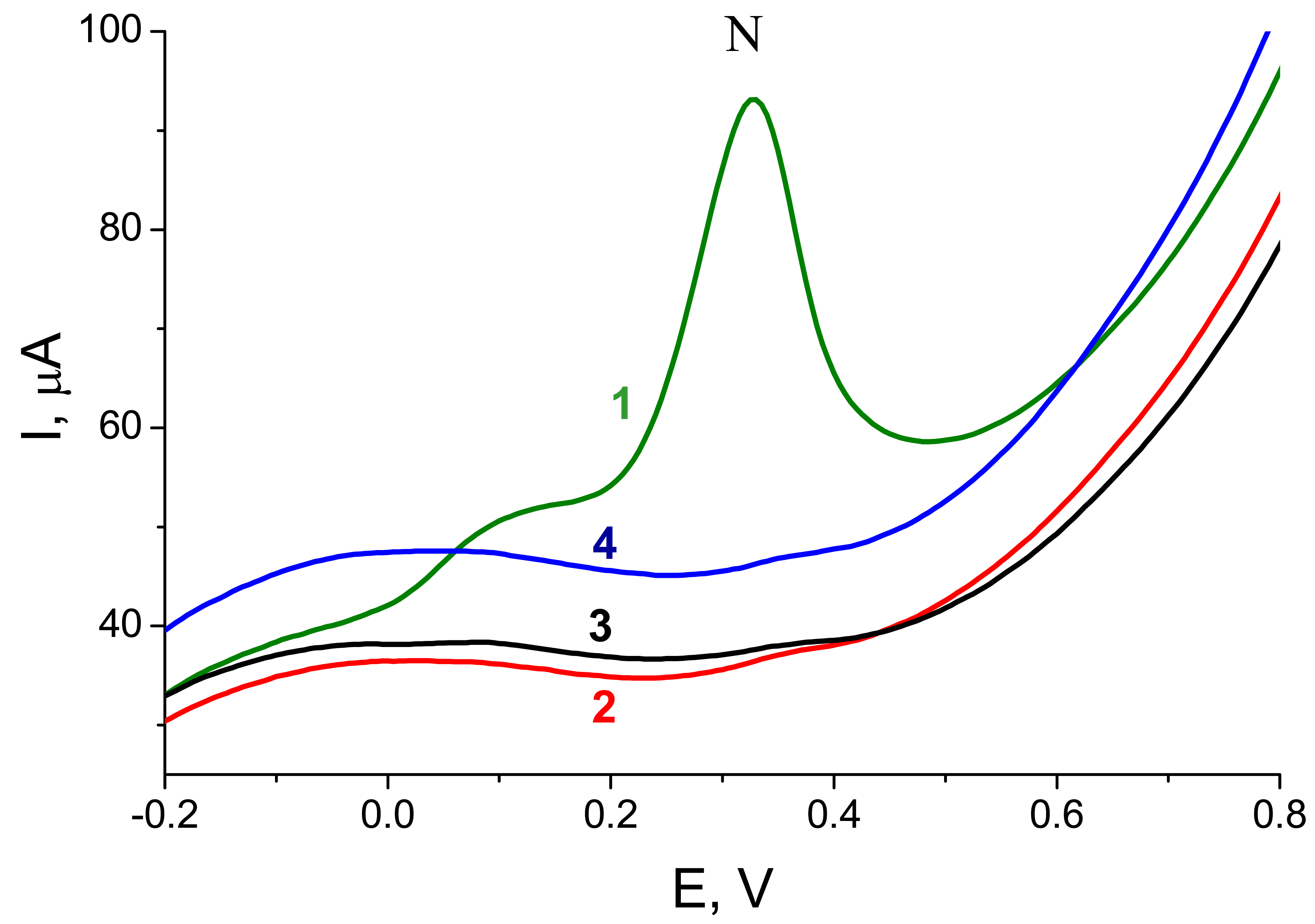
Figure 2C shows responses obtained via the procedure shown in
Fig. 1B for a biotinylated ODN (
Pp53) or for dU
bioTP. While the ODN applied at the electrode in 0.4 μM solution yielded a well defined peak N, practically no signal was observed for dU
bioTP concentration by 4 orders of magnitude higher (1 mM). This striking difference was in accord with different adsorbabilities of the two species at the electrode surface. As shown previously (reviewed in [
37,
38]), monomeric nucleic acid components, in contrast to oligonucleotides, polynucleotides and natural nucleic acids, are not adsorbed at electrode surfaces strongly enough to resist medium exchange. This difference makes it possible to analyze (strongly adsorbing) nucleic acids in mixtures with (weakly adsorbing) bases, nucleosides or nucleotides by means of adsorptive transfer stripping (
ex situ) voltammetry without any significant interference of the monomeric species. Here, the lack of signal of the dU
bioTP made it in principle possible to analyze PEX reaction mixtures using the electrochemical enzyme-linked assay without removal of the unreacted dNTPs.
2.3.1 PEX-based analysis of PCR products and monitoring of gene expression
The PEX-based technique was used to detect the
frp53 amplicon. The probe primer
primp53 was mixed with the PCR product, complete dNTP mix (involving dU
bioTP instead of dTTP) and a thermostable DNA polymerase. The mixture was subjected to a thermal cycle during which the PCR-amplified double-stranded DNA fragment was denatured, the probe primer hybridized with the template and the primer extension proceeded. The cycle was repeated ten times to enrich the sample for the biotin-labeled DNA. (It should be noted that the biotinylated dNTP can in principle be added to the PCR mixture, resulting in global modification of the amplicon molecules. Nevertheless, probing a specific sequence segment within the amplified DNA fragment (see
Table I) is advantageous since it makes the PCR-based assays less prone to producing false positives due to erroneous amplification of non-specific products.) Finally, the PEX product was applied at the SPCE and the detection procedure was conducted according to
Figure 1B. As shown in
Figure 5B, an intense signal was detected for the primer
primp53 and target template
frp53 (involving a sequence complementary to the primer, see
Table I). Signal arising from the PEX experiment was by an order of magnitude more intense than that arising from hybridization of the
frp53 amplicon with the
Pp53 probe at the SPCE surface (while the amounts of the
frp53 fragments used per experiment were comparable,
Fig. 5B). Enhancement of the signal intensity was in accord with incorporation of multiple biotin tags per probe primer hybridized with the target template; in the hybridization assay, only one biotin moiety per hybridization event was collected.
In next experiment, the RT-PCR products prepared for the tobacco tissues were analyzed by the PEX technique. Again, a strong signal was observed for
rbcL-expressing green leaves (
Fig. 6). For the non-green plant material, including the solid callus culture (
Fig. 4) as well as TBY-2 cell suspension culture [
39], only negligible background responses comparable to blank PEX mixture (with no target template added), were detected. Differentiation between the expressing and non-expressing tissues was in the PEX technique even better than in the hybridization assay (
Fig. 4) due to specific enhancement of the positive signal.
3. Experimental Section
3.1 Materials
Synthetic ODNs (see
Table I) were purchased from VBC Biotech, random nonamers used for reverse transcription experiments from Sigma. Plasmid pT77 bearing wild type
p53 cDNA insert [
40] (used as primary template for PCR amplification of the
frp53 fragment) was isolated from
E. coli cells using Qiagen Plasmid Purification Kit and linearized with
Eco RI restrictase (Takara). Total genomic DNA from the tobacco leaves, solid callus culture or the TBY-2 suspension culture [
39] was isolated as described ([
1] and references therein). Total RNA from the same plant material was isolated using RNeasy Plant Mini Kit (Qiagen), including treatment with RNase-free DNase I (DNase I Set, Qiagen).
Pfu DNA polymerase and Streptavidin alkaline phosphate conjugate were obtained from Promega, DyNAzyme™ II DNA Polymerase from Finnzymes (Finland), reverse transcriptase SuperScript II from Invitrogen, unmodified nucleoside triphosphates (dATP, dTTP, dCTP and dGTP) from Sigma, Biotin-16-dUTP (dU
bioTP) from Roche, and 1-naphtyl phosphate disodium salt from Sigma. Other chemicals were of analytical grade.
3.2 PCR and RT-PCR
Amplification of the frp53 fragment: 500 ng of the pT77 template was mixed with p53-for and p53-rev primers (0.5 μM each), Pfu DNA polymerase (3 U) and mix of standard dNTPs (125 μM each) in a total volume of 100 μl. The PCR involved 30 cycles (denaturation 94 °C/90 s, annealing 60 °C/120 s, polymerization 72 °C/180 s). The same procedure, using rbcL-for and rbcL-rev primers, was used for amplification of the frrbcL fragment from the total tobacco genomic DNA.
RT-PCR: Reverse transcription of the total tobacco RNA was performed according to manufacturer's instructions. PCR amplification of the rbcL reverse transcript was conducted as above using the rbcL-for and rbcL-rev primers.
For the DNA hybridization and PEX experiments, the PCR products were purified using QIAquick PCR Purification Kit (Qiagen).
3.3 Primer extension
0.7 μM primer (primp53 or primrbcL) was mixed with 300 ng of the respective template, 1 U of DyNAzyme II DNA polymerase, and a mix of dATP, dCTP, dGTP and dUbioTP (125 μM each) in a total volume of 30 μl. The reaction was conducted in 10 thermal cycles (94 °C/90 s, 60 °C/120 s, 72 °C/180 s).
3.4 Preparation of the screen-printed carbon electrodes
The SPCEs were prepared by coating of the carbon ink (Gwent C 10903P14, UK) through a stencil, using squeegee of the printing device (SP-200, MPM), onto an inert laser pre-etched ceramic support (Coors Ceramic). The resulting plates were dried at 60 °C for 1h. Active surface area of the SPCE sensors was delimited by coating the adjacent part of the SPCE stripe by nail-varnish.
3.5 Electrochemical enzyme-linked assays
3.5.1 Hybridization at the SPCE surface
Target DNAs were adsorbed at the active SPCE surface from 6-μL drops of solutions containing either 0.3 M NaCl, 10 mM Tris-HCl, pH 7.5 (buffer H; used for adsorption of synthetic ODNs) or 20 mM NaOH + 180 mM NaCl (used for the PCR-amplified DNA fragments). Accumulation time was always 120 s. Unoccupied SPCE surface was then blocked by incubation of the electrode in strirred solution of 2 % bovine serum albumin (BSA) in PBS (0.28 M NaCl, 5.5 mM KCl, 24 mM NaHPO
4, 3.5 mM KH
2PO
4, pH 7.4) for 120 s. After rinsing by PBS, 6 μL of the probe solution (5 μg mL
-1) in buffer H was applied at the electrode surface for 120 s. Then the electrode was rinsed by PBS and 6-μL of SALP solution (100-times diluted stock in PBS containing 2 % BSA) was applied at the SPCE for 120 s. The modified electrode was then washed in PBS containing 0.05 % of Tween 20 for 30 s and then in PBS for another 30 s. Finally, the electrode was placed into voltammetric cell containing 3 mL of background electrolyte (0.5 M K
2CO
3 and 0.5 M NaHCO
3, pH 9.5) containing 5 mM 1-naphtyl phosphate. Enzymatically produced 1-naphtol was detected after a short incubation period using the voltammetric peak N (
Fig. 2). Each experiment involved two voltammetric scans with the same sensor.The first was recorded after 20 s of the electrode incubation in the substrate solution and the other, recorded after another 60 s, was evaluated as the analytical signal [
13].
3.5.2 Analysis of biotinylated PEX products
The experiments were carried out as above but hybridization with the biotinylated probe was omitted. Briefly, the biotinylated DNA was adsorbed at the SPCE, followed by surface blocking by BSA, binding of SALP and production of the 1-naphthol indicator (see
Fig. 1 for schemes of the procedures).
3.6 Voltammetric measurements
All measurements were performed with a CHI440 Electrochemical Workstation (CH Instruments, Inc., USA) connected to a three-electrode system (with the SPCE as working, Ag/AgCl/3M KCl as reference and platinum wire as counter electrode). The electroactive indicator 1-naphtol was detected using linear sweep voltammetry (LSV) in 0.5 M K2CO3 and 0.5 M NaHCO3, pH 9.5, with initial potential -0.5 V, end potential +0.9 V, scan rate 1 V s-1, potential step 5 mV.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}