Inhibition of NADPH Oxidase by Apocynin Attenuates Progression of Atherosclerosis

Abstract
:1. Introduction
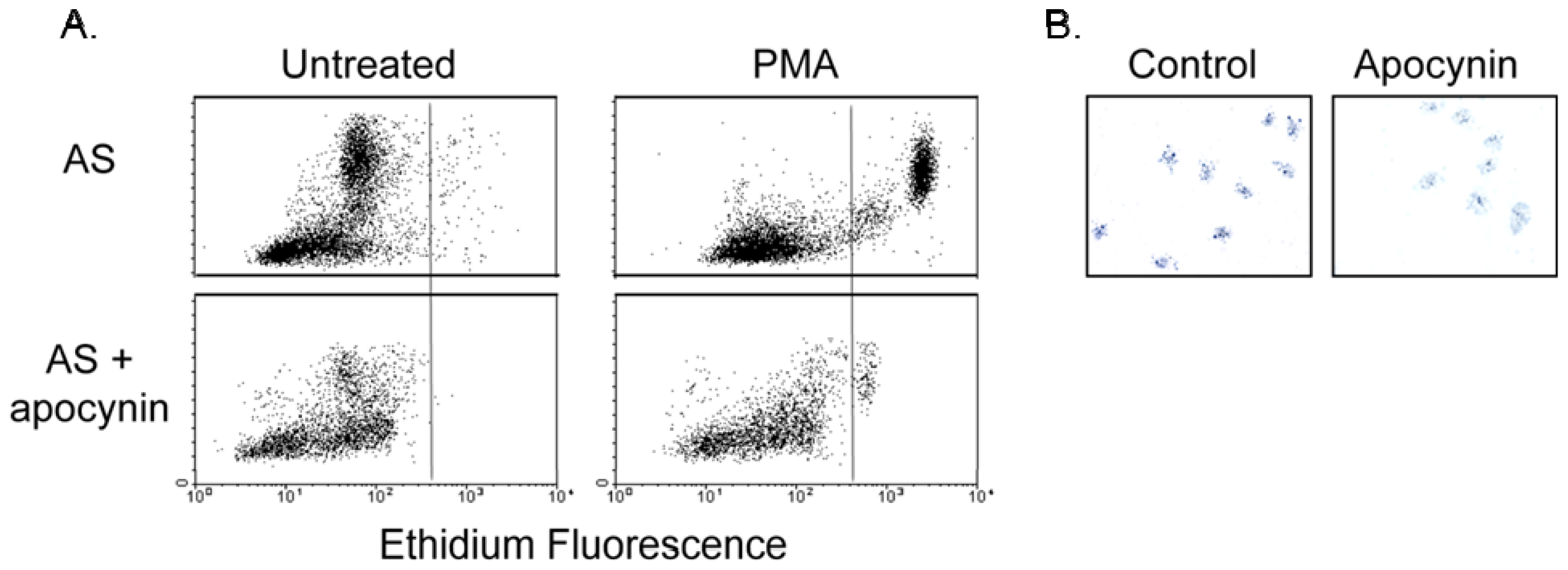
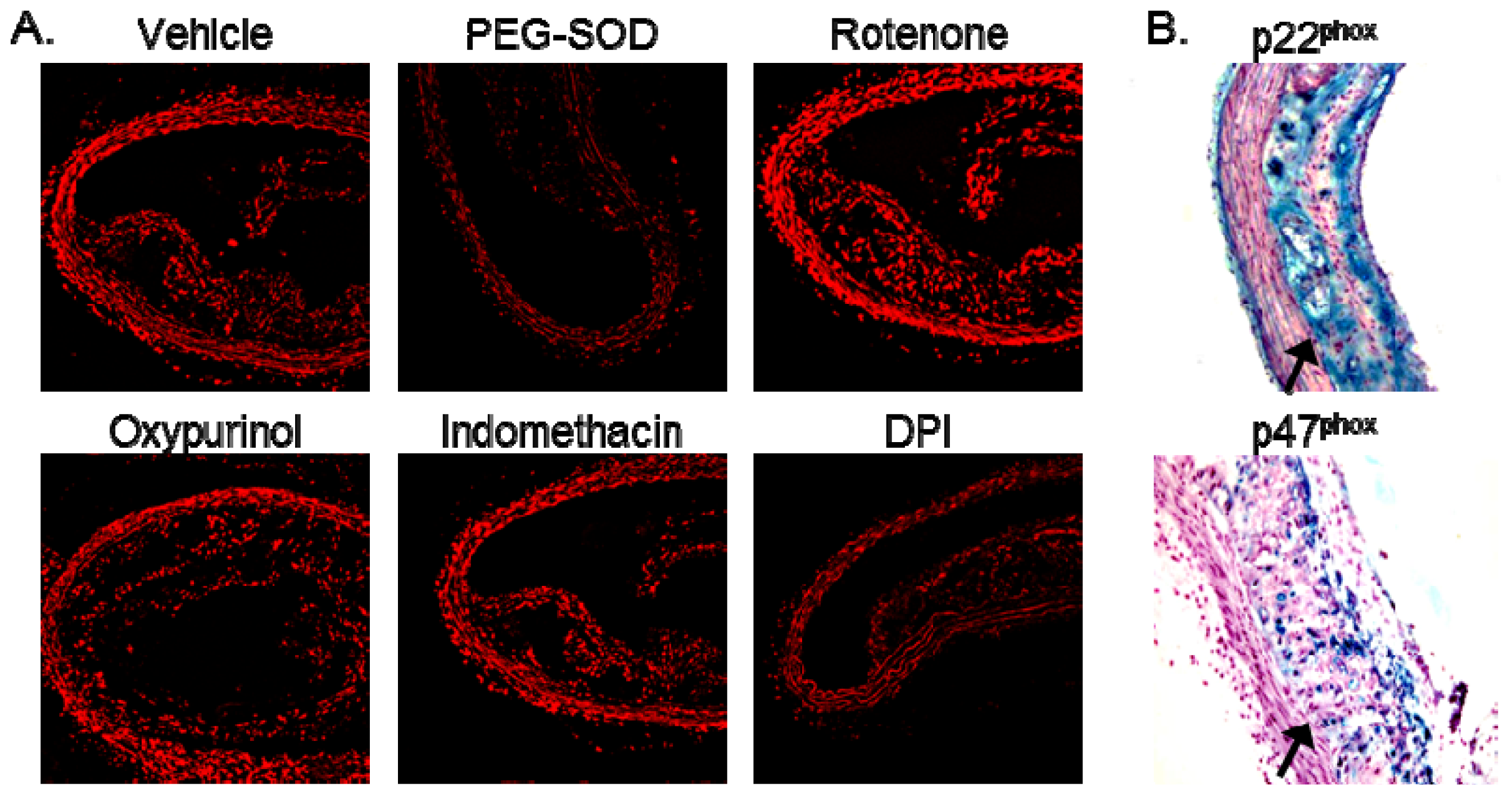
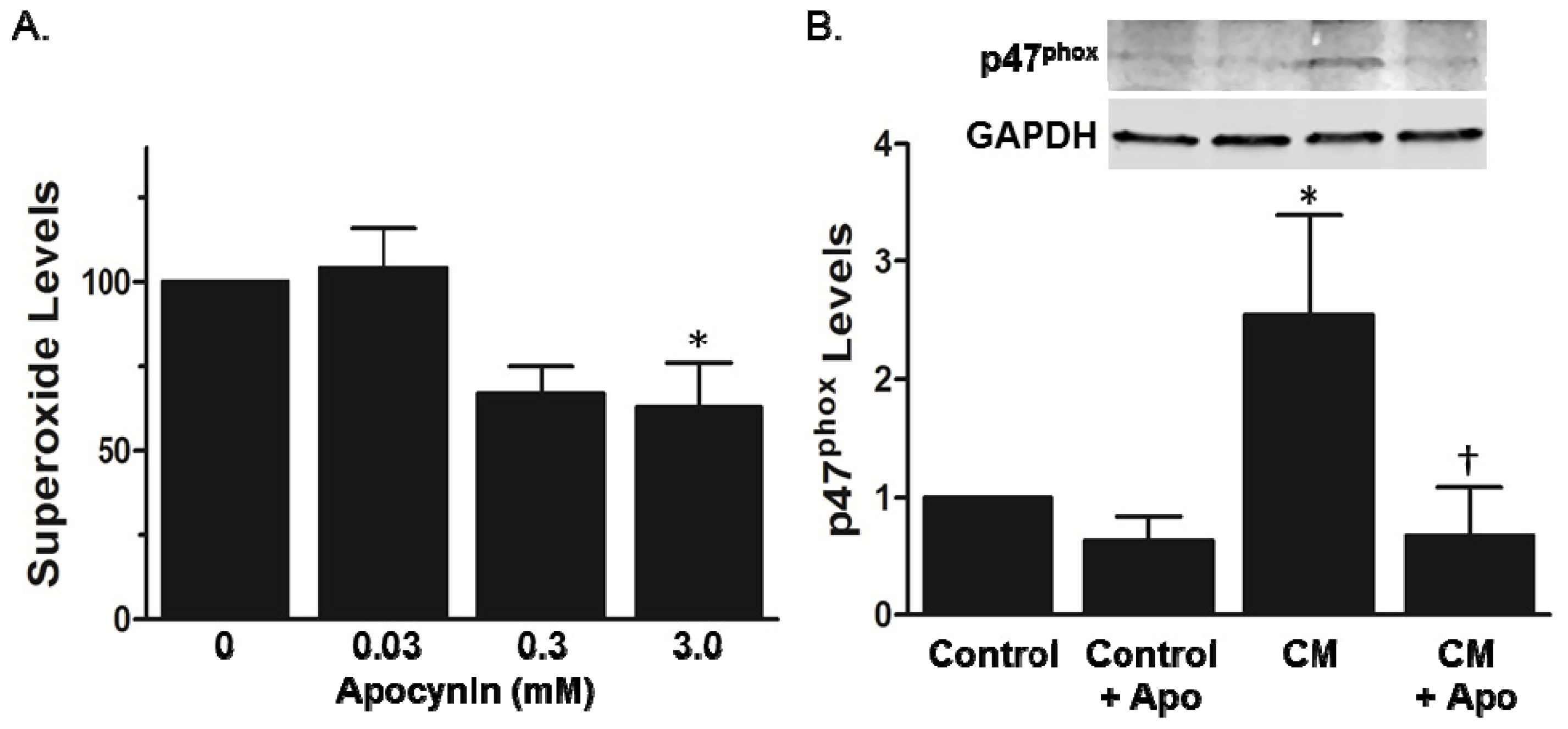
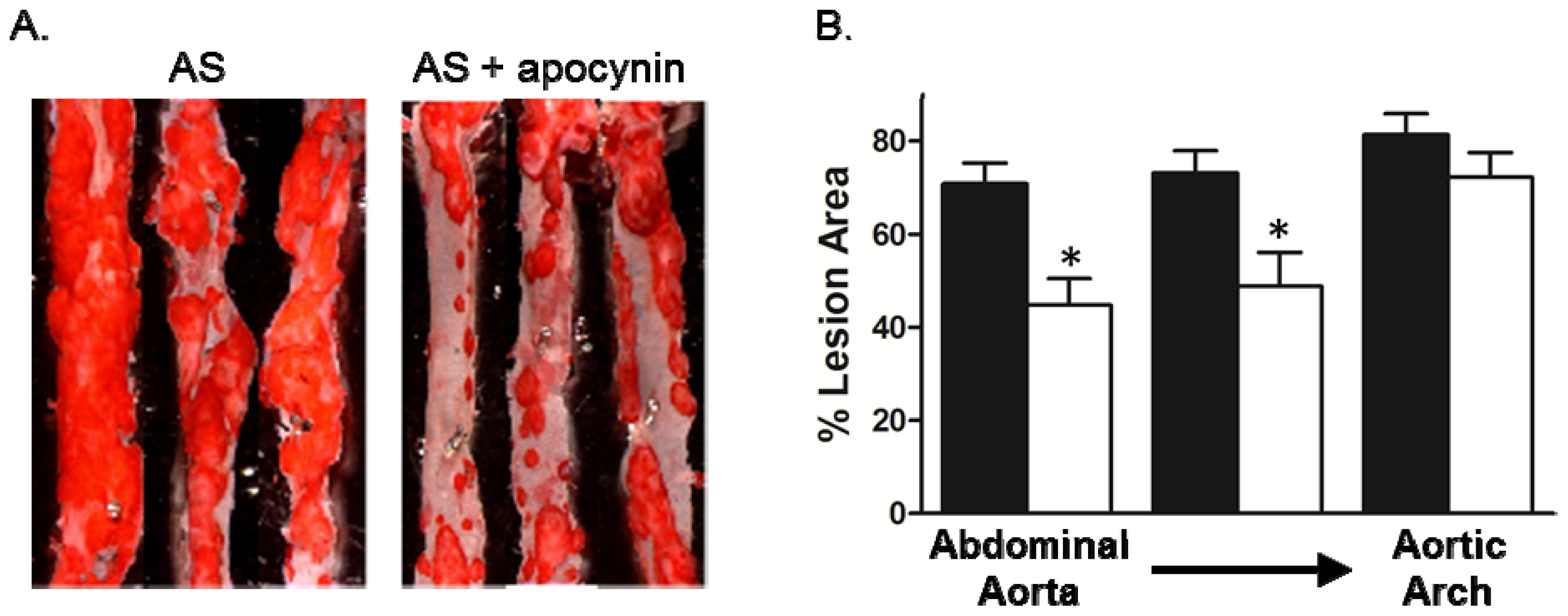
2. Results and Discussion
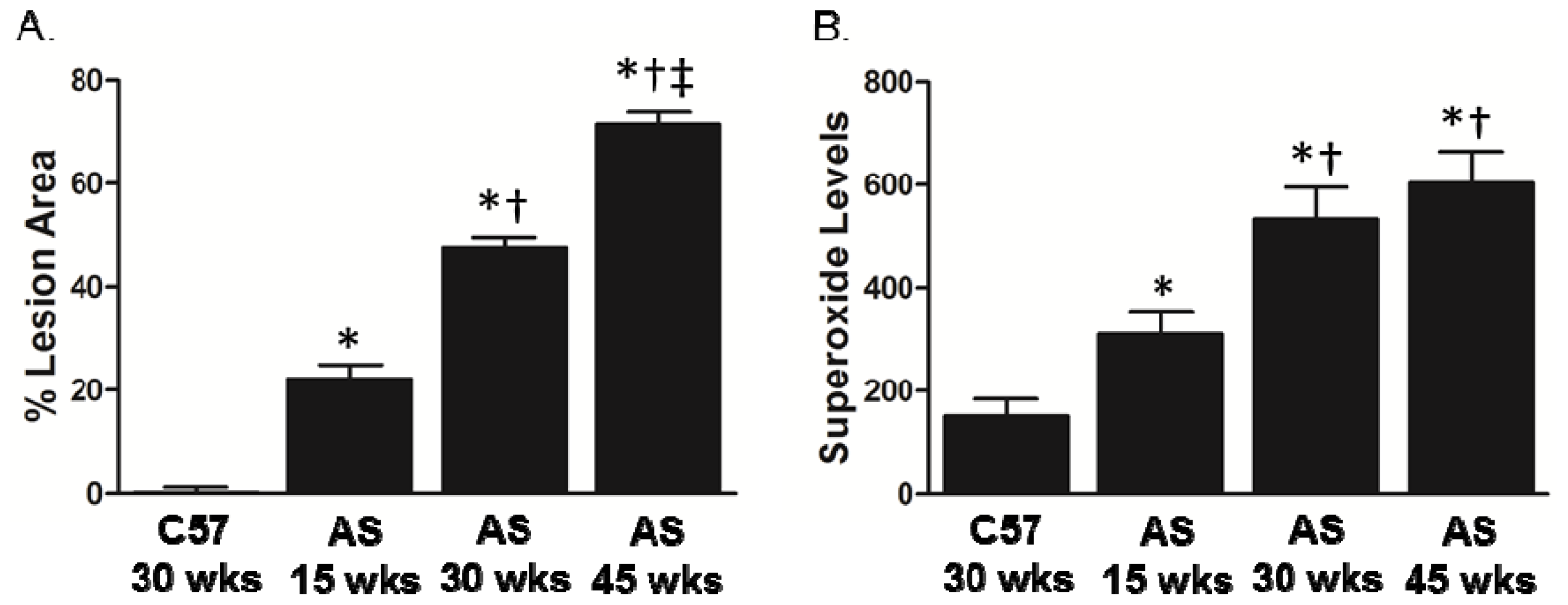
2.1. Association between Lesion Size and Superoxide Levels in Atherosclerotic Mice
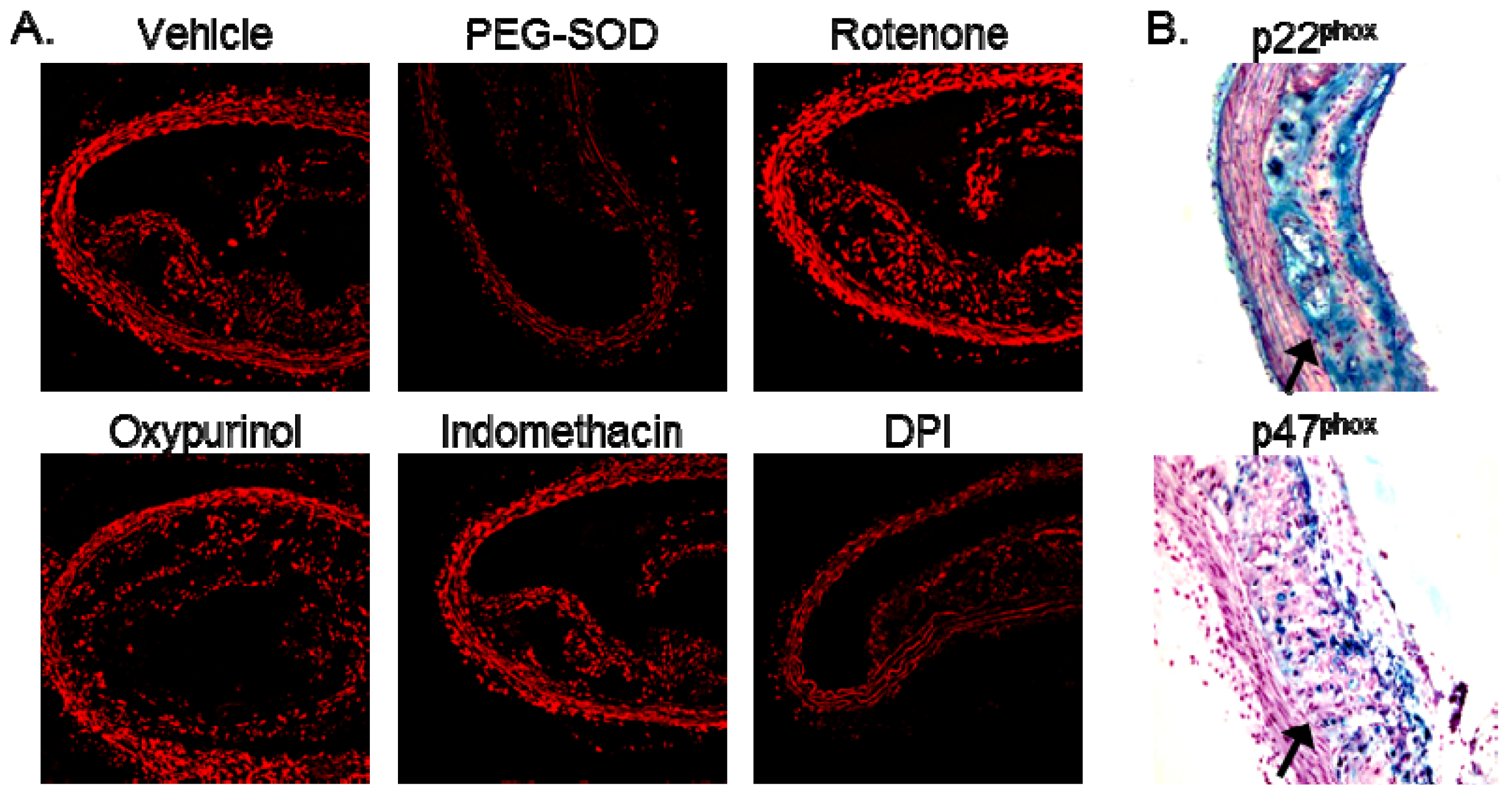
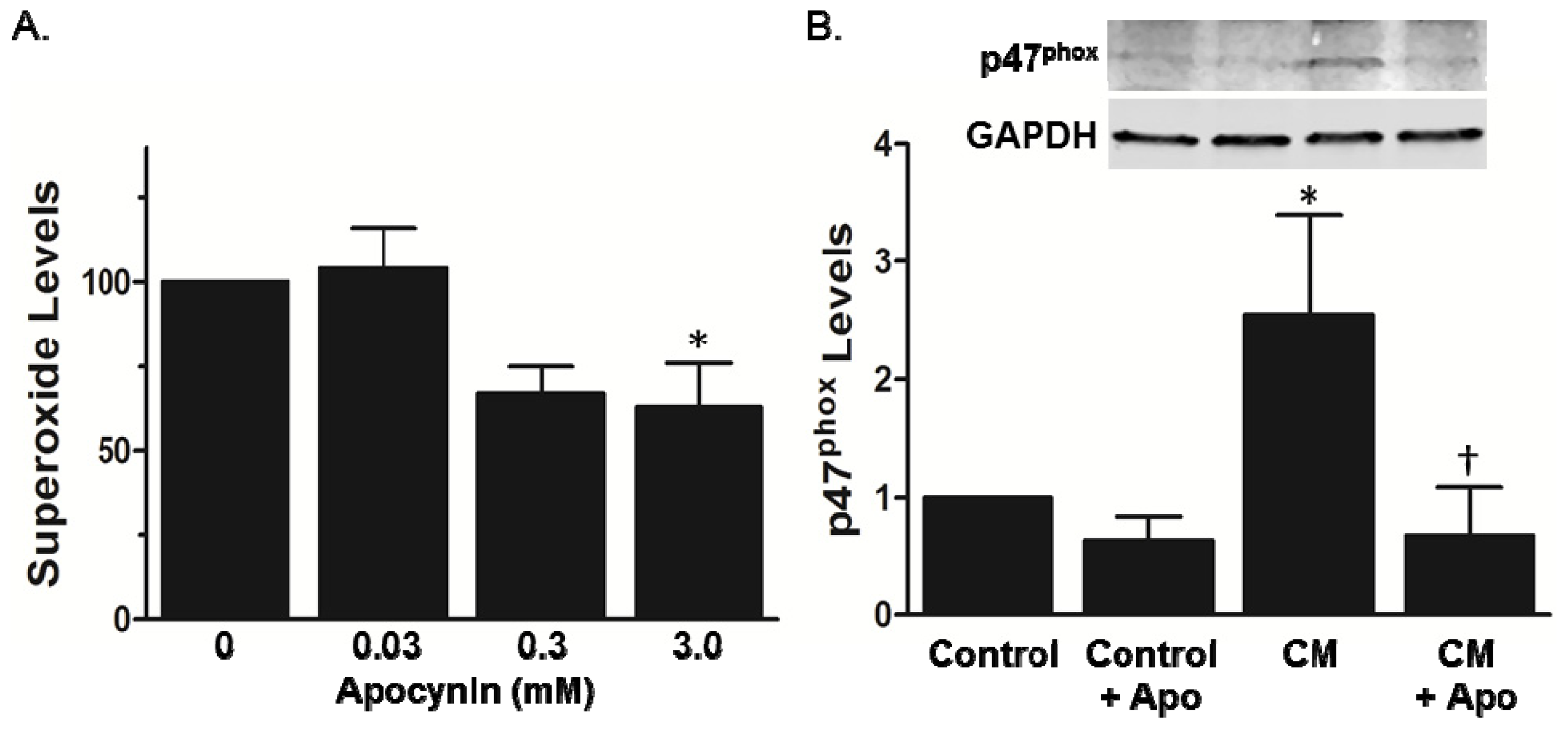
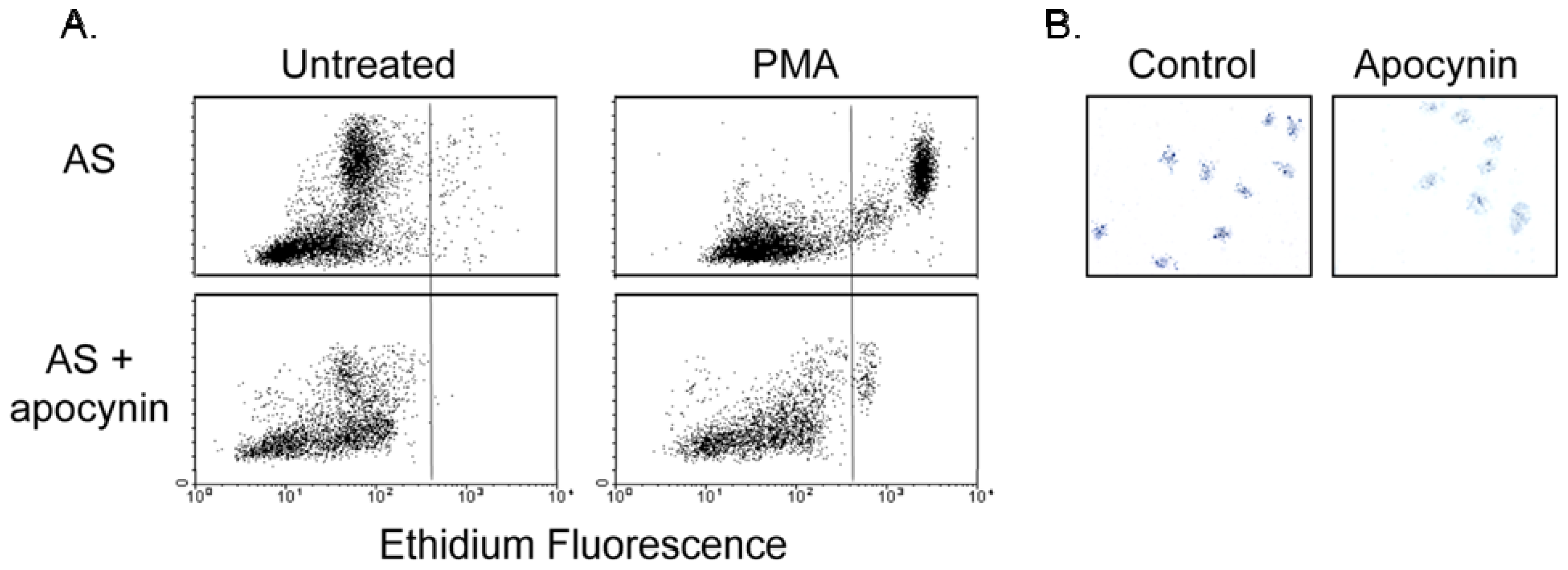
2.2. Increased Superoxide in AS Aorta Is Derived from NADPH Oxidase
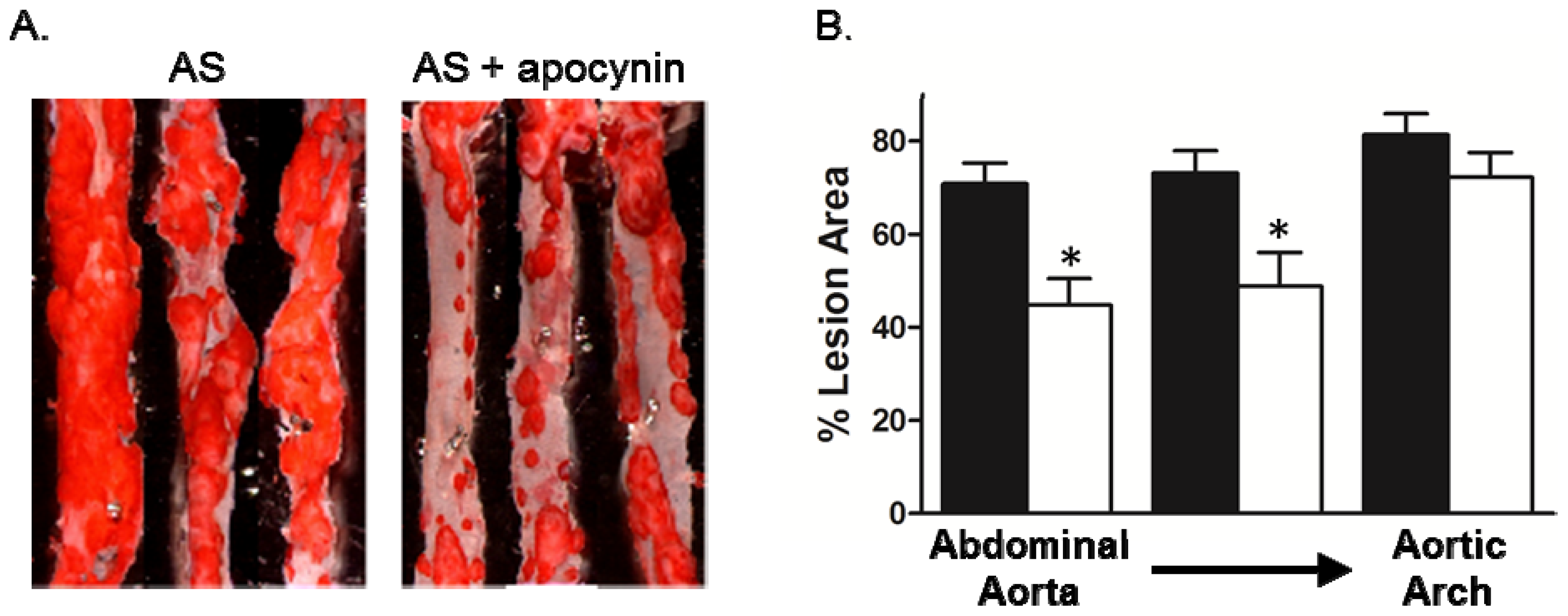
2.3. Effect of Apocynin on Atherosclerosis in the Aorta
3. Experimental Section
3.1. Animals and Tissue Preparation
3.2. Total Cholesterol Measurements
3.3. Detection of ROS
3.3.1. DHE Staining
3.3.2. Lucigenin-Enhanced Chemiluminescence
3.4. p47phox Membrane Translocation
3.5. Lesion Assessment
3.6. Immunohistochemistry
3.7. Respiratory Burst Measurement
3.7.1. Flow Cytometry
3.7.2. NBT Assay
3.8. Statistical Analysis
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Lassegue, B.; Griendling, K.K. NADPH oxidases: Functions and pathologies in the vasculature. Arterioscler. Thromb. Vasc. Biol 2010, 30, 653–661. [Google Scholar]
- Azumi, H.; Inoue, N.; Takeshita, S.; Rikitake, Y.; Kawashima, S.; Hayashi, Y.; Itoh, H.; Yokoyama, M. Expression of NADH/NADPH oxidase p22phox in human coronary arteries. Circulation 1999, 100, 1494–1498. [Google Scholar]
- Sorescu, D.; Weiss, D.; Lassegue, B.; Clempus, R.E.; Szocs, K.; Sorescu, G.P.; Valppu, L.; Quinn, M.T.; Lambeth, J.D.; Vega, J.D.; et al. Superoxide production and expression of nox family proteins in human atherosclerosis. Circulation 2002, 105, 1429–1435. [Google Scholar]
- Guzik, T.J.; Sadowski, J.; Guzik, B.; Jopek, A.; Kapelak, B.; Przybylowski, P.; Wierzbicki, K.; Korbut, R.; Harrison, D.G.; Channon, K.M. Coronary artery superoxide production and nox isoform expression in human coronary artery disease. Arterioscler. Thromb. Vasc. Biol 2006, 26, 333–339. [Google Scholar]
- Guzik, T.J.; Chen, W.; Gongora, M.C.; Guzik, B.; Lob, H.E.; Mangalat, D.; Hoch, N.; Dikalov, S.; Rudzinski, P.; Kapelak, B.; Sadowski, J.; Harrison, D.G. Calcium-dependent NOX5 nicotinamide adenine dinucleotide phosphate oxidase contributes to vascular oxidative stress in human coronary artery disease. J. Am. Coll. Cardiol 2008, 52, 1803–1809. [Google Scholar]
- Madrigal-Matute, J.; Fernandez-Garcia, C.E.; Gomez-Guerrero, C.; Lopez-Franco, O.; Munoz-Garcia, B.; Egido, J.; Blanco-Colio, L.M.; Martin-Ventura, J.L. HSP90 inhibition by 17-DMAG attenuates oxidative stress in experimental atherosclerosis. Cardiovasc. Res 2012, 95, 116–123. [Google Scholar]
- Kirk, E.A.; Dinauer, M.C.; Rosen, H.; Chait, A.; Heinecke, J.W.; LeBoeuf, R.C. Impaired superoxide production due to a deficiency in phagocyte NADPH oxidase fails to inhibit atherosclerosis in mice. Arterioscler. Thromb. Vasc. Biol 2000, 20, 1529–1535. [Google Scholar]
- Barry-Lane, P.A.; Patterson, C.; van der Merwe, M.; Hu, Z.Y.; Holland, S.M.; Yeh, E.T.H.; Runge, M.S. p47phox is required for atherosclerotic lesion progression in ApoE(−/−) mice. J. Clin. Invest 2001, 108, 1513–1522. [Google Scholar]
- Vendrov, A.E.; Hakim, Z.S.; Madamanchi, N.R.; Rojas, M.; Madamanchi, C.; Runge, M.S. Atherosclerosis is attenuated by limiting superoxide generation in both macrophages and vessel wall cells. Arterioscler. Thromb. Vasc. Biol 2007, 27, 2714–2721. [Google Scholar]
- Judkins, C.P.; Diep, H.; Broughton, B.R.; Mast, A.E.; Hooker, E.U.; Miller, A.A.; Selemidis, S.; Dusting, G.J.; Sobey, C.G.; Drummond, G.R. Direct evidence of a role for Nox2 in superoxide production, reduced nitric oxide bioavailability, and early atherosclerotic plaque formation in ApoE−/− mice. Am. J. Physiol. Heart Circ. Physiol 2010, 298, H24–H32. [Google Scholar]
- Sheehan, A.L.; Carrell, S.; Johnson, B.; Stanic, B.; Banfi, B.; Miller, F.J., Jr. Role for Nox1 NADPH oxidase in atherosclerosis. Atherosclerosis 2011, 216, 321–326. [Google Scholar]
- Douglas, G.; Bendall, J.K.; Crabtree, M.J.; Tatham, A.L.; Carter, E.E.; Hale, A.B.; Channon, K.M. Endothelial-specific Nox2 overexpression increases vascular superoxide and macrophage recruitment in ApoE(−)/(−) mice. Cardiovasc. Res 2012, 94, 20–29. [Google Scholar]
- Thomas, M.; Gavrila, D.; McCormick, M.L.; Miller, F.J., Jr; Daugherty, A.; Cassis, L.A.; Dellsperger, K.C.; Weintraub, N.L. Deletion of p47phox attenuates angiotensin II-induced abdominal aortic aneurysm formation in apolipoprotein E-deficient mice. Circulation 2006, 114, 404–413. [Google Scholar]
- Hsich, E.; Segal, B.H.; Pagano, P.J.; Rey, F.E.; Paigen, B.; Deleonardis, J.; Hoyt, R.F.; Holland, S.M.; Finkel, T. Vascular effects following homozygous disruption of p47(phox): An essential component of NADPH oxidase. Circulation 2000, 101, 1234–1236. [Google Scholar]
- Gray, S.P.; di Marco, E.; Okabe, J.; Szyndralewiez, C.; Heitz, F.; Montezano, A.C.; de Haan, J.B.; Koulis, C.; El-Osta, A.; Andrews, K.L.; et al. NADPH oxidase 1 plays a key role in diabetes mellitus-accelerated atherosclerosis. Circulation 2013, 127, 1888–1902. [Google Scholar]
- Miller, F.J., Jr; Sharp, W.J.; Fang, X.; Oberley, L.W.; Oberley, T.D.; Weintraub, N.L. Oxidative stress in human abdominal aortic aneurysms: A potential mediator of aneurysmal remodeling. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 560–565. [Google Scholar]
- Stolk, J.; Hiltermann, T.J.; Dijkman, J.H.; Verhoeven, A.J. Characteristics of the inhibition of NADPH oxidase activation in neutrophils by apocynin, a methoxy-substituted catechol. Am. J. Respir. Cell Mol. Biol 1994, 11, 95–102. [Google Scholar]
- Barbieri, S.S.; Cavalca, V.; Eligini, S.; Brambilla, M.; Caiani, A.; Tremoli, E.; Colli, S. Apocynin prevents cyclooxygenase 2 expression in human monocytes through NADPH oxidase and glutathione redox-dependent mechanisms. Free Radic. Biol. Med 2004, 37, 156–165. [Google Scholar]
- Hathaway, C.A.; Heistad, D.D.; Piegors, D.J.; Miller, F.J., Jr. Regression of atherosclerosis in monkeys reduces vascular superoxide levels. Circ. Res. 2002, 90, 277–283. [Google Scholar]
- Miller, F.J., Jr; Gutterman, D.D.; Rios, C.D.; Heistad, D.D.; Davidson, B.L. Superoxide production in vascular smooth muscle contributes to oxidative stress and impaired relaxation in atherosclerosis. Circ. Res. 1998, 82, 1298–1305. [Google Scholar]
- Heumuller, S.; Wind, S.; Barbosa-Sicard, E.; Schmidt, H.H.; Busse, R.; Schroder, K.; Brandes, R.P. Apocynin is not an inhibitor of vascular NADPH oxidases but an antioxidant. Hypertension 2008, 51, 211–217. [Google Scholar]
- Lassegue, B.; San Martin, A.; Griendling, K.K. Biochemistry, physiology, and pathophysiology of NADPH oxidases in the cardiovascular system. Circ. Res 2012, 110, 1364–1390. [Google Scholar]
- Drummond, G.R.; Selemidis, S.; Griendling, K.K.; Sobey, C.G. Combating oxidative stress in vascular disease: NADPH oxidases as therapeutic targets. Nat. Rev. Drug Discov 2011, 10, 453–471. [Google Scholar]
- Johnson, D.K.; Schillinger, K.J.; Kwait, D.M.; Hughes, C.V.; McNamara, E.J.; Ishmael, F.; O’Donnell, R.W.; Chang, M.M.; Hogg, M.G.; Dordick, J.S.; et al. Inhibition of NADPH oxidase activation in endothelial cells by ortho-methoxy-substituted catechols. Endothelium 2002, 9, 191–203. [Google Scholar]
- Cheret, C.; Gervais, A.; Lelli, A.; Colin, C.; Amar, L.; Ravassard, P.; Mallet, J.; Cumano, A.; Krause, K.H.; Mallat, M. Neurotoxic activation of microglia is promoted by a nox1-dependent NADPH oxidase. J. Neurosci 2008, 28, 12039–12051. [Google Scholar]
- Banfi, B.; Clark, R.A.; Steger, K.; Krause, K.H. Two novel proteins activate superoxide generation by the NADPH oxidase NOX1. J. Biol. Chem 2003, 278, 3510–3513. [Google Scholar]
- Wu, M.C.; Ho, H.I.; Lee, T.W.; Wu, H.L.; Lo, J.M. In vivo examination of 111In-bis-5HT-DTPA to target myeloperoxidase in atherosclerotic ApoE knockout mice. J. Drug Target 2012, 20, 605–614. [Google Scholar]
- Liu, C.; Desikan, R.; Ying, Z.; Gushchina, L.; Kampfrath, T.; Deiuliis, J.; Wang, A.; Xu, X.; Zhong, J.; Rao, X.; et al. Effects of a novel pharmacologic inhibitor of myeloperoxidase in a mouse atherosclerosis model. PLoS One 2012, 7, e50767. [Google Scholar]
- Castor, L.R.; Locatelli, K.A.; Ximenes, V.F. Pro-oxidant activity of apocynin radical. Free Radic. Biol. Med 2010, 48, 1636–1643. [Google Scholar]
- Kinoshita, H.; Matsumura, T.; Ishii, N.; Fukuda, K.; Senokuchi, T.; Motoshima, H.; Kondo, T.; Taketa, K.; Kawasaki, S.; Hanatani, S.; et al. Apocynin suppresses the progression of atherosclerosis in apoE-deficient mice by inactivation of macrophages. Biochem. Biophys. Res. Commun 2013, 431, 124–130. [Google Scholar]
- Streeter, J.; Thiel, W.; Brieger, K.; Miller, F.J., Jr. Opportunity Nox: The future of NADPH oxidases as therapeutic targets in cardiovascular disease. Cardiovasc. Ther. 2013, 31, 125–137. [Google Scholar]
- Xu, S.; Shriver, A.S.; Jagadeesha, D.K.; Chamseddine, A.; Szocs, K.; Weintraub, N.L.; Griendling, K.K.; Bhalla, R.; Miller, F.J., Jr. Increased expression of Nox1 in neointimal smooth muscle cells promotes activation of matrix metalloproteinase-9. J. Vasc. Res. 2012, 49, 242–248. [Google Scholar]
- Vuorte, J.; Jansson, S.E.; Repo, H. Standardization of a flow cytometric assay for phagocyte respiratory burst activity. Scand. J. Immunol 1996, 43, 329–334. [Google Scholar]
- Ochs, H.D.; Igo, R.P. The NBT slide test: A simple screening method for detecting chronic granulomatous disease and female carriers. J. Pediatr 1973, 83, 77–82. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Body weight (grams) | Cholesterol (mg/dL) | |
|---|---|---|
| C57BL/6 | 30 ± 1 | 91 ± 5 |
| AS (15 weeks) | 31 ± 2 | 749 ± 148 |
| AS (30–35 weeks) | 28 ± 1 | 596 ± 52 |
| AS + apocynin (34–35 weeks) | 28 ± 1 | 592 ± 155 |
© 2013 by the authors; licensee MDPI, Basel, Switzerland This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Kinkade, K.; Streeter, J.; Miller, F.J. Inhibition of NADPH Oxidase by Apocynin Attenuates Progression of Atherosclerosis. Int. J. Mol. Sci. 2013, 14, 17017-17028. https://doi.org/10.3390/ijms140817017
Kinkade K, Streeter J, Miller FJ. Inhibition of NADPH Oxidase by Apocynin Attenuates Progression of Atherosclerosis. International Journal of Molecular Sciences. 2013; 14(8):17017-17028. https://doi.org/10.3390/ijms140817017
Chicago/Turabian StyleKinkade, Kara, Jennifer Streeter, and Francis J. Miller. 2013. "Inhibition of NADPH Oxidase by Apocynin Attenuates Progression of Atherosclerosis" International Journal of Molecular Sciences 14, no. 8: 17017-17028. https://doi.org/10.3390/ijms140817017