The Role of Antioxidation and Immunomodulation in Postnatal Multipotent Stem Cell-Mediated Cardiac Repair


{kind=link}
Abstract
:1. Introduction
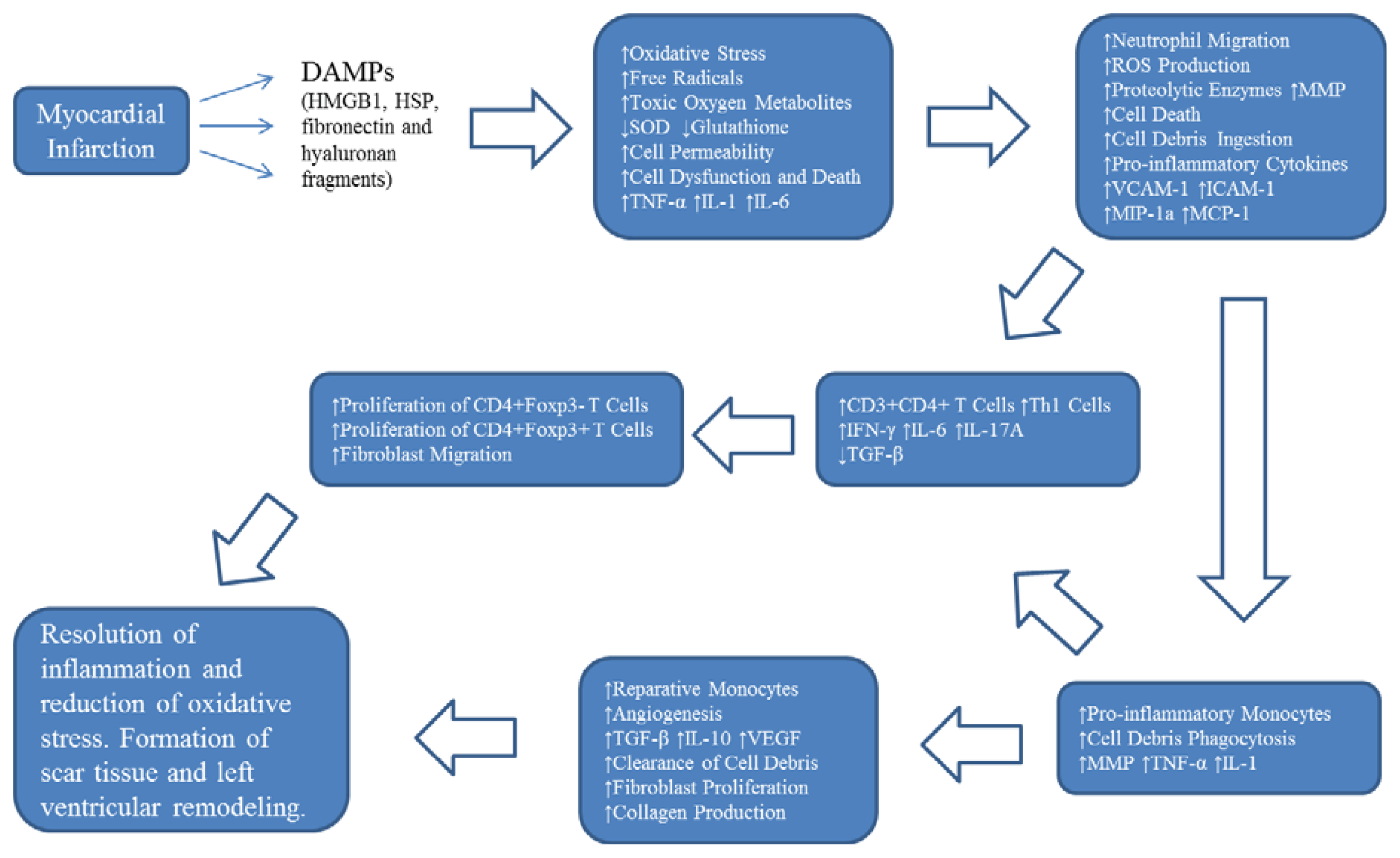
2. Oxidative Stress and Inflammation Following Myocardial Infarction
3. Cellular Antioxidant Level Represents a Major Determinant in the Cardiac Regenerative Capacity of Stem Cells
4. Mesenchymal Stem Cells as Immunomodulators in Cardiac Repair
5. Human Blood Vessel-Derived Stem Cells for Cardiac Repair and Regeneration
6. The Role of Human Pericytes in Antioxidation and Immunomodulation
7. Conclusions
Acknowledgments
Conflict of Interest
References
- Alwan, A. Global Status Report on Noncommunicable Diseases 2010, 1st ed; World Health Organization: Geneva, Switzerland, 2011; pp. 9–11. [Google Scholar]
- Mathers, C.D.; Loncar, D. Projections of global mortality and burden of disease from 2002 to 2030. PLoS Med 2006, 3, e442. [Google Scholar]
- Arslan, F.; de Kleijn, D.P.; Pasterkamp, G. Innate immune signaling in cardiac ischemia. Nat. Rev. Cardiol 2011, 8, 292–300. [Google Scholar]
- Coggins, M.; Rosenzweig, A. The fire within: Cardiac inflammatory signaling in health and disease. Circ. Res 2012, 110, 116–125. [Google Scholar]
- Frangogiannis, N.G. Regulation of the inflammatory response in cardiac repair. Circ. Res 2012, 110, 159–173. [Google Scholar]
- Swirski, F.K.; Nahrendorf, M. Leukocyte behavior in atherosclerosis, myocardial infarction, and heart failure. Science 2013, 339, 161–166. [Google Scholar]
- Ptaszek, L.M.; Mansour, M.; Ruskin, J.N.; Chien, K.R. Towards regenerative therapy for cardiac disease. Lancet 2012, 379, 933–942. [Google Scholar]
- Choudry, F.A.; Mathur, A. Stem cell therapy in cardiology. Regener. Med 2011, 6, 17–23. [Google Scholar]
- Anversa, P.; Kajstura, J.; Rota, M.; Leri, A. Regenerating new heart with stem cells. J. Clin. Invest 2013, 123, 62–70. [Google Scholar]
- Matzinger, P. The danger model: A renewed sense of self. Sci. Signal 2002, 296, 301–305. [Google Scholar]
- Sha, Y.; Zmijewski, J.; Xu, Z.; Abraham, E. HMGB1 develops enhanced proinflammatory activity by binding to cytokines. J. Immunol 2008, 180, 2531–2537. [Google Scholar]
- Yang, H.; Hreggvidsdottir, H.S.; Palmblad, K.; Wang, H.; Ochani, M.; Li, J.; Lu, B.; Chavan, S.; Rosas-Ballina, M.; Al-Abed, Y. A critical cysteine is required for HMGB1 binding to Toll-like receptor 4 and activation of macrophage cytokine release. Proc. Natl. Acad. Sci. USA 2010, 107, 11942–11947. [Google Scholar]
- Tsung, A.; Sahai, R.; Tanaka, H.; Nakao, A.; Fink, M.P.; Lotze, M.T.; Yang, H.; Li, J.; Tracey, K.J.; Geller, D.A. The nuclear factor HMGB1 mediates hepatic injury after murine liver ischemia-reperfusion. J. Exp. Med 2005, 201, 1135–1143. [Google Scholar]
- Schiraldi, M.; Raucci, A.; Muñoz, L.M.; Livoti, E.; Celona, B.; Venereau, E.; Apuzzo, T.; de Marchis, F.; Pedotti, M.; Bachi, A. HMGB1 promotes recruitment of inflammatory cells to damaged tissues by forming a complex with CXCL12 and signaling via CXCR4. J. Exp. Med 2012, 209, 551–563. [Google Scholar]
- Kim, S.; Stice, J.P.; Chen, L.; Jung, J.S.; Gupta, S.; Wang, Y.; Baumgarten, G.; Trial, J.; Knowlton, A.A. Extracellular heat shock protein 60, cardiac myocytes, and apoptosis. Circ. Res 2009, 105, 1186–1195. [Google Scholar]
- Li, Y.; Si, R.; Feng, Y.; Chen, H.H.; Zou, L.; Wang, E.; Zhang, M.; Warren, H.S.; Sosnovik, D.E.; Chao, W. Myocardial ischemia activates an injurious innate immune signaling via cardiac heat shock protein 60 and Toll-like receptor 4. J. Biol. Chem 2011, 286, 31308–31319. [Google Scholar]
- Taylor, K.R.; Yamasaki, K.; Radek, K.A.; Nardo, A.D.; Goodarzi, H.; Golenbock, D.; Beutler, B.; Gallo, R.L. Recognition of hyaluronan released in sterile injury involves a unique receptor complex dependent on Toll-like receptor 4, CD44, and MD-2. Sci. Signal 2007, 282, 18265–18275. [Google Scholar]
- Arslan, F.; Smeets, M.B.; Vis, P.W.R.; Karper, J.C.; Quax, P.H.; Bongartz, L.G.; Peters, J.H.; Hoefer, I.E.; Doevendans, P.A.; Pasterkamp, G. Lack of fibronectin-EDA promotes survival and prevents adverse remodeling and heart function deterioration after myocardial infarctionnovelty and significance. Circ. Res 2011, 108, 582–592. [Google Scholar]
- Medzhitov, R.; Preston-Hurlburt, P.; Janeway, C.A. A human homologue of the drosophila Toll protein signals activation of adaptive immunity. Nature 1997, 388, 394–397. [Google Scholar]
- Oyama, J.; Blais, C., Jr; Liu, X.; Pu, M.; Kobzik, L.; Kelly, R.A.; Bourcier, T. Reduced myocardial ischemia-reperfusion injury in Toll-like receptor 4-deficient mice. Circulation 2004, 109, 784–789. [Google Scholar]
- Riad, A.; Jäger, S.; Sobirey, M.; Escher, F.; Yaulema-Riss, A.; Westermann, D.; Karatas, A.; Heimesaat, M.M.; Bereswill, S.; Dragun, D. Toll-like receptor-4 modulates survival by induction of left ventricular remodeling after myocardial infarction in mice. J. Immunol 2008, 180, 6954–6961. [Google Scholar]
- Shishido, T.; Nozaki, N.; Yamaguchi, S.; Shibata, Y.; Nitobe, J.; Miyamoto, T.; Takahashi, H.; Arimoto, T.; Maeda, K.; Yamakawa, M. Toll-like receptor-2 modulates ventricular remodeling after myocardial infarction. Circulation 2003, 108, 2905–2910. [Google Scholar]
- Akira, S.; Takeda, K. Toll-like receptor signalling. Nat. Rev. Immunol 2004, 4, 499–511. [Google Scholar]
- Chen, C.; Kono, H.; Golenbock, D.; Reed, G.; Akira, S.; Rock, K.L. Identification of a key pathway required for the sterile inflammatory response triggered by dying cells. Nat. Med 2007, 13, 851–856. [Google Scholar]
- Diepenhorst, G.M.; van Gulik, T.M.; Hack, C.E. Complement-mediated ischemia-reperfusion injury: Lessons learned from animal and clinical studies. Annu. Surg 2009, 249, 889–899. [Google Scholar]
- Banz, Y.; Rieben, R. Role of complement and perspectives for intervention in ischemia-reperfusion damage. Annu. Med 2012, 44, 205–217. [Google Scholar]
- Zhang, H.; Qin, G.; Liang, G.; Li, J.; Barrington, R.A.; Liu, D. C5aR-mediated myocardial ischemia/reperfusion injury. Biochem. Biophys. Res. Commun 2007, 357, 446–452. [Google Scholar]
- Van der Pals, J.; Koul, S.; Andersson, P.; Götberg, M.; Ubachs, J.; Kanski, M.; Arheden, H.; Olivecrona, G.; Larsson, B.; Erlinge, D. Treatment with the C5a receptor antagonist ADC-1004 reduces myocardial infarction in a porcine ischemia-reperfusion model. BMC Cardiovasc. Disord 2010, 10, 45. [Google Scholar]
- Ferrari, R.; Guardigli, G.; Mele, D.; Percoco, G.; Ceconi, C.; Curello, S. Oxidative stress during myocardial ischaemia and heart failure. Curr. Pharm. Des 2004, 10, 1699–1711. [Google Scholar]
- Elahi, M.M.; Kong, Y.X.; Matata, B.M. Oxidative stress as a mediator of cardiovascular disease. Oxid. Med. Cell. Longevity 2009, 2, 259–269. [Google Scholar]
- Tsutsui, H.; Kinugawa, S.; Matsushima, S. Oxidative stress and heart failure. Am. J. Physiol 2011, 301, H2181–H2190. [Google Scholar]
- Palazzo, A.J.; Jones, S.P.; Anderson, D.C.; Granger, D.N.; Lefer, D.J. Coronary endothelial P-selectin in pathogenesis of myocardial ischemia-reperfusion injury. Am. J. Physiol 1998, 275, H1865–H1872. [Google Scholar]
- Ivetic, A. Signals regulating l-selectin-dependent leucocyte adhesion and transmigration. Int. J. Biochem. Cell Biol 2013, 45, 550–555. [Google Scholar]
- Sellak, H.; Franzini, E.; Hakim, J.; Pasquier, C. Reactive oxygen species rapidly increase endothelial ICAM-1 ability to bind neutrophils without detectable upregulation. Blood 1994, 83, 2669–2677. [Google Scholar]
- Briaud, S.A.; Ding, Z.; Michael, L.H.; Entman, M.L.; Daniel, S.; Ballantyne, C.M. Leukocyte trafficking and myocardial reperfusion injury in ICAM-1/P-selectin-knockout mice. Am. J. Physiol 2001, 280, H60–H67. [Google Scholar]
- Williams, M.R.; Azcutia, V.; Newton, G.; Alcaide, P.; Luscinskas, F.W. Emerging mechanisms of neutrophil recruitment across endothelium. Trends Immunol 2011, 32, 461–469. [Google Scholar]
- Entman, M.L.; Youker, K.; Shoji, T.; Kukielka, G.; Shappell, S.B.; Taylor, A.A.; Smith, C. Neutrophil induced oxidative injury of cardiac myocytes. A compartmented system requiring CD11b/CD18-ICAM-1 adherence. J. Clin. Invest 1992, 90, 1335–1345. [Google Scholar]
- Soehnlein, O.; Weber, C.; Lindbom, L. Neutrophil granule proteins tune monocytic cell function. Trends Immunol 2009, 30, 538–546. [Google Scholar]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med 2007, 204, 3037–3047. [Google Scholar]
- Leuschner, F.; Rauch, P.J.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med 2012, 209, 123–137. [Google Scholar]
- Gregory, C.D.; Devitt, A. The macrophage and the apoptotic cell: An innate immune interaction viewed simplistically? Immunology 2004, 113, 1–14. [Google Scholar]
- Martinez, F.O.; Helming, L.; Gordon, S. Alternative activation of macrophages: An immunologic functional perspective. Annu. Rev. Immunol 2009, 27, 451–483. [Google Scholar]
- Geissmann, F.; Manz, M.G.; Jung, S.; Sieweke, M.H.; Merad, M.; Ley, K. Development of monocytes, macrophages, and dendritic cells. Science 2010, 327, 656–661. [Google Scholar]
- Anzai, A.; Anzai, T.; Nagai, S.; Maekawa, Y.; Naito, K.; Kaneko, H.; Sugano, Y.; Takahashi, T.; Abe, H.; Mochizuki, S. Regulatory role of dendritic cells in postinfarction healing and left ventricular remodeling clinical perspective. Circulation 2012, 125, 1234–1245. [Google Scholar]
- Zhu, J.; Paul, W.E. Peripheral CD4+ T cell differentiation regulated by networks of cytokines and transcription factors. Immunol. Rev 2010, 238, 247–262. [Google Scholar]
- Locksley, R.M. Nine lives: Plasticity among T helper cell subsets. J. Exp. Med 2009, 206, 1643–1646. [Google Scholar]
- Zhou, L.; Chong, M.; Littman, D.R. Plasticity of CD4 T cell lineage differentiation. Immunity 2009, 30, 646–655. [Google Scholar]
- Zhang, N.; Bevan, M.J. CD8+ T cells: Foot soldiers of the immune system. Immunity 2011, 35, 161–168. [Google Scholar]
- Maisel, A.; Cesario, D.; Baird, S.; Rehman, J.; Haghighi, P.; Carter, S. Experimental autoimmune myocarditis produced by adoptive transfer of splenocytes after myocardial infarction. Circ. Res 1998, 82, 458–463. [Google Scholar]
- Varda-Bloom, N.; Leor, J.; Ohad, D.G.; Hasin, Y.; Amar, M.; Fixler, R.; Battler, A.; Eldar, M.; Hasin, D. Cytotoxic t lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J. Mol. Cell. Cardiol 2000, 32, 2141–2149. [Google Scholar]
- Ávalos, A.M.; Apablaza, F.A.; Quiroz, M.; Toledo, V.; Peña, J.P.; Michea, L.; Irarrazabal, C.E.; Carrión, F.A.; Figueroa, F.E. Il-17a levels increase in the infarcted region of the left ventricle in a rat model of myocardial infarction. Biol. Res 2012, 45, 193–200. [Google Scholar]
- Yan, X.; Shichita, T.; Katsumata, Y.; Matsuhashi, T.; Ito, H.; Ito, K.; Anzai, A.; Endo, J.; Tamura, Y.; Kimura, K. Deleterious effect of the IL-23/IL-17a axis and γδt cells on left ventricular remodeling after myocardial infarction. J. Am. Heart Assoc 2012, 1, e004408. [Google Scholar]
- Hofmann, U.; Beyersdorf, N.; Weirather, J.; Podolskaya, A.; Bauersachs, J.; Ertl, G.; Kerkau, T.; Frantz, S. Activation of CD4 T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice clinical perspective. Circulation 2012, 125, 1652–1663. [Google Scholar]
- Dobaczewski, M.; Xia, Y.; Bujak, M.; Gonzalez-Quesada, C.; Frangogiannis, N.G. CCR5 signaling suppresses inflammation and reduces adverse remodeling of the infarcted heart, mediating recruitment of regulatory t cells. Am. J. Pathol 2010, 176, 2177–2187. [Google Scholar]
- Cheng, X.; Liao, Y.; Ge, H.; Li, B.; Zhang, J.; Yuan, J.; Wang, M.; Liu, Y.; Guo, Z.; Chen, J. TH1/TH2 functional imbalance after acute myocardial infarction: coronary arterial inflammation or myocardial inflammation. J. Clin. Immunol 2005, 25, 246–253. [Google Scholar]
- Zhao, Z.; Wu, Y.; Cheng, M.; Ji, Y.; Yang, X.; Liu, P.; Jia, S.; Yuan, Z. Activation of Th17/Th1 and Th1, but not Th17, is associated with the acute cardiac event in patients with acute coronary syndrome. Atherosclerosis 2011, 217, 518–524. [Google Scholar]
- Van den Borne, S.W.M.; Diez, J.; Blankesteijn, W.M.; Verjans, J.; Hofstra, L.; Narula, J. Myocardial remodeling after infarction: The role of myofibroblasts. Nat. Rev. Cardiol 2009, 7, 30–37. [Google Scholar]
- Hausenloy, D.J.; Yellon, D.M. Myocardial ischemia-reperfusion injury: A neglected therapeutic target. J. Clin. Invest 2013, 123, 92–100. [Google Scholar]
- Segers, V.F.; Lee, R.T. Stem-cell therapy for cardiac disease. Nature 2008, 451, 937–942. [Google Scholar]
- Haider, H.K.; Ashraf, M. Preconditioning and stem cell survival. J. Cardiovas. Transl. Res 2010, 3, 89–102. [Google Scholar]
- Deutsch, M.; Sturzu, A.; Wu, S.M. At a crossroad cell therapy for cardiac repair. Circ. Res 2013, 112, 884–890. [Google Scholar]
- Oshima, H.; Payne, T.R.; Urish, K.L.; Sakai, T.; Ling, Y.; Gharaibeh, B.; Tobita, K.; Keller, B.B.; Cummins, J.H.; Huard, J. Differential myocardial infarct repair with muscle stem cells compared to myoblasts. Mol. Therapy 2005, 12, 1130–1141. [Google Scholar]
- Okada, M.; Payne, T.R.; Zheng, B.; Oshima, H.; Momoi, N.; Tobita, K.; Keller, B.B.; Phillippi, J.A.; Péault, B.; Huard, J. Myogenic endothelial cells purified from human skeletal muscle improve cardiac function after transplantation into infarcted myocardium. J. Am. Coll. Cardiol 2008, 52, 1869–1880. [Google Scholar]
- Urish, K.L.; Vella, J.B.; Okada, M.; Deasy, B.M.; Tobita, K.; Keller, B.B.; Cao, B.; Piganelli, J.D.; Huard, J. Antioxidant levels represent a major determinant in the regenerative capacity of muscle stem cells. Mol. Biol. Cell 2009, 20, 509–520. [Google Scholar]
- Drowley, L.; Okada, M.; Beckman, S.; Vella, J.; Keller, B.; Tobita, K.; Huard, J. Cellular antioxidant levels influence muscle stem cell therapy. Mol. Therapy 2010, 18, 1865–1873. [Google Scholar]
- Mangi, A.A.; Noiseux, N.; Kong, D.; He, H.; Rezvani, M.; Ingwall, J.S.; Dzau, V.J. Mesenchymal stem cells modified with AKT prevent remodeling and restore performance of infarcted hearts. Nat. Med 2003, 9, 1195–1201. [Google Scholar]
- Jiang, S.; Haider, H.K.; Idris, N.M.; Salim, A.; Ashraf, M. Supportive interaction between cell survival signaling and angiocompetent factors enhances donor cell survival and promotes angiomyogenesis for cardiac repair. Circ. Res 2006, 99, 776–784. [Google Scholar]
- Hassan, F.; Meduru, S.; Taguchi, K.; Kuppusamy, M.L.; Mostafa, M.; Kuppusamy, P.; Khan, M. Carvedilol enhances mesenchymal stem cell therapy for myocardial infarction via inhibition of caspase-3 expression. J. Pharmacol. Exp. Ther 2012, 343, 62–71. [Google Scholar]
- DeSantiago, J.; Bare, D.J.; Banach, K. Ischemia-reperfusion injury protection by mesenchymal stem cell derived antioxidant capacity. Stem Cells Dev. 2013. [Google Scholar] [CrossRef]
- Pittenger, M.F.; Mackay, A.M.; Beck, S.C.; Jaiswal, R.K.; Douglas, R.; Mosca, J.D.; Moorman, M.A.; Simonetti, D.W.; Craig, S.; Marshak, D.R. Multilineage potential of adult human mesenchymal stem cells. Science 1999, 284, 143–147. [Google Scholar]
- Toma, C.; Pittenger, M.F.; Cahill, K.S.; Byrne, B.J.; Kessler, P.D. Human mesenchymal stem cells differentiate to a cardiomyocyte phenotype in the adult Murine heart. Circulation 2002, 105, 93–98. [Google Scholar]
- Jiang, Y.; Jahagirdar, B.N.; Reinhardt, R.L.; Schwartz, R.E.; Keene, C.D.; Ortiz-Gonzalez, X.R.; Reyes, M.; Lenvik, T.; Lund, T.; Blackstad, M. Pluripotency of mesenchymal stem cells derived from adult marrow. Nature 2002, 418, 41–49. [Google Scholar] [Green Version]
- Amado, L.C.; Saliaris, A.P.; Schuleri, K.H.; John, M.S.; Xie, J.; Cattaneo, S.; Durand, D.J.; Fitton, T.; Kuang, J.Q.; Stewart, G. Cardiac repair with intramyocardial injection of allogeneic mesenchymal stem cells after myocardial infarction. Proc. Natl. Acad. Sci. USA 2005, 102, 11474–11479. [Google Scholar]
- Miyahara, Y.; Nagaya, N.; Kataoka, M.; Yanagawa, B.; Tanaka, K.; Hao, H.; Ishino, K.; Ishida, H.; Shimizu, T.; Kangawa, K. Monolayered mesenchymal stem cells repair scarred myocardium after myocardial infarction. Nat. Med 2006, 12, 459–465. [Google Scholar]
- Gebler, A.; Zabel, O.; Seliger, B. The immunomodulatory capacity of mesenchymal stem cells. Trends Mol. Med 2012, 18, 128–134. [Google Scholar]
- Huang, X.; Sun, Z.; Miyagi, Y.; Kinkaid, H.M.; Zhang, L.; Weisel, R.D.; Li, R. Differentiation of allogeneic mesenchymal stem cells induces immunogenicity and limits their long-term benefits for myocardial repairclinical perspective. Circulation 2010, 122, 2419–2429. [Google Scholar]
- Duffy, M.M.; Pindjakova, J.; Hanley, S.A.; McCarthy, C.; Weidhofer, G.A.; Sweeney, E.M.; English, K.; Shaw, G.; Murphy, J.M.; Barry, F.P. Mesenchymal stem cell inhibition of T-helper 17 cell-differentiation is triggered by cell-cell contact and mediated by prostaglandin E2 via the ep4 receptor. Eur. J. Immunol 2011, 41, 2840–2851. [Google Scholar]
- Shi, Y.; Hu, G.; Su, J.; Li, W.; Chen, Q.; Shou, P.; Xu, C.; Chen, X.; Huang, Y.; Zhu, Z. Mesenchymal stem cells: A new strategy for immunosuppression and tissue repair. Cell Res 2010, 20, 510–518. [Google Scholar]
- DelaRosa, O.; Lombardo, E.; Beraza, A.; Mancheño-Corvo, P.; Ramirez, C.; Menta, R.; Rico, L.; Camarillo, E.; García, L.; Abad, J.L. Requirement of IFN-γ–mediated indoleamine 2,3-dioxygenase expression in the modulation of lymphocyte proliferation by human adipose-derived stem cells. Tissue Eng, Part A 2009, 15, 2795–2806. [Google Scholar]
- Chabannes, D.; Hill, M.; Merieau, E.; Rossignol, J.; Brion, R.; Soulillou, J.P.; Anegon, I.; Cuturi, M.C. A role for heme oxygenase-1 in the immunosuppressive effect of adult rat and human mesenchymal stem cells. Blood 2007, 110, 3691–3694. [Google Scholar]
- Uccelli, A.; Moretta, L.; Pistoia, V. Mesenchymal stem cells in health and disease. Nat. Rev. Immunol 2008, 8, 726–736. [Google Scholar]
- Bernardo, M.E.; Fibbe, W.E. Safety and efficacy of mesenchymal stromal cell therapy in autoimmune disorders. Annu. N.Y. Acad. Sci 2012, 1266, 107–117. [Google Scholar]
- Williams, A.R.; Hare, J.M. Mesenchymal stem cells biology, pathophysiology, translational findings, and therapeutic implications for cardiac disease. Circ. Res 2011, 109, 923–940. [Google Scholar]
- Ranganath, S.H.; Levy, O.; Inamdar, M.S.; Karp, J.M. Harnessing the mesenchymal stem cell secretome for the treatment of cardiovascular disease. Cell 2012, 10, 244–258. [Google Scholar]
- Cassatella, M.A.; Mosna, F.; Micheletti, A.; Lisi, V.; Tamassia, N.; Cont, C.; Calzetti, F.; Pelletier, M.; Pizzolo, G.; Krampera, M. Toll-like receptor-3-activated human mesenchymal stromal cells significantly prolong the survival and function of neutrophils. Stem Cells 2011, 29, 1001–1011. [Google Scholar]
- Raffaghello, L.; Bianchi, G.; Bertolotto, M.; Montecucco, F.; Dallegri, F.; Ottonello, L.; Pistoia, V. Human mesenchymal stem cells inhibit neutrophil apoptosis: A model for neutrophil preservation in the bone marrow niche. Stem Cells 2008, 26, 151–162. [Google Scholar]
- Henning, R.J.; Shariff, M.; Eadula, U.; Alvarado, F.; Vasko, M.; Sanberg, P.R.; Sanberg, C.D.; Delostia, V. Human cord blood mononuclear cells decrease cytokines and inflammatory cells in acute myocardial infarction. Stem Cells Dev 2008, 17, 1207–1220. [Google Scholar]
- Dayan, V.; Yannarelli, G.; Billia, F.; Filomeno, P.; Wang, X.; Davies, J.E.; Keating, A. Mesenchymal stromal cells mediate a switch to alternatively activated monocytes/macrophages after acute myocardial infarction. Basic Res. Cardiol 2011, 106, 1299–1310. [Google Scholar]
- Maggini, J.; Mirkin, G.; Bognanni, I.; Holmberg, J.; Piazzón, I.M.; Nepomnaschy, I.; Costa, H.; Cañones, C.; Raiden, S.; Vermeulen, M. Mouse bone marrow-derived mesenchymal stromal cells turn activated macrophages into a regulatory-like profile. PLoS One 2010, 5, e9252. [Google Scholar]
- Adutler-Lieber, S.; Ben-Mordechai, T.; Naftali-Shani, N.; Asher, E.; Loberman, D.; Raanani, E.; Leor, J. Human macrophage regulation via interaction with cardiac adipose tissue-derived mesenchymal stromal cells. J. Cardiovasc. Pharmacol. Ther 2013, 18, 78–86. [Google Scholar]
- Kim, J.; Hematti, P. Mesenchymal stem cell-educated macrophages: A novel type of alternatively activated macrophages. Exp. Hematol 2009, 37, 1445–1453. [Google Scholar]
- François, M.; Romieu-Mourez, R.; Li, M.; Galipeau, J. Human MSC suppression correlates with cytokine induction of indoleamine 2, 3-dioxygenase and bystander m2 macrophage differentiation. Mol. Ther 2011, 20, 187–195. [Google Scholar]
- Burchfield, J.S.; Iwasaki, M.; Koyanagi, M.; Urbich, C.; Rosenthal, N.; Zeiher, A.M.; Dimmeler, S. Interleukin-10 from transplanted bone marrow mononuclear cells contributes to cardiac protection after myocardial infarction. Circ. Res 2008, 103, 203–211. [Google Scholar]
- English, K.; Ryan, J.; Tobin, L.; Murphy, M.; Barry, F.; Mahon, B. Cell contact, prostaglandin E2 and transforming growth factor beta 1 play non-redundant roles in human mesenchymal stem cell induction of CD4 CD25 highforkhead box P3 regulatory T cells. Clin. Exp. Immunol 2009, 156, 149–160. [Google Scholar]
- Del Papa, B.; Sportoletti, P.; Cecchini, D.; Rosati, E.; Balucani, C.; Baldoni, S.; Fettucciari, K.; Marconi, P.; Martelli, M.F.; Falzetti, F. Notch1 modulates mesenchymal stem cells mediated regulatory T-cell induction. Eur. J. Immunol 2013, 43, 182–187. [Google Scholar]
- Di Ianni, M.; del Papa, B.; de Ioanni, M.; Moretti, L.; Bonifacio, E.; Cecchini, D.; Sportoletti, P.; Falzetti, F.; Tabilio, A. Mesenchymal cells recruit and regulate t regulatory cells. Exp. Hematol 2008, 36, 309–318. [Google Scholar]
- Matsumoto, K.; Ogawa, M.; Suzuki, J.; Hirata, Y.; Nagai, R.; Isobe, M. Regulatory T lymphocytes attenuate myocardial infarction-induced ventricular remodeling in mice. Int. Heart J 2011, 52, 382–387. [Google Scholar]
- Tang, T.; Yuan, J.; Zhu, Z.; Zhang, W.; Xiao, H.; Xia, N.; Yan, X.; Nie, S.; Liu, J.; Zhou, S. Regulatory T cells ameliorate cardiac remodeling after myocardial infarction. Basic Res. Cardiol 2012, 107, 1–17. [Google Scholar]
- Lai, R.C.; Arslan, F.; Lee, M.M.; Sze, N.S.K.; Choo, A.; Chen, T.S.; Salto-Tellez, M.; Timmers, L.; Lee, C.N.; el Oakley, R.M. Exosome secreted by msc reduces myocardial ischemia/reperfusion injury. Stem Cell Res 2010, 4, 214–222. [Google Scholar]
- Arslan, F.; Lai, R.C.; Smeets, M.B.; Akeroyd, L.; Choo, A.; Aguor, E.N.; Timmers, L.; van Rijen, H.V.; Doevendans, P.A.; Pasterkamp, G. Mesenchymal stem cell-derived exosomes increase ATP levels, decrease oxidative stress and activate PI3K/Akt pathway to enhance myocardial viability and prevent adverse remodeling after myocardial ischemia/reperfusion injury. Stem Cell Res 2013, 10, 301–312. [Google Scholar]
- Kumar, V.; Abbas, A.K.; Fausto, N.; Aster, J.C. Robbins & Cotran Pathologic Basis of Disease, 8th ed; Elsevier Health Sciences: Philadelphia, PA, USA, 2009; pp. 512–514. [Google Scholar]
- Armulik, A.; Genové, G.; Betsholtz, C. Pericytes: Developmental, physiological, and pathological perspectives, problems, and promises. Dev. Cell 2011, 21, 193–215. [Google Scholar]
- Caplan, A.I. All MSCs are pericytes? Cell Stem Cell 2008, 3, 229–230. [Google Scholar]
- Chen, C.; Montelatici, E.; Crisan, M.; Corselli, M.; Huard, J.; Lazzari, L.; Péault, B. Perivascular multi-lineage progenitor cells in human organs: Regenerative units, cytokine sources or both? Cytokine Growth Factor Rev 2009, 20, 429–434. [Google Scholar]
- Chen, C.; Corselli, M.; Péault, B.; Huard, J. Human blood-vessel-derived stem cells for tissue repair and regeneration. Biomed. Res. Int. 2012, 2012. [Google Scholar] [CrossRef]
- Crisan, M.; Yap, S.; Casteilla, L.; Chen, C.; Corselli, M.; Park, T.S.; Andriolo, G.; Sun, B.; Zheng, B.; Zhang, L. A perivascular origin for mesenchymal stem cells in multiple human organs. Cell Stem Cell 2008, 3, 301–313. [Google Scholar]
- Zheng, B.; Cao, B.; Crisan, M.; Sun, B.; Li, G.; Logar, A.; Yap, S.; Pollett, J.B.; Drowley, L.; Cassino, T. Prospective identification of myogenic endothelial cells in human skeletal muscle. Nat. Biotechnol 2007, 25, 1025–1034. [Google Scholar]
- Corselli, M.; Chen, C.; Sun, B.; Yap, S.; Rubin, J.P.; Péault, B. The tunica adventitia of human arteries and veins as a source of mesenchymal stem cells. Stem Cells Dev 2012, 21, 1299–1308. [Google Scholar]
- Zheng, B.; Li, G.; Chen, W.C.; Deasy, B.M.; Pollett, J.B.; Sun, B.; Drowley, L.; Gharaibeh, B.; Usas, A.; Péault, B. Human myogenic endothelial cells exhibit chondrogenic and osteogenic potentials at the clonal level. J. Orthop. Res 2013, 31, 1089–1095. [Google Scholar]
- Zheng, B.; Chen, C.; Li, G.; Thompson, S.D.; Poddar, M.; Peault, B.; Huard, J. Isolation of myogenic stem cells from cultures of cryopreserved human skeletal muscle. Cell Transplant 2012, 21, 1087–1093. [Google Scholar]
- Rucker, H.K.; Wynder, H.J.; Thomas, W.E. Cellular mechanisms of CNS pericytes. Brain Res. Bull 2000, 51, 363–369. [Google Scholar]
- Hellström, M.; Gerhardt, H.; Kalén, M.; Li, X.; Eriksson, U.; Wolburg, H.; Betsholtz, C. Lack of pericytes leads to endothelial hyperplasia and abnormal vascular morphogenesis. J. Cell Biol 2001, 153, 543–554. [Google Scholar]
- Von Tell, D.; Armulik, A.; Betsholtz, C. Pericytes and vascular stability. Exp. Cell Res 2006, 312, 623–629. [Google Scholar]
- Armulik, A.; Abramsson, A.; Betsholtz, C. Endothelial/pericyte interactions. Circ. Res 2005, 97, 512–523. [Google Scholar]
- Gaengel, K.; Genové, G.; Armulik, A.; Betsholtz, C. Endothelial-mural cell signaling in vascular development and angiogenesis. Arterioscler. Thromb Vasc. Biol 2009, 29, 630–638. [Google Scholar]
- Dulmovits, B.M.; Herman, I.M. Microvascular remodeling and wound healing: A role for pericytes. Int. J. Biochem. Cell Biol 2012, 44, 1800–1812. [Google Scholar]
- Chen, C.; Okada, M.; Proto, J.D.; Gao, X.; Sekiya, N.; Beckman, S.A.; Corselli, M.; Crisan, M.; Saparov, A.; Tobita, K. Human pericytes for ischemic heart repair. Stem Cells 2013, 31, 305–316. [Google Scholar]
- He, W.; Nieponice, A.; Soletti, L.; Hong, Y.; Gharaibeh, B.; Crisan, M.; Usas, A.; Peault, B.; Huard, J.; Wagner, W.R. Pericyte-based human tissue engineered vascular grafts. Biomaterials 2010, 31, 8235–8244. [Google Scholar]
- Campagnolo, P.; Cesselli, D.; Zen, A.A.H.; Beltrami, A.P.; Kränkel, N.; Katare, R.; Angelini, G.; Emanueli, C.; Madeddu, P. Human adult vena saphena contains perivascular progenitor cells endowed with clonogenic and proangiogenic potential. Circulation 2010, 121, 1735–1745. [Google Scholar]
- Katare, R.; Riu, F.; Mitchell, K.; Gubernator, M.; Campagnolo, P.; Cui, Y.; Fortunato, O.; Avolio, E.; Cesselli, D.; Beltrami, A.P. Transplantation of human pericyte progenitor cells improves the repair of infarcted heart through activation of an angiogenic program involving micro-RNA-132 novelty and significance. Circ. Res 2011, 109, 894–906. [Google Scholar]
- Okada, M.; Payne, T.R.; Drowley, L.; Jankowski, R.J.; Momoi, N.; Beckman, S.; Chen, W.C.; Keller, B.B.; Tobita, K.; Huard, J. Human skeletal muscle cells with a slow adhesion rate after isolation and an enhanced stress resistance improve function of ischemic hearts. Mol. Ther 2012, 20, 138–145. [Google Scholar]
- Quaegebeur, A.; Lange, C.; Carmeliet, P. The neurovascular link in health and disease: Molecular mechanisms and therapeutic implications. Neuron 2011, 71, 406–424. [Google Scholar]
- Stark, K.; Eckart, A.; Haidari, S.; Tirniceriu, A.; Lorenz, M.; von Brühl, M.; Gärtner, F.; Khandoga, A.G.; Legate, K.R.; Pless, R. Capillary and arteriolar pericytes attract innate leukocytes exiting through venules and “instruct” them with pattern-recognition and motility programs. Nat. Immunol 2012, 14, 41–51. [Google Scholar]
- Alon, R.; Nourshargh, S. Learning in motion: Pericytes instruct migrating innate leukocytes. Nat. Immunol 2012, 14, 14–15. [Google Scholar]
- Maier, C.L.; Pober, J.S. Human placental pericytes poorly stimulate and actively regulate allogeneic CD4 T Cell responses. Arterioscler. Thromb Vasc. Biol 2011, 31, 183–189. [Google Scholar]
- Verbeek, M.M.; Westphal, J.R.; Ruiter, D.J.; de Waal, R. T lymphocyte adhesion to human brain pericytes is mediated via very late Antigen-4/vascular cell adhesion Molecule-1 interactions. J. Immunol 1995, 154, 5876–5884. [Google Scholar]
- Pober, J.S.; Tellides, G. Participation of blood vessel cells in human adaptive immune responses. Trends Immunol 2012, 33, 49–57. [Google Scholar]
- Tu, Z.; Li, Y.; Smith, D.S.; Sheibani, N.; Huang, S.; Kern, T.; Lin, F. Retinal pericytes inhibit activated t cell proliferation. Invest. Ophthalmol. Vis. Sci 2011, 52, 9005–9010. [Google Scholar]
- Chen, C.-W.; Saparov, A. University of Pittsburgh: Pittsburgh, PA, USA, Unpublished work; 2013.
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Saparov, A.; Chen, C.-W.; Beckman, S.A.; Wang, Y.; Huard, J. The Role of Antioxidation and Immunomodulation in Postnatal Multipotent Stem Cell-Mediated Cardiac Repair. Int. J. Mol. Sci. 2013, 14, 16258-16279. https://doi.org/10.3390/ijms140816258
Saparov A, Chen C-W, Beckman SA, Wang Y, Huard J. The Role of Antioxidation and Immunomodulation in Postnatal Multipotent Stem Cell-Mediated Cardiac Repair. International Journal of Molecular Sciences. 2013; 14(8):16258-16279. https://doi.org/10.3390/ijms140816258
Chicago/Turabian StyleSaparov, Arman, Chien-Wen Chen, Sarah A. Beckman, Yadong Wang, and Johnny Huard. 2013. "The Role of Antioxidation and Immunomodulation in Postnatal Multipotent Stem Cell-Mediated Cardiac Repair" International Journal of Molecular Sciences 14, no. 8: 16258-16279. https://doi.org/10.3390/ijms140816258