Glycyrrhetinic Acid Liposomes Containing Mannose-Diester Lauric Diacid-Cholesterol Conjugate Synthesized by Lipase-Catalytic Acylation for Liver-Specific Delivery

Abstract
:1. Introduction
2. Results and Discussion
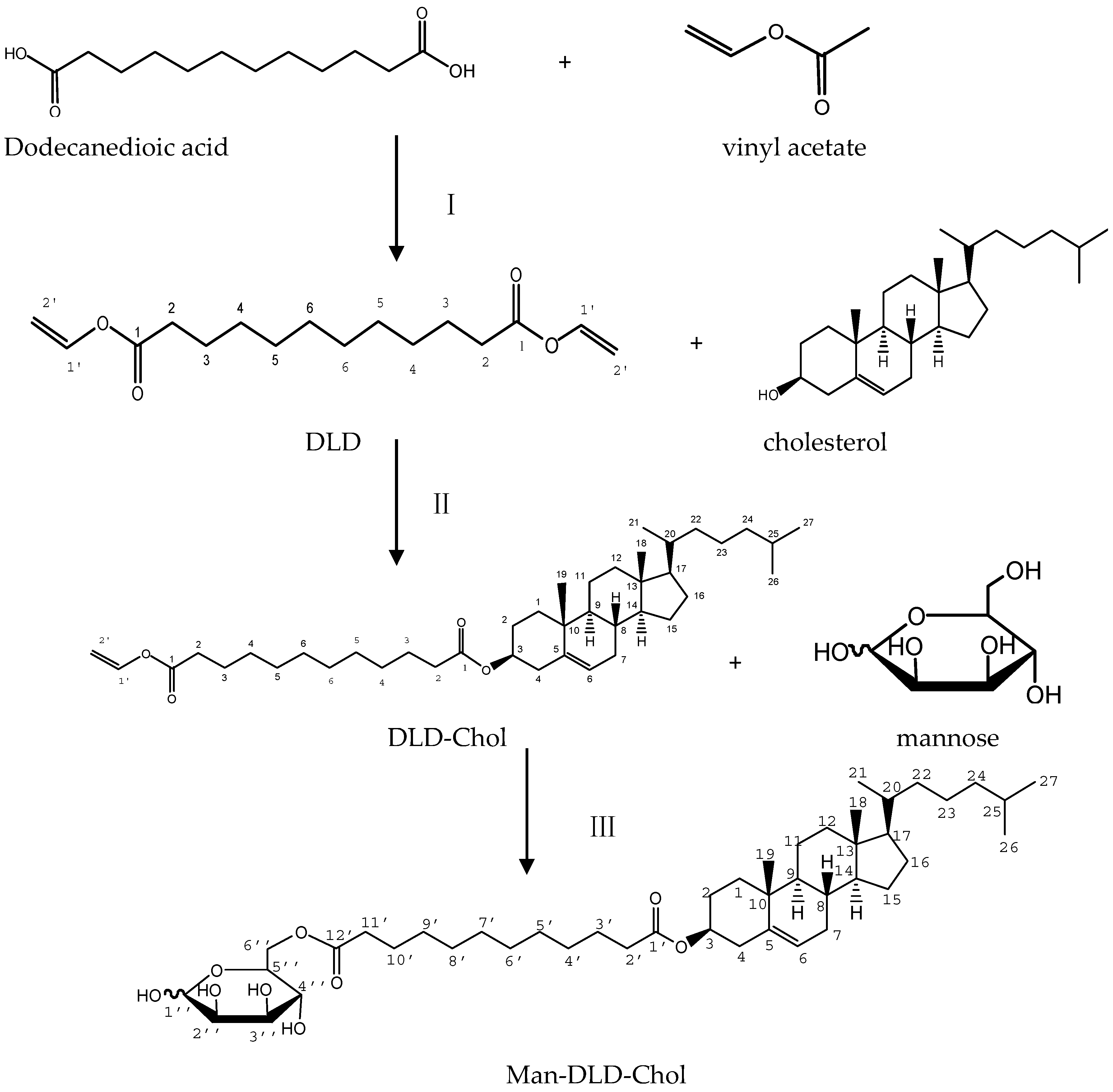
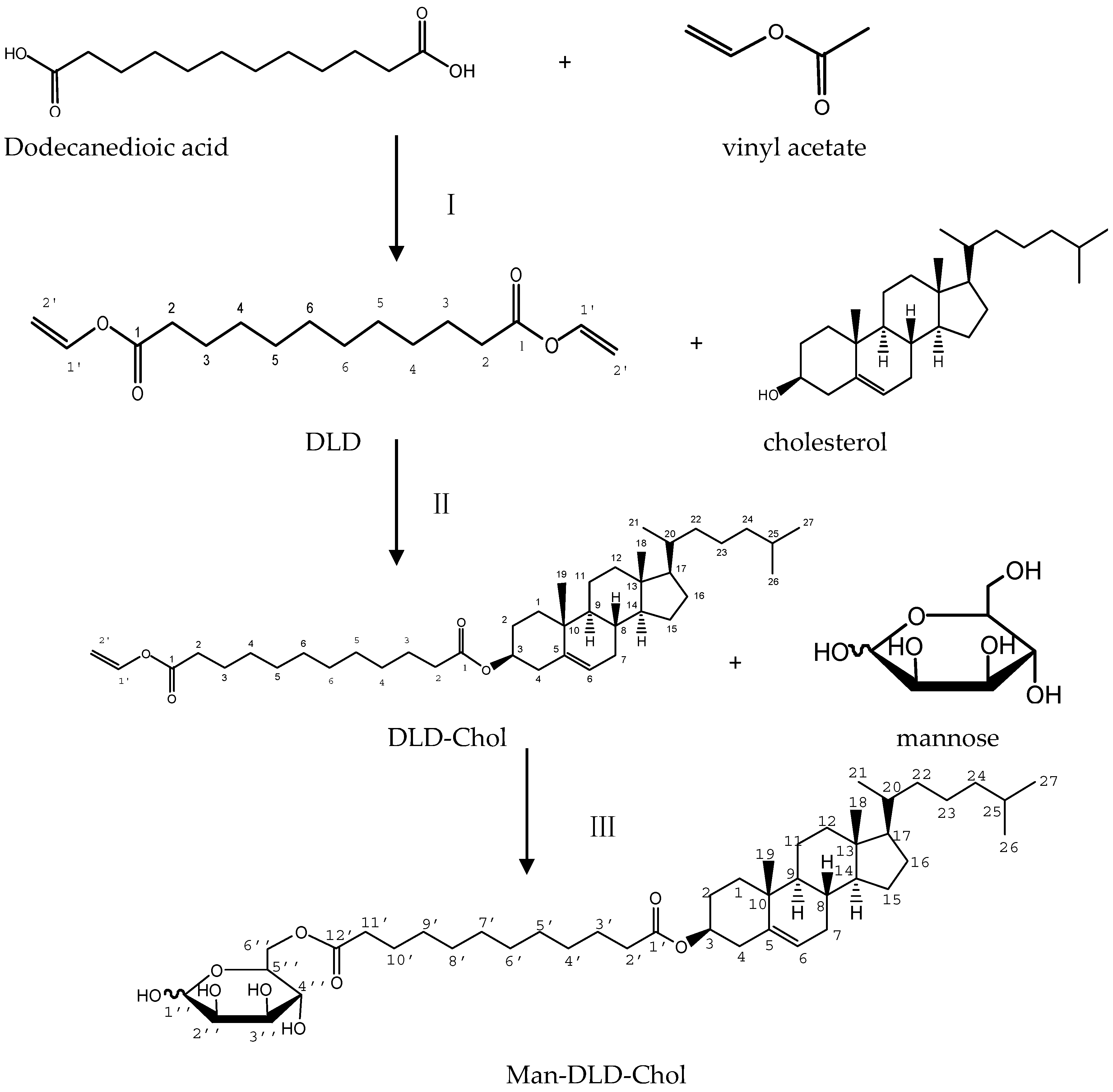
2.1. Synthesis of Man-DLD-Chol
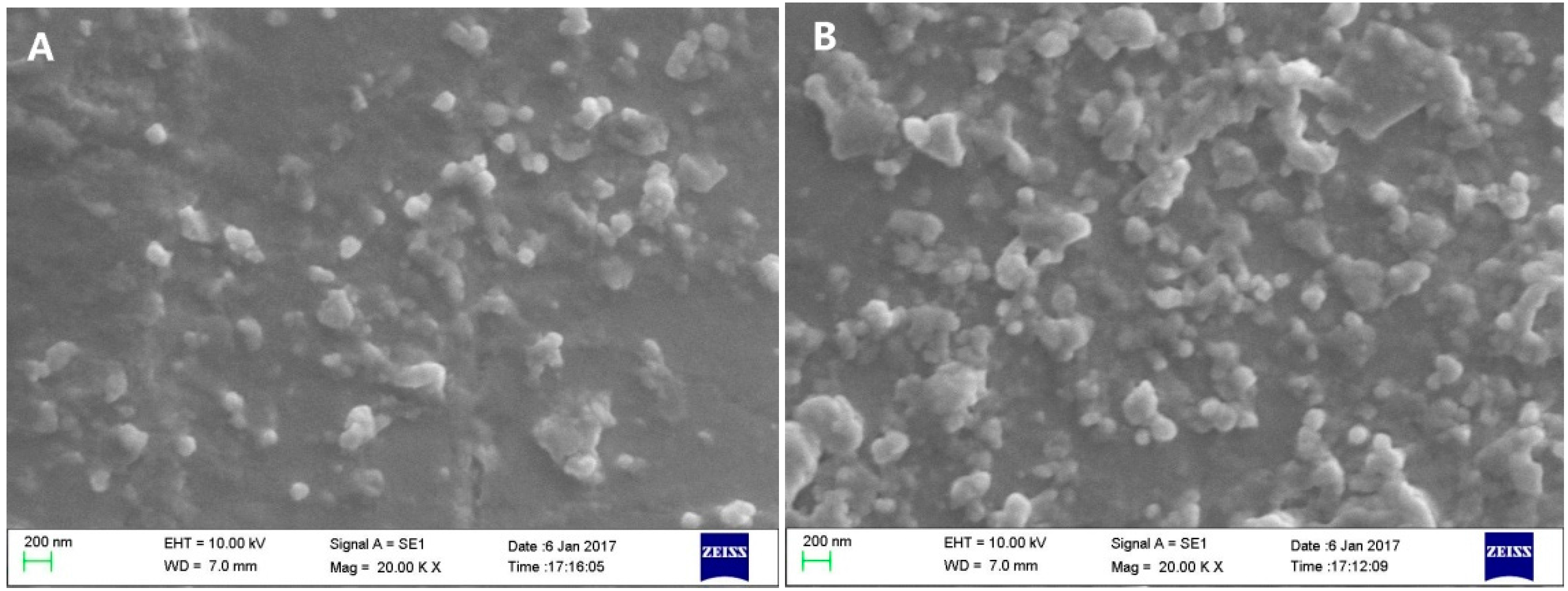
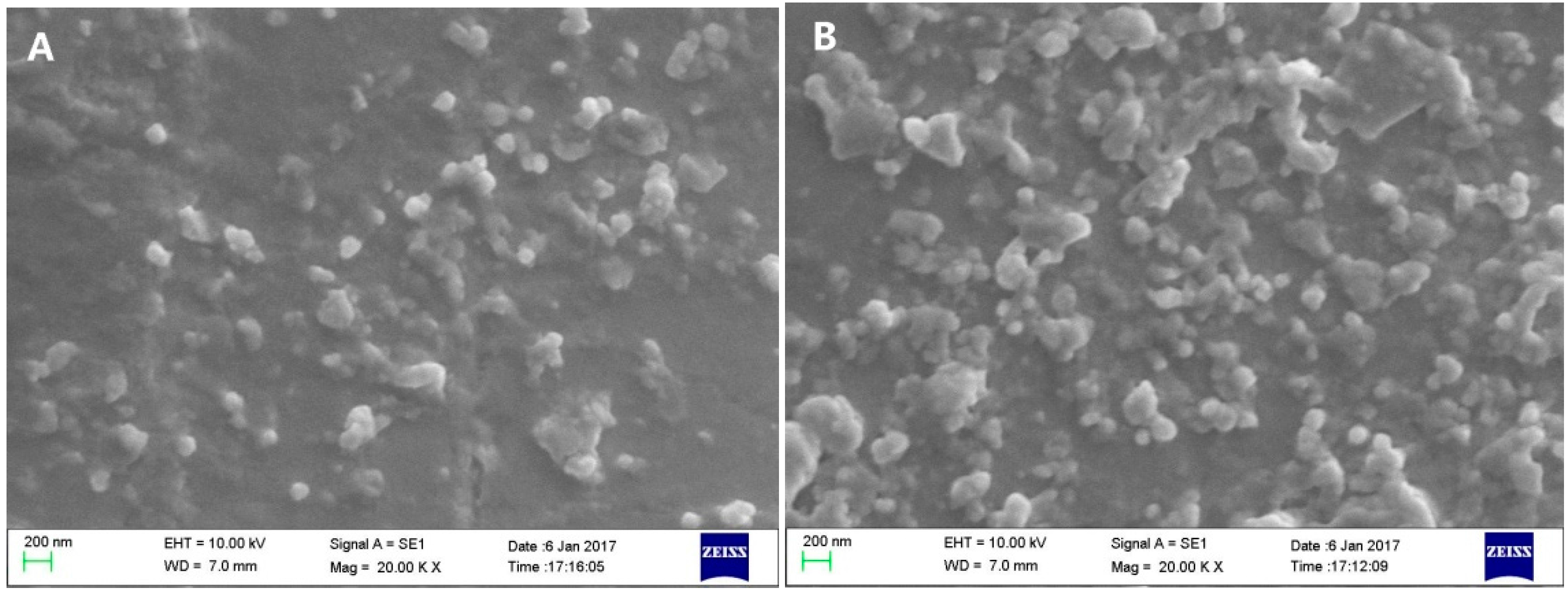
2.2. Characterization of Liposomes
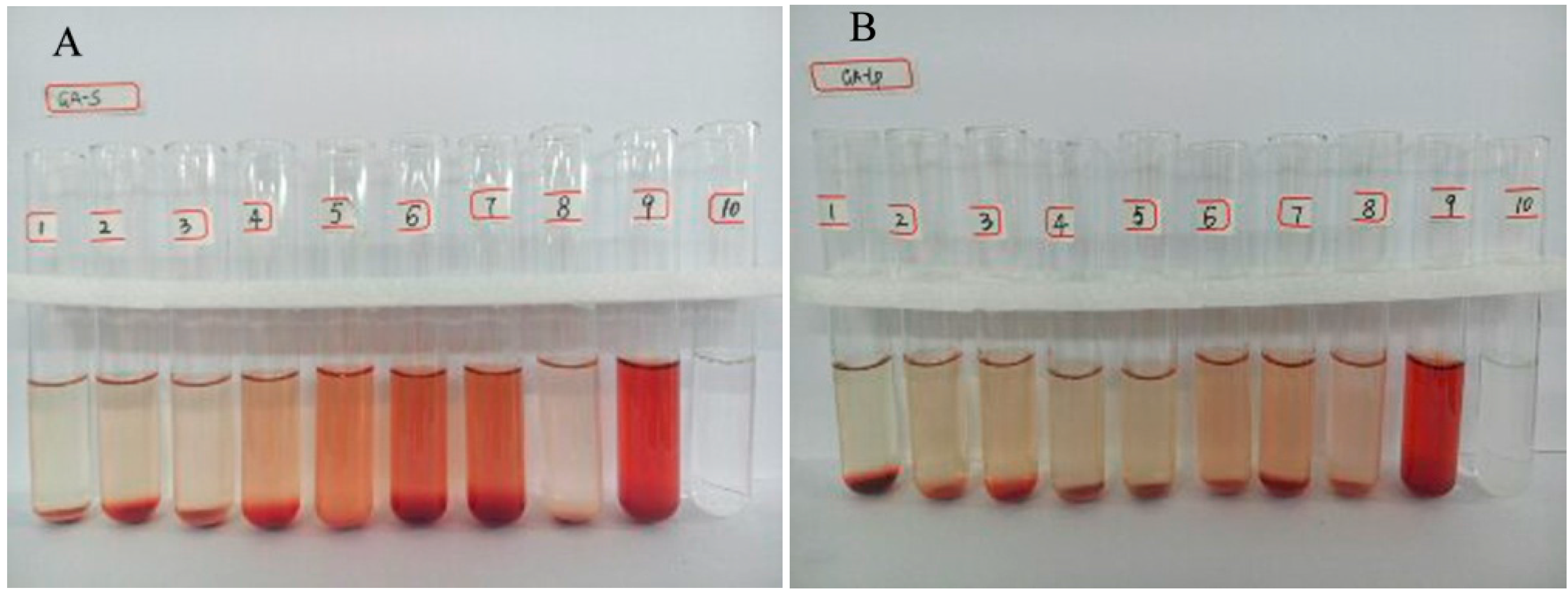
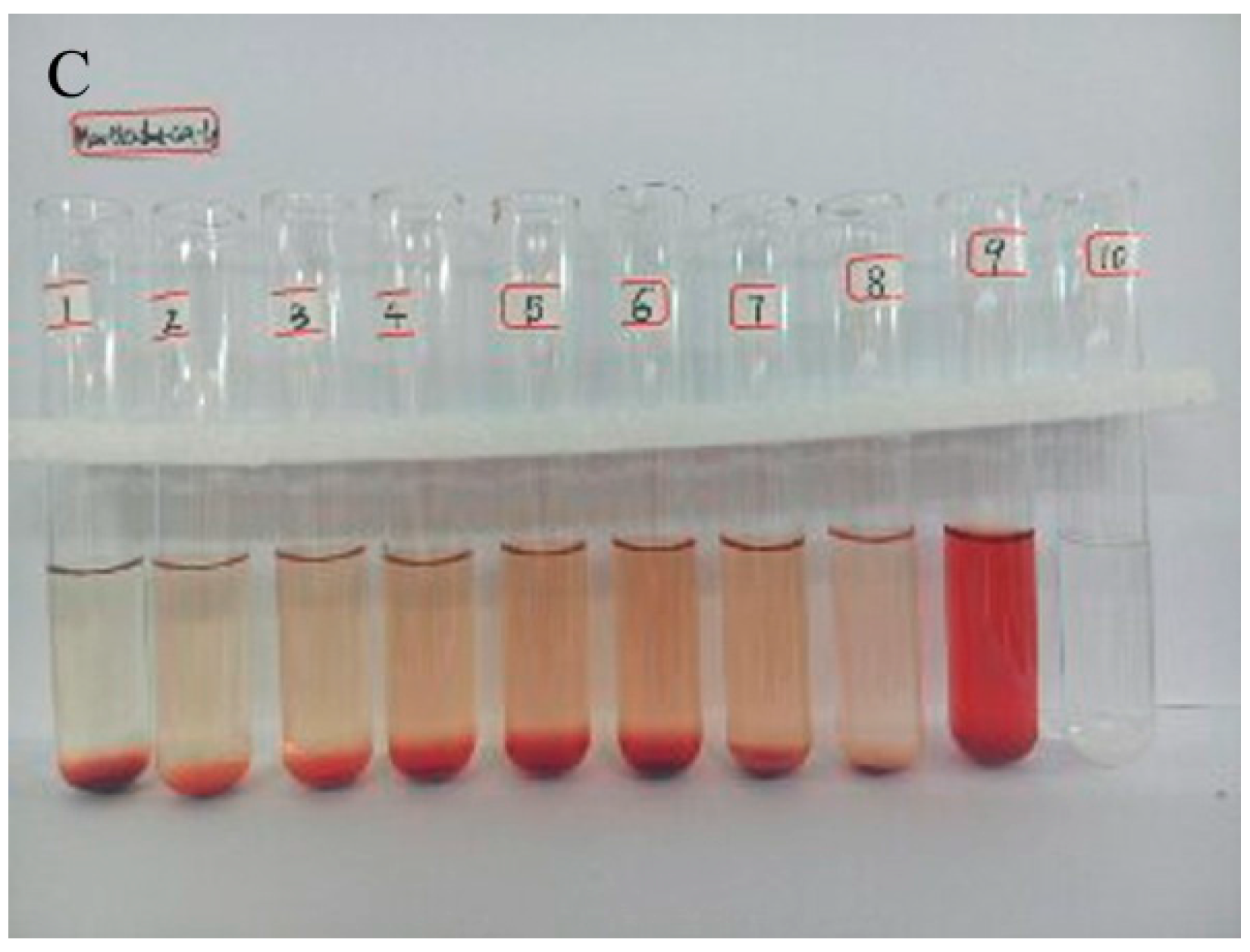
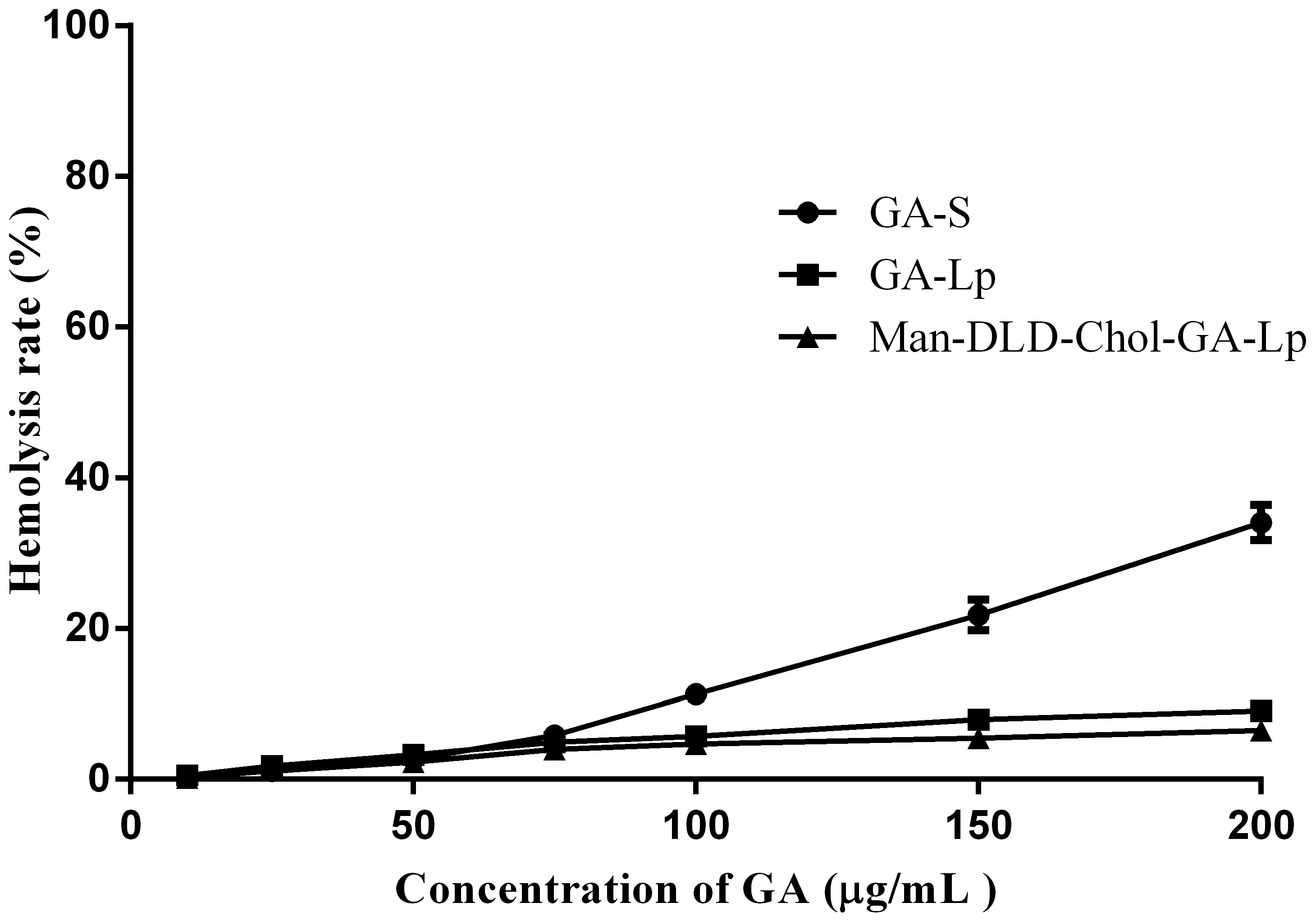
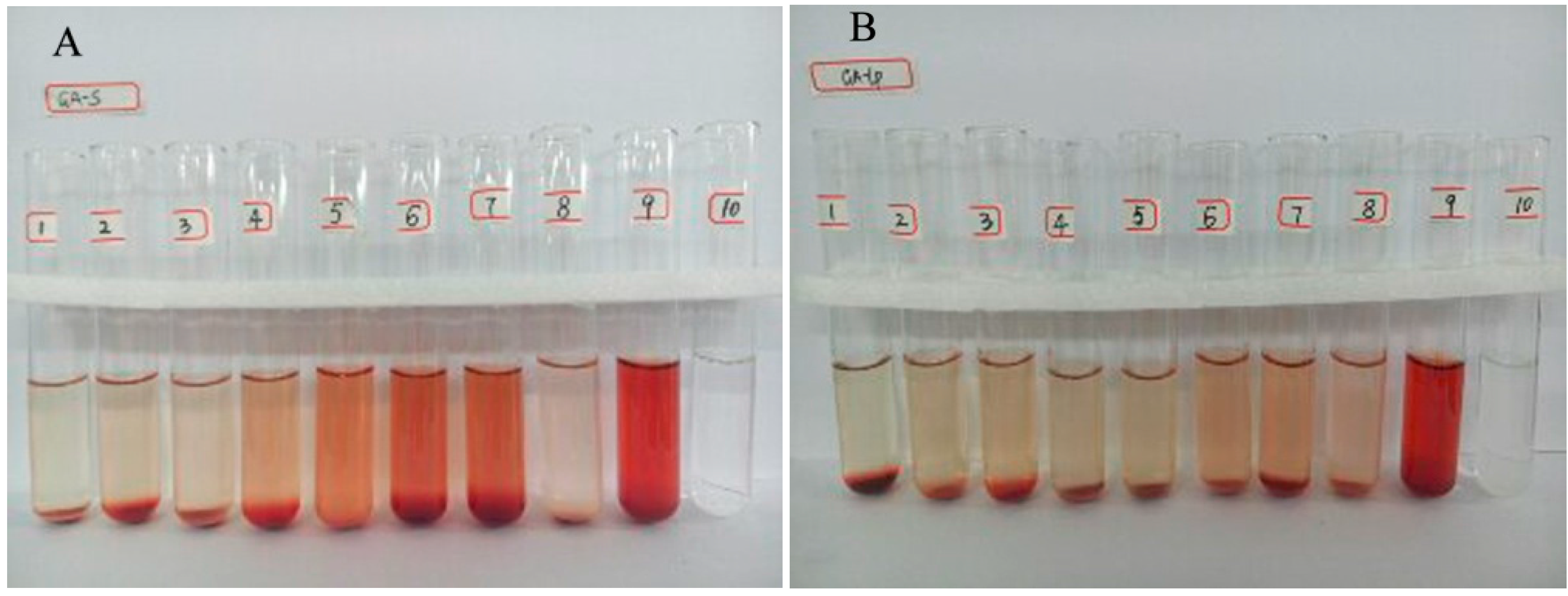
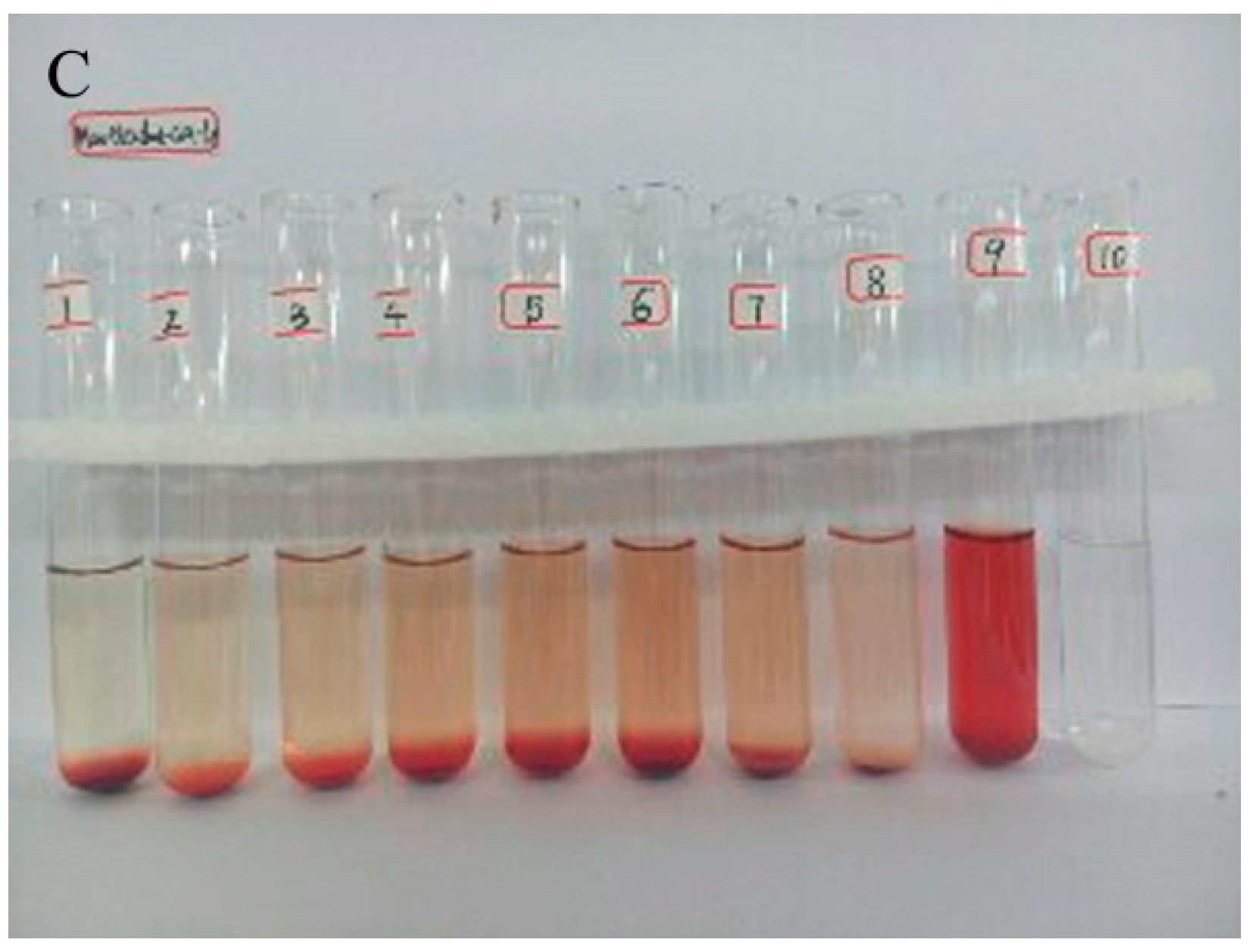
2.3. Hemolytic Study
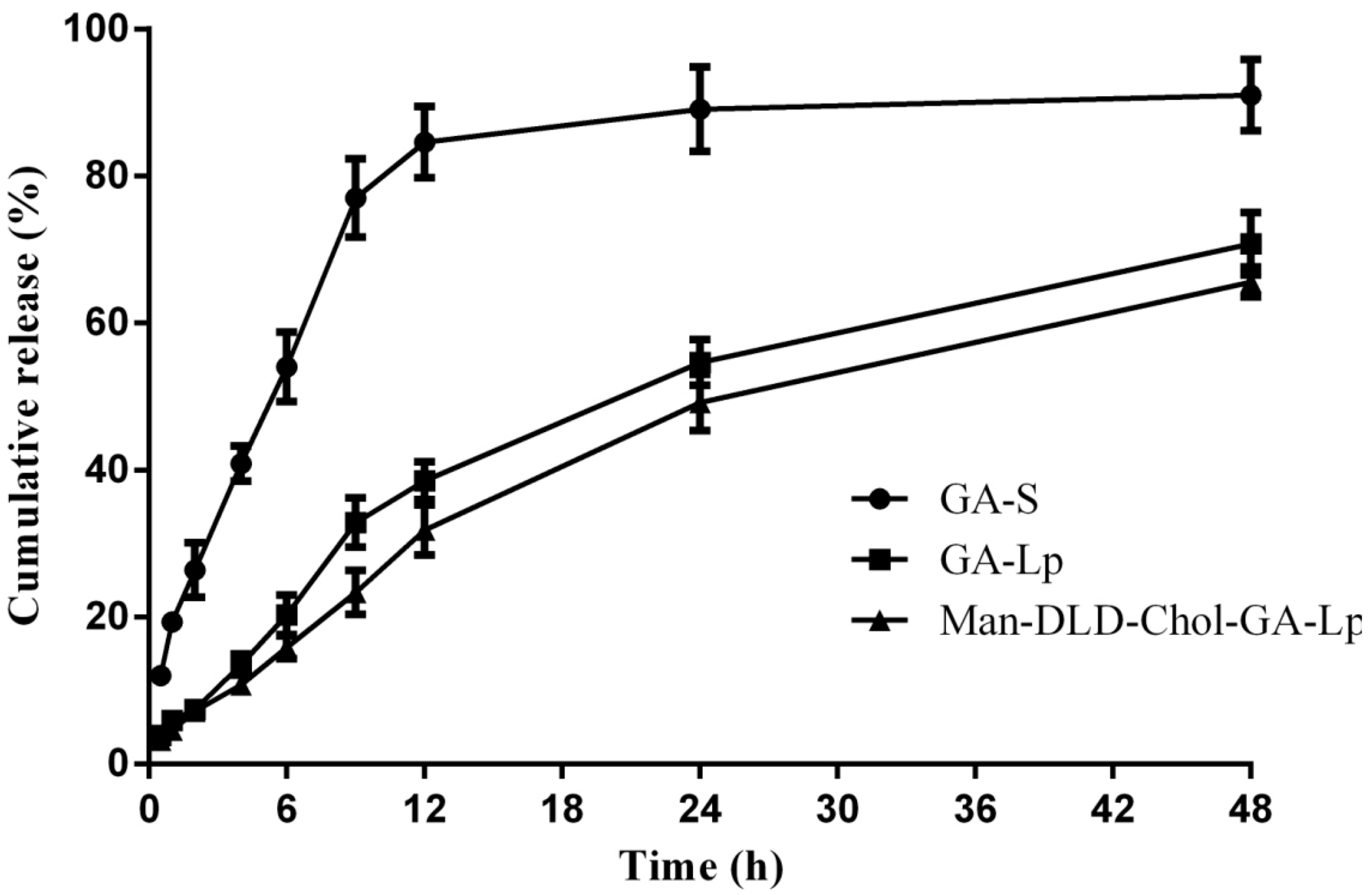
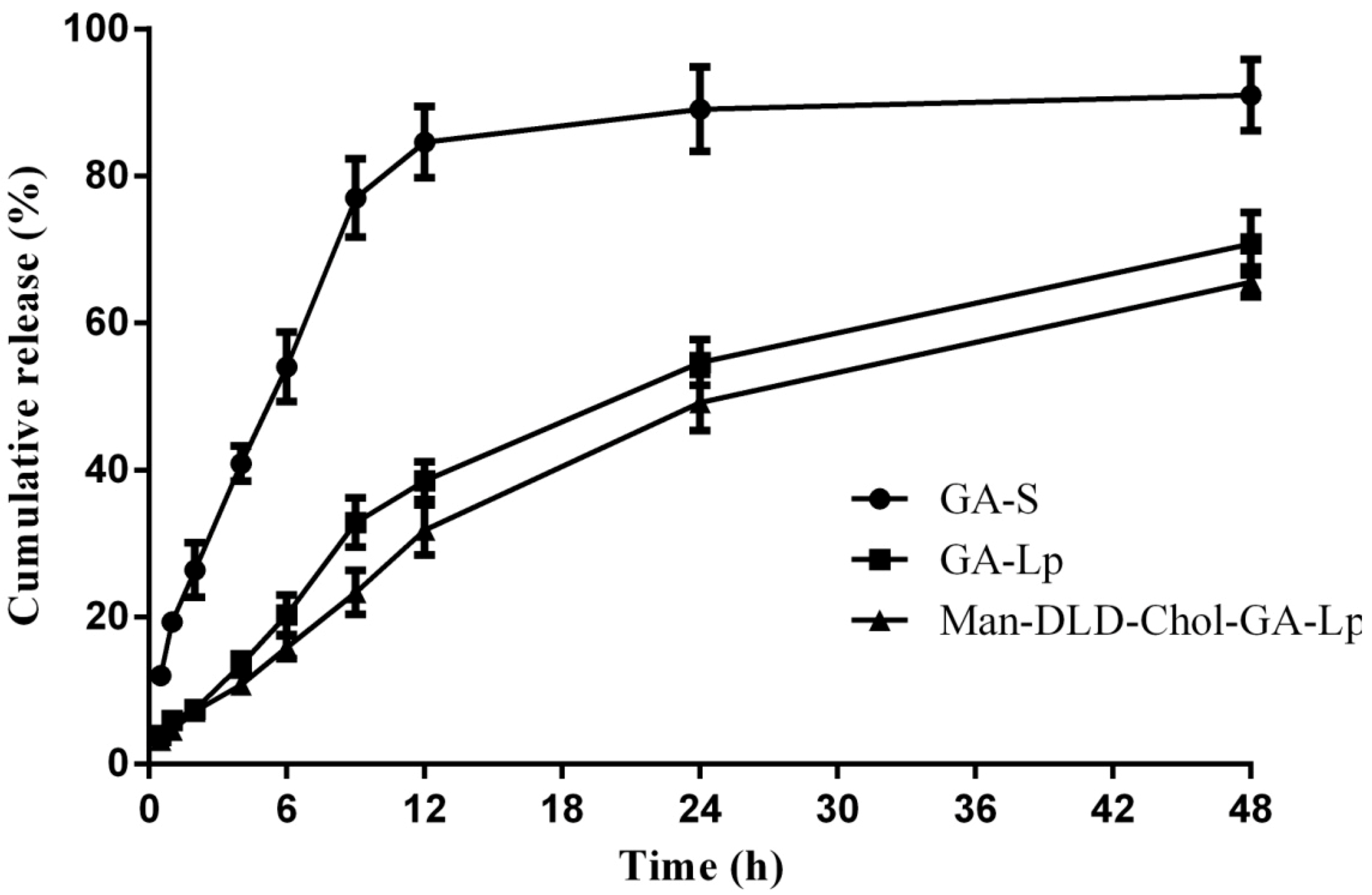
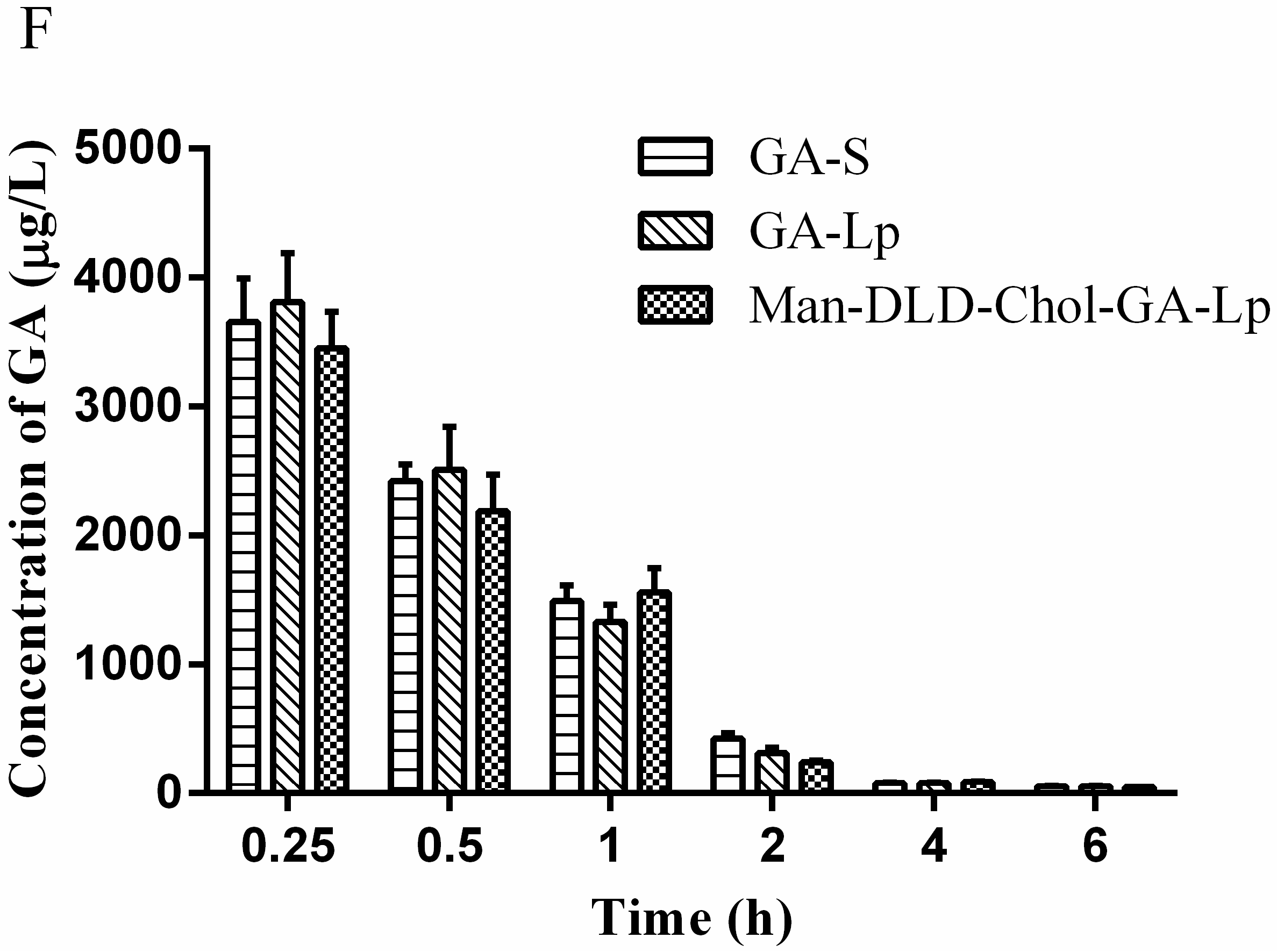
2.4. Drug Release of Liposome In Vitro
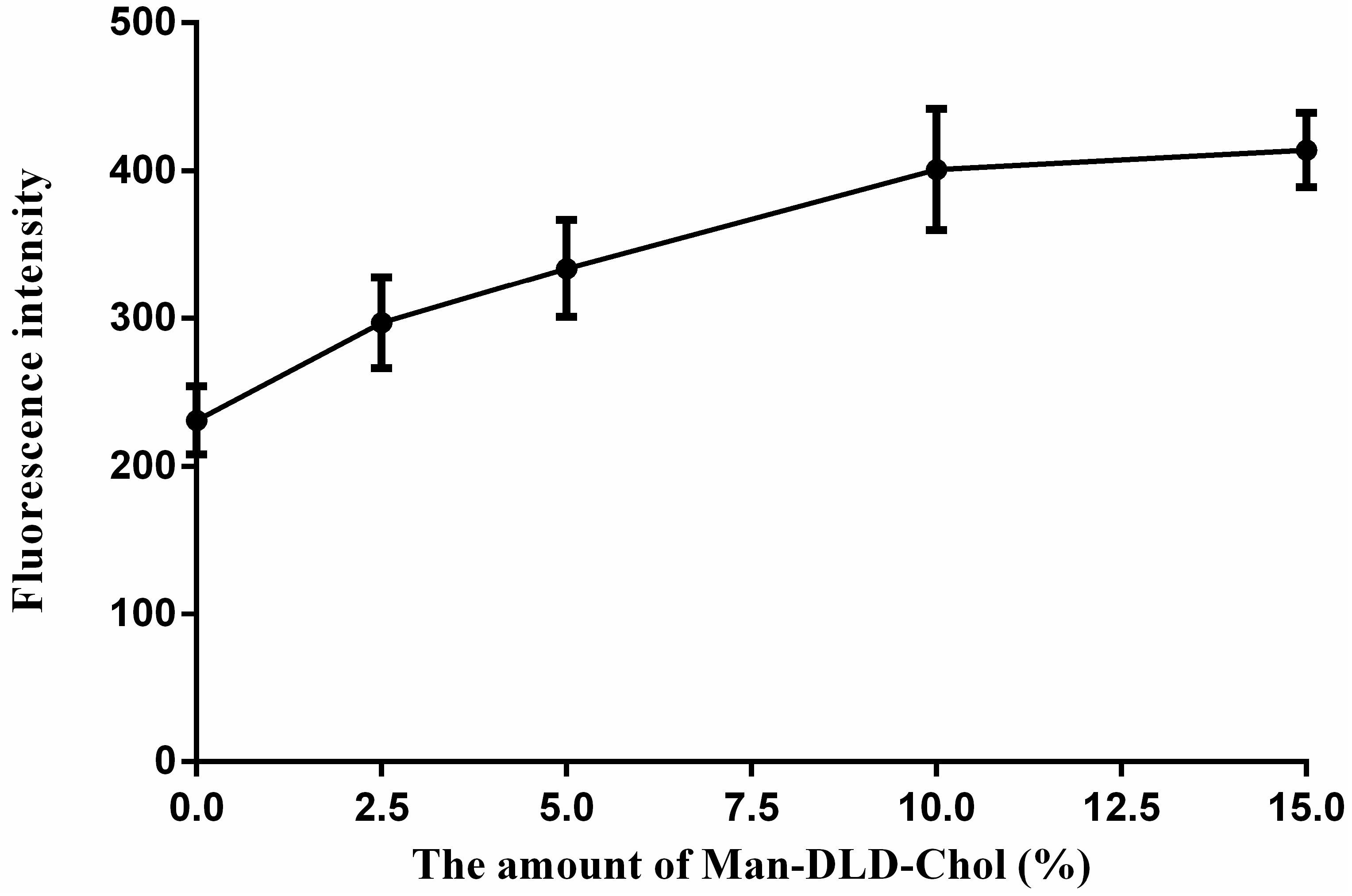
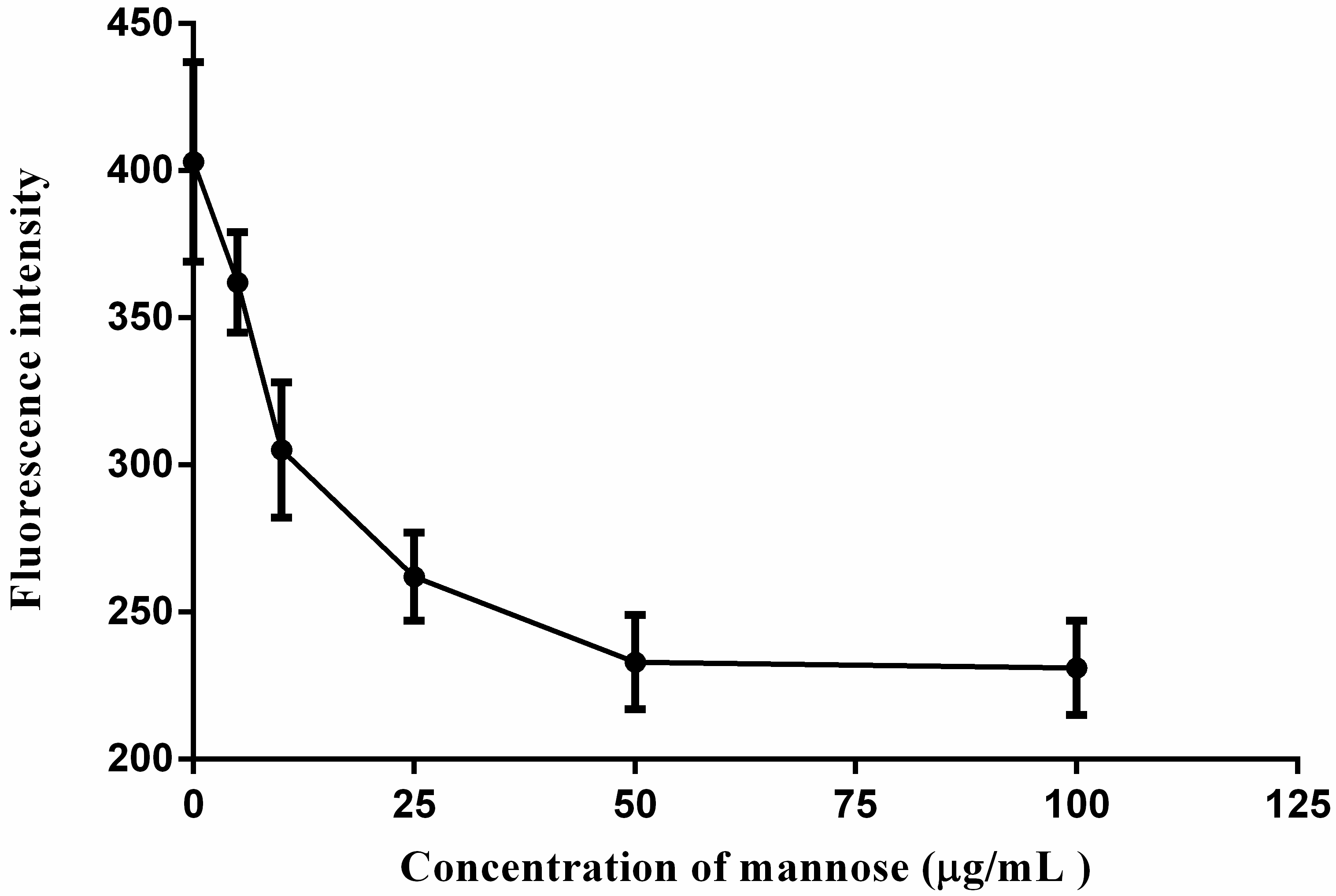
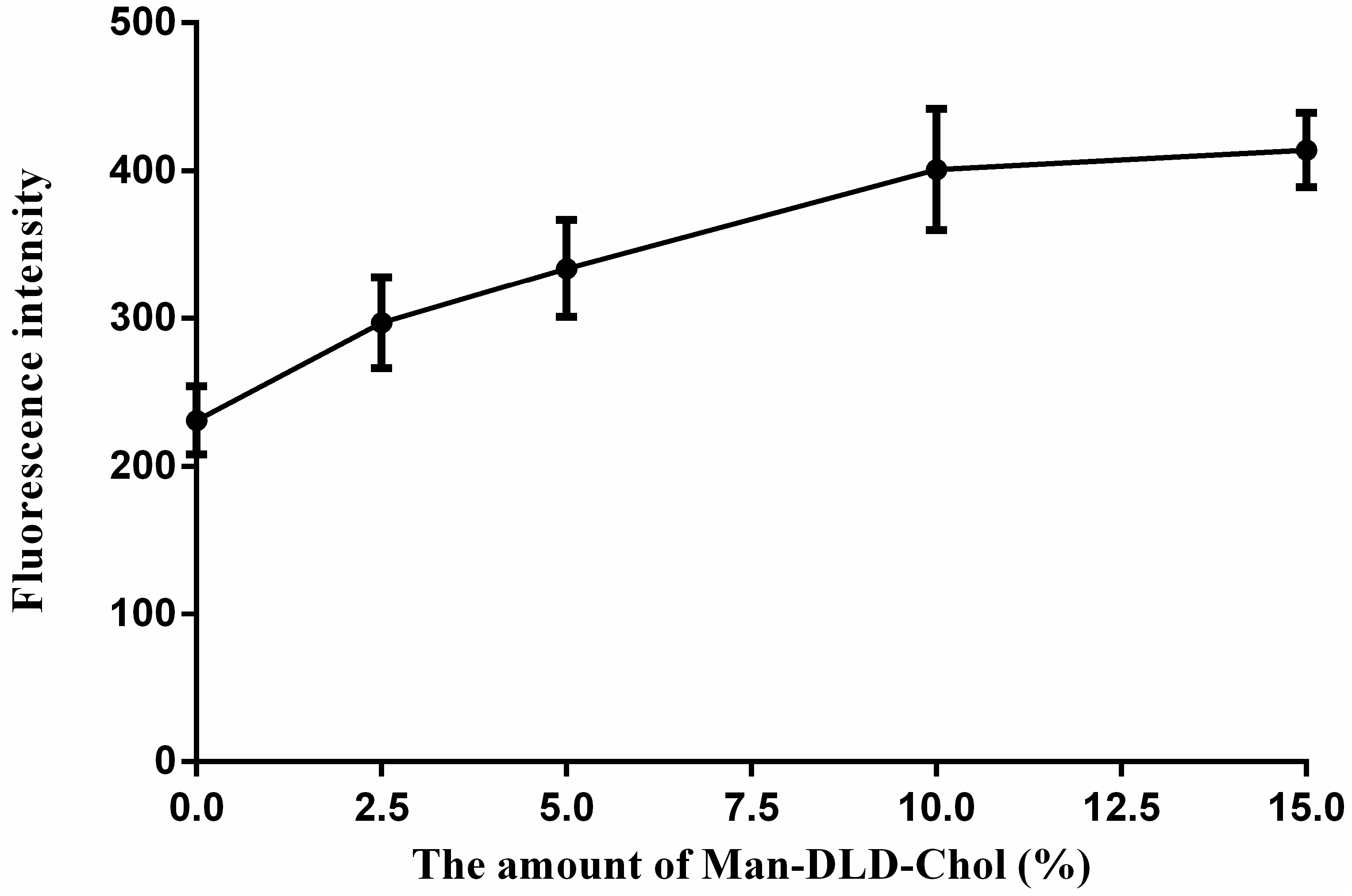
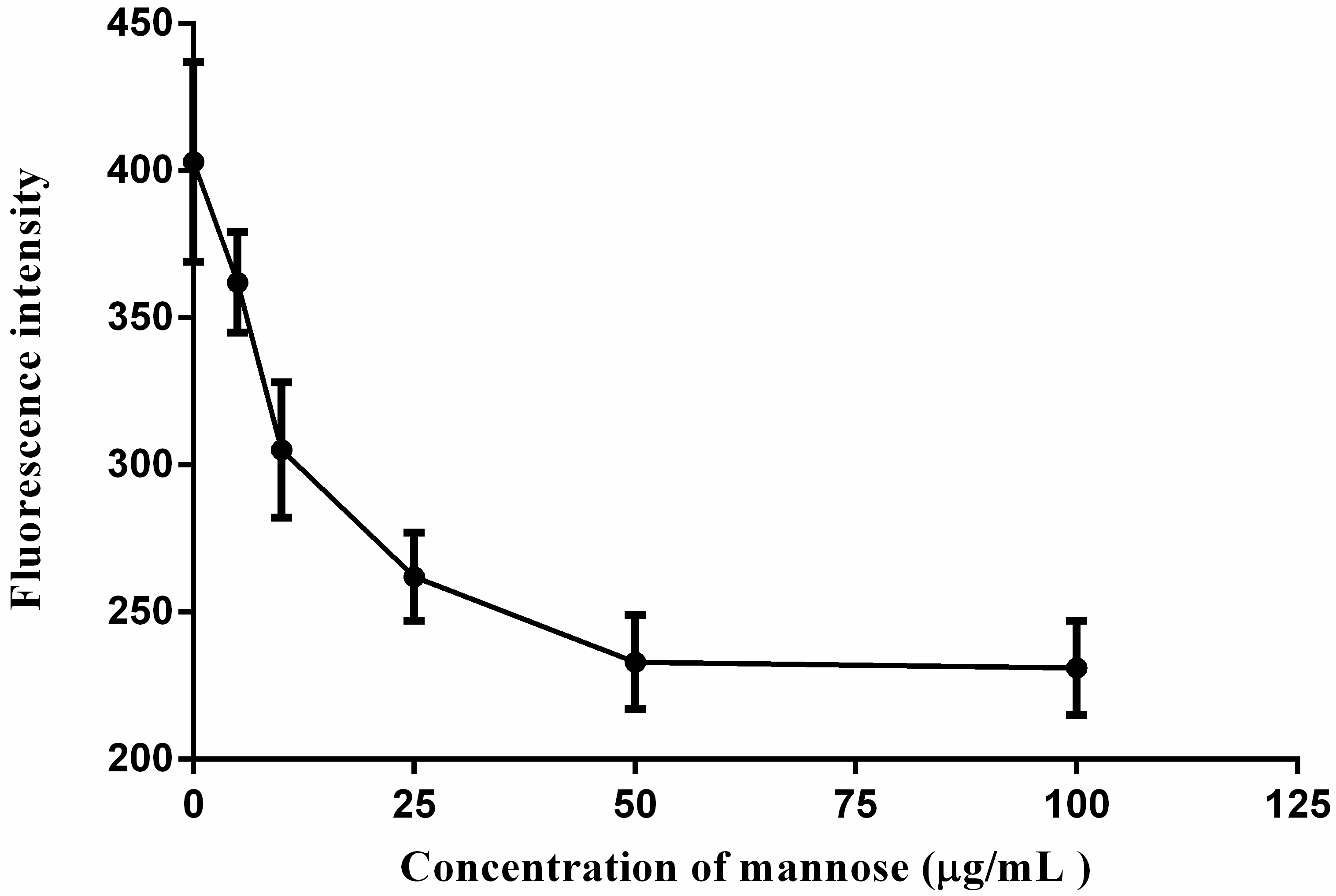
2.5. Cellular Uptakes
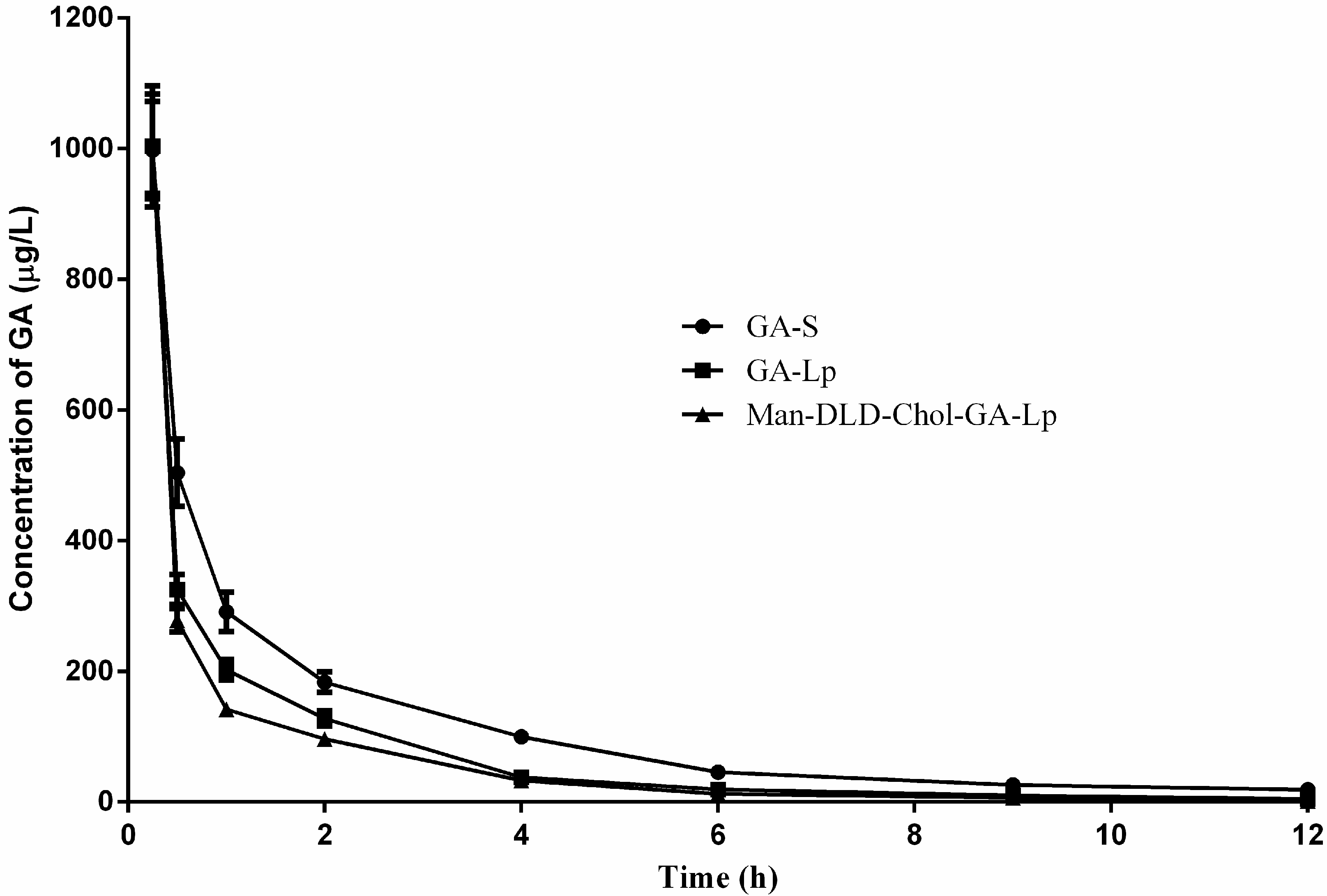
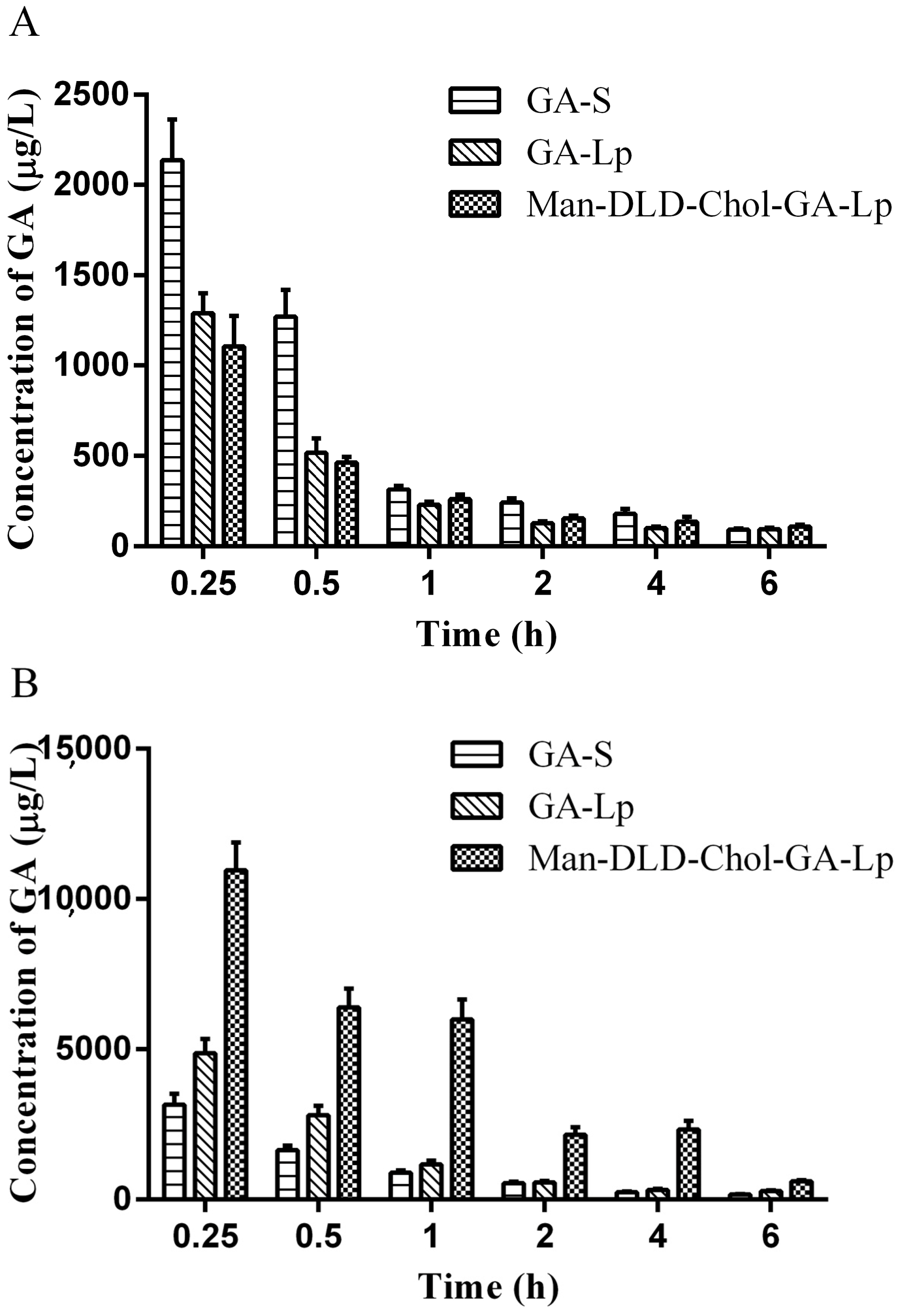
2.6. Pharmacokinetics Study
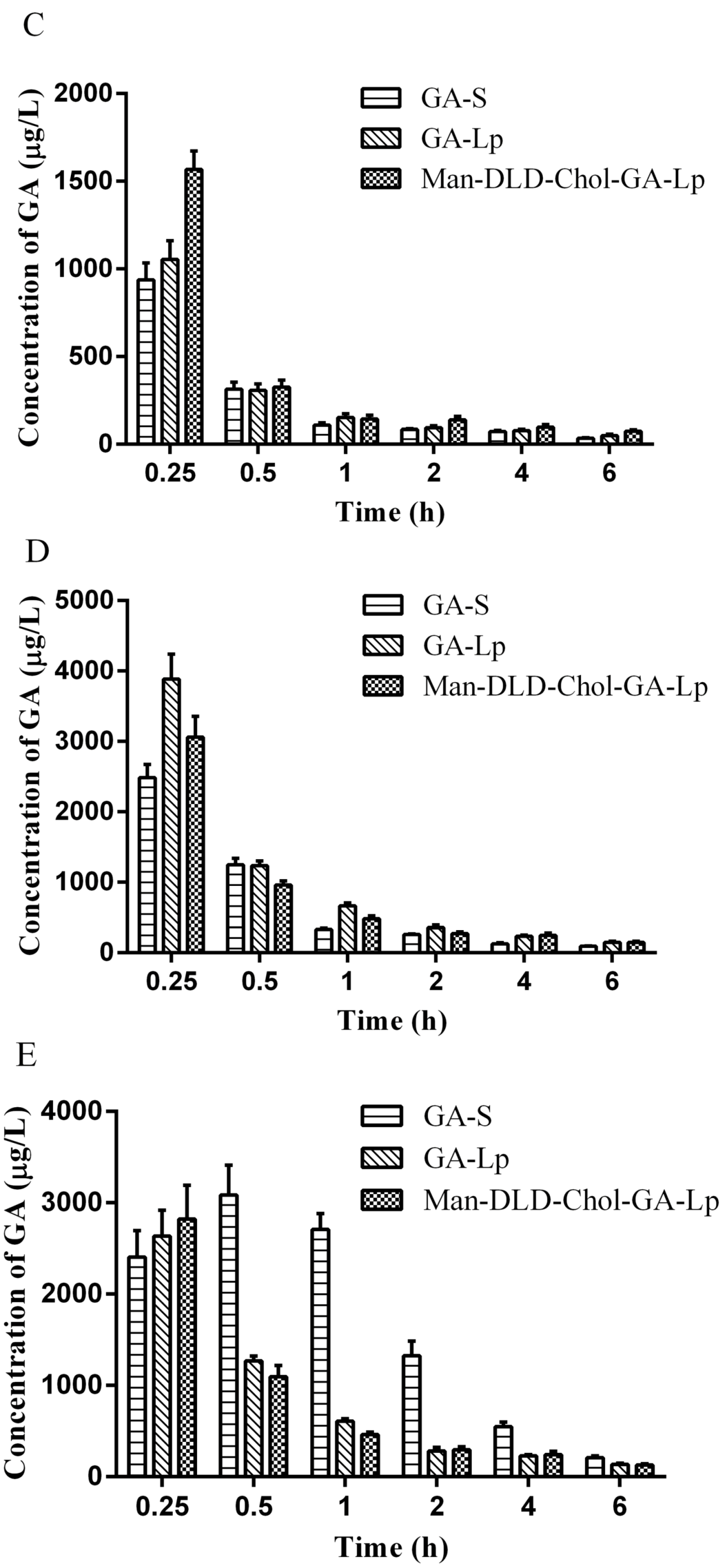
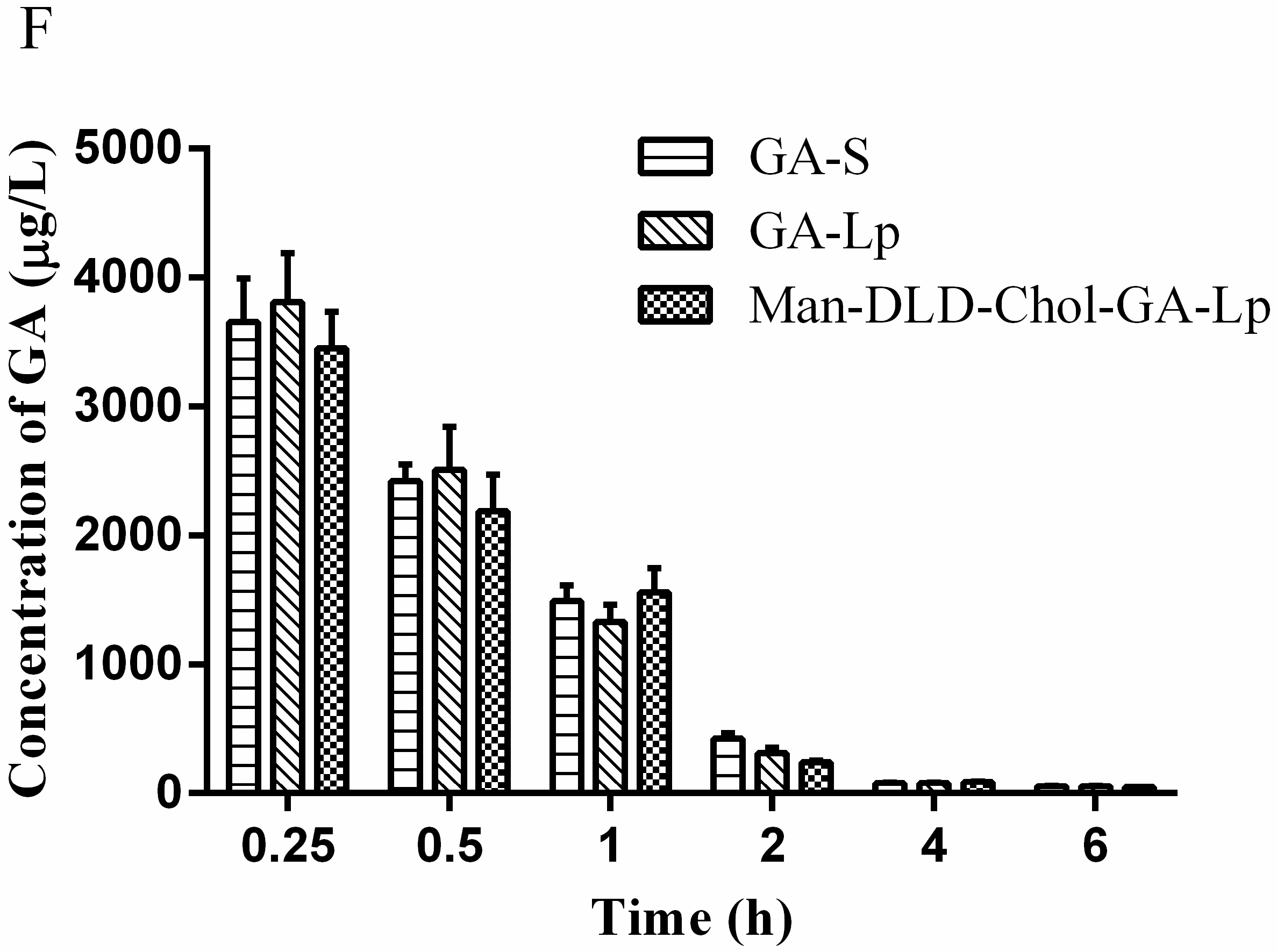
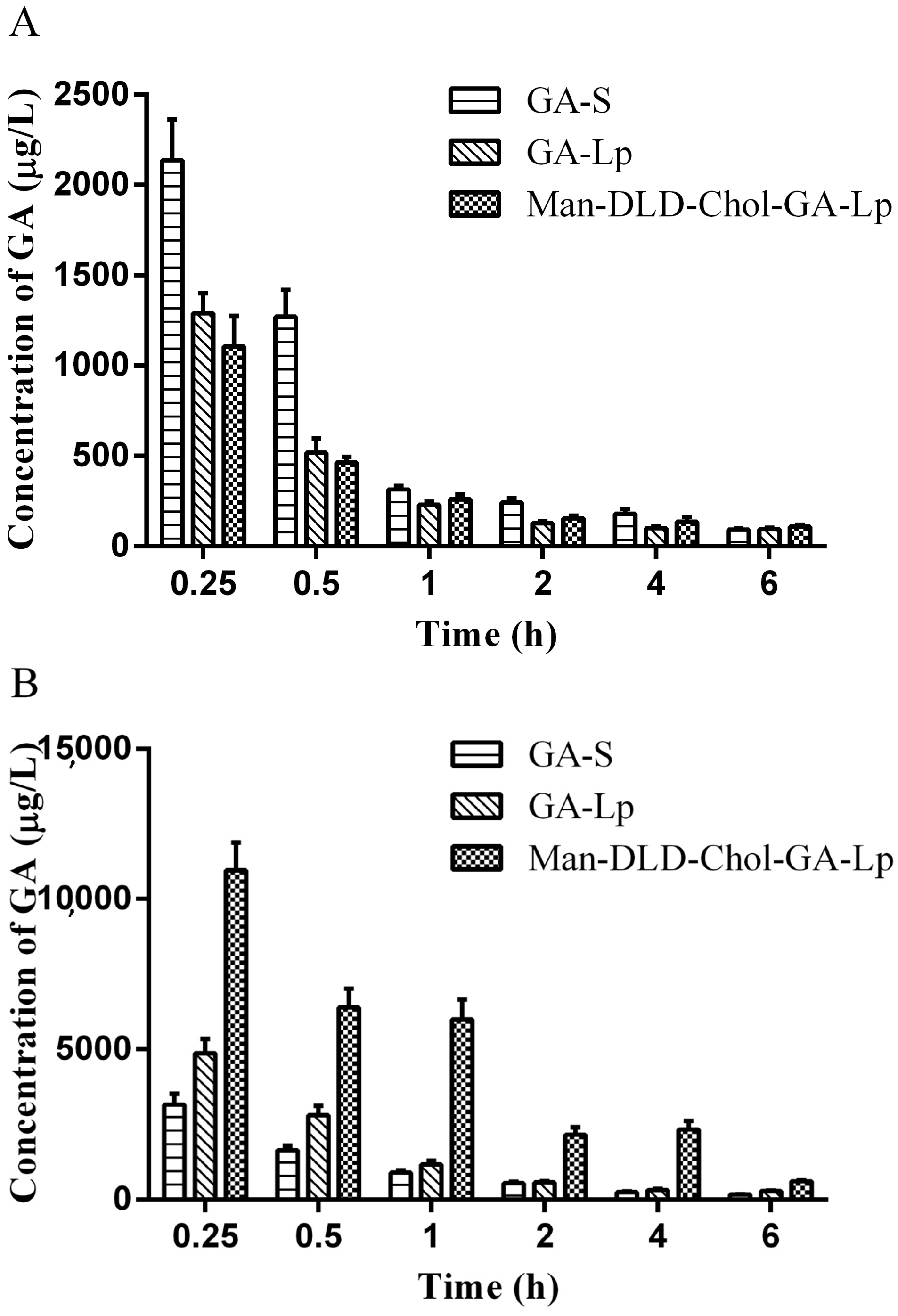
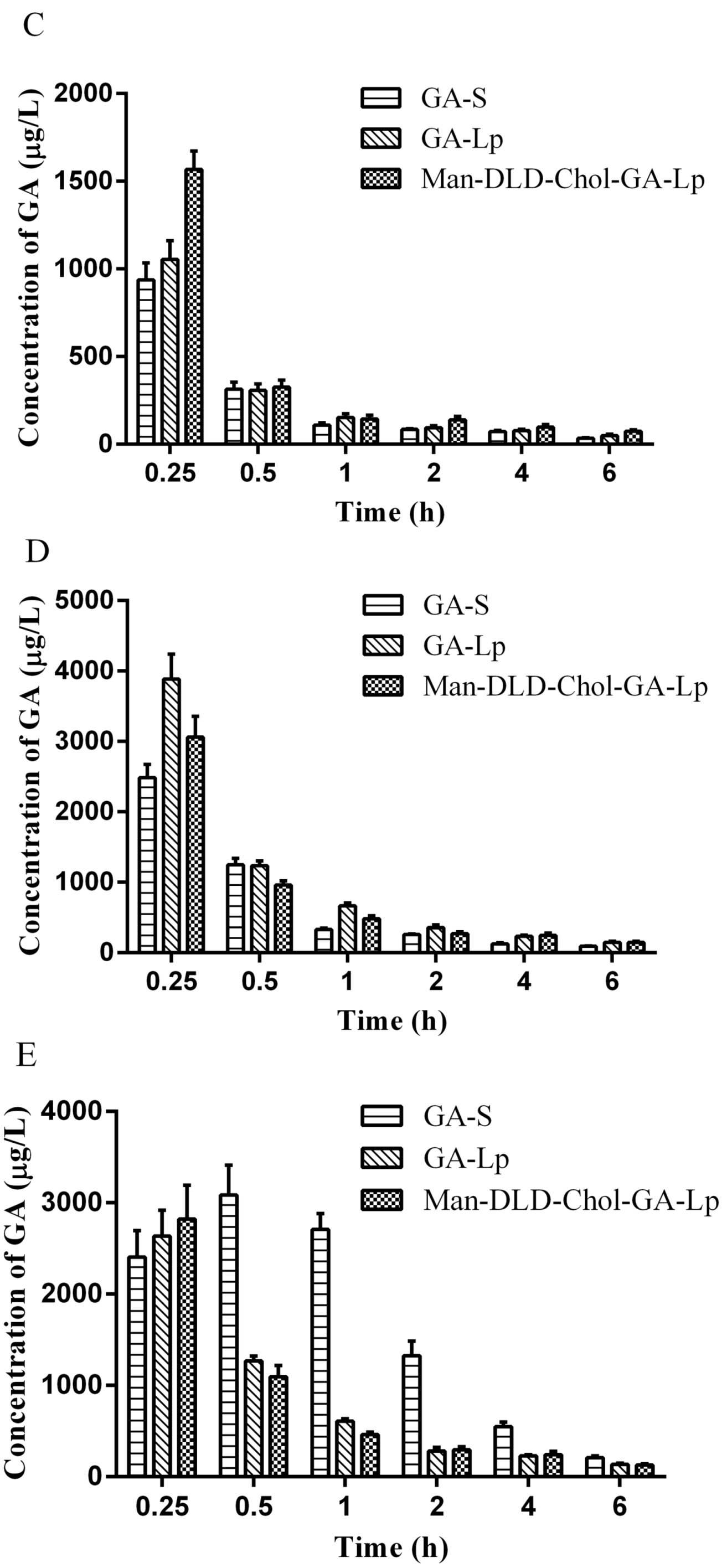
2.7. Tissue Distribution Study
3. Materials and Methods
3.1. Materials
3.2. Synthesis of Man-DLD-Chol
3.3. Liposome Preparation
3.4. Characterization of Liposome
3.5. Hemolytic Study
3.6. Drug Release from Liposome In Vitro
3.7. Cellular Uptakes
3.8. Pharmacokinetic Studies
3.9. Tissue Distributions
3.10. Statistical Analysis
4. Conclusions
Supplementary Materials
Acknowledgments
Author contributions
Conflicts of Interest
References
- Lavanchy, D. Chronic viral hepatitis as a public health issue in the world. Best Pract. Res. Clin. Gastroenterol. 2008, 22, 991–1008. [Google Scholar] [CrossRef] [PubMed]
- Bhatti, A.H.; Dar, F.S.; Waheed, A.; Shafique, K.; Sultan, F.; Shah, N.H. Hepatocellular Carcinoma in Pakistan: National Trend and Global Perspective. Gastroenterol. Res. Pract. 2016, 2016, 5942306. [Google Scholar] [CrossRef]
- Osada, S.; Saji, S.; Kuno, T. Clinical significance of combination study of apoptotic factors and proliferating cell nuclear antigen in estimating the prognosis of hepatocellular carcinoma. J. Surg. Oncol. 2004, 85, 48–54. [Google Scholar] [CrossRef]
- Chen, W.; Zheng, R.S.; Baade, P.D.; Zhang, S.W.; Zeng, H.M.; Bray, F.; Jemal, A.; Yu, X.Q.; He, J. Cancer statistics in China, 2015. CA Cancer J. Clin. 2016, 66, 115–132. [Google Scholar] [CrossRef] [PubMed]
- Jaracz, S.; Chen, J.; Kuznetsova, L.V.; Ojima, I. Recent advances in tumor-targeting anticancer drug conjugates. Bioorg. Med. Chem. 2005, 13, 5043–5054. [Google Scholar] [CrossRef] [PubMed]
- Kiso, Y.; Tohkin, M.; Hikino, H.; Hattori, M.; Sakamoto, T.; Namba, T. Mechanism of antihepatotoxic activity of glycyrrhizin. I: Effect on free radical generation and lipid peroxidation. Planta Med. 1984, 50, 298–302. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.G.; You, S.J.; Moon, A.R.; Chung, Y.C.; Kang, K.; Chun, H.K. Hepatoprotective effects of 18beta-glycyrrhetinic acid on carbon tetrachloride-induced liver injury: Inhibition of cytochrome P450 2E1 expression. Pharmacol. Res. 2002, 46, 221–227. [Google Scholar] [CrossRef]
- Shim, S.B.; Kim, N.J.; Kim, D.H. Beta-glucuronidase inhibitory activity and hepatoprotective effect of 18beta–glycyrrhetinic acid from the rhizomes of glycyrrhiza uralensis. Planta Med. 2000, 66, 40–43. [Google Scholar] [CrossRef] [PubMed]
- Kuang, P.H.; Zhao, W.X.; Su, W.X.; Zhang, L.; Liu, J.M.; Ren, G.L.; Yim, Z.Y.; Wang, X.M. 18beta-Glycyrrhetinic acid inhibits hepatocellular carcinoma development by reversing hepatic stellate cell-mediated immunosuppression in mice. Int. J. Cancer 2013, 132, 1831–1841. [Google Scholar] [CrossRef] [PubMed]
- Satomi, Y.; Nishino, H.; Shibata, S. Glycyrrhetinic acid and related compounds induce G1 arrest and apoptosis in human hepatocellular carcinoma HepG2. Anticancer Res. 2005, 25, 4043–4047. [Google Scholar] [PubMed]
- Cao, L.; Ding, W.; Jia, R.; Du, J.L.; Wang, T.; Zhang, C.Y.; Gu, Z.Y.; Yin, G.J. Anti-inflammatory and hepatoprotective effects of glycyrrhetinic acid on CCl4-induced damage in precision-cut liver slices from Jian carp (Cyprinus carpio var. jian) through inhibition of the nf-kƁ pathway. Fish Shellfish Immun. 2017, 64, 234–242. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.M.; Lee, C.K. Antiviral activity of glycyrrhetinic acid on MA-104 cell infection of the K-21 Korea human rotavirus isolate. J. Exp. Biomed. Sci. 2006, 12, 209–215. [Google Scholar]
- Abe, N.; Ebina, T.; Ishida, N. Interferon induction by glycyrrhizin and glycyrrhetinic acid in mice. Microbiol. Immunol. 1982, 26, 535–539. [Google Scholar] [CrossRef] [PubMed]
- Serra, A.; Vehlinger, D.E.; Ferrari, P.; Dick, B.; Frey, B.M.; Frey, F.J.; Vogt, B. Glycyrrhetinic acid decreases plasma potassium concentrations in patients in patients with anuria. J. Am. Soc. Nephrol. 2002, 13, 191–196. [Google Scholar] [PubMed]
- Allen, T.M.; Cullis, P.R. Liposomal drug delivery systems: From concept to clinical applications. Adv. Drug Deliv. Rev. 2013, 65, 36–48. [Google Scholar] [CrossRef] [PubMed]
- Vijaykumar, N.; Sandeep, K. Recent Advances in Liposomal Drug Delivery: A Review. Pharm. Nanotechnol. 2015, 3, 25–55. [Google Scholar]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef] [PubMed]
- Xiao, B.; Wang, X.Y.; Qiu, Z.Y.; Man, J.; Zhou, L.; Wan, Y.; Zhang, S.M. A dual-functionally modified chitosan derivative for efficient liver-targeted geng delivery. J. Biomed. Res. A 2013, 101, 1888–1897. [Google Scholar] [CrossRef]
- Jia, H.J.; Jia, F.Y.; Zhu, B.J.; Zhang, W.P. Preparation and characterization of Glycyrrhetinic-acid loaded PEG-modified liposome based on PEG-7 glyceryl cocoate. Eur. J. Lipid Sci. Technol. 2017, 119. [Google Scholar] [CrossRef]
- Lu, Y.; Li, J.; Wang, G.J. In vitro and in vivo evaluation of mPEG-PLA modified liposomes loaded glycyrrhetinic acid. Int. J. Pharm. 2008, 356, 274–281. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.E.; Xu, Y.Q.; Chan, H.F.; Fang, X.B.; He, C.W.; Chen, M.W. Glycyrrhetinic Acid Mediated Drug Delivery Carriers for Hepatocellular Carcinoma Therapy. Mol. Pharm. 2016, 13, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Stang, E.; Kindberg, G.M.; Berg, T.; Roos, N. Endocytosis mediated by the mannose receptor in liver endothelial cells. A immunocytochemical study. Eur. J. Cell Biol. 1990, 52, 67–76. [Google Scholar] [PubMed]
- Diebold, S.S.; Plank, C.; Cotton, M.; Wanger, E.; Zenke, M. Mannose receptor-mediated gene delivery into antigen presenting dendritic cells. Somat. Cell Mol. Genet. 2002, 27, 65–74. [Google Scholar] [CrossRef] [PubMed]
- Ashwell, G.; Harford, J. Carbohydrate-Specific Receptors of the Liver. Annu. Rev. Biochem. 1982, 51, 531–554. [Google Scholar] [CrossRef] [PubMed]
- Taylor, M.E.; Conary, J.T.; Lennartz, M.R.; Stahl, P.D.; Drickmer, K. Primary structure of the mannose receptor contains multiple motifs resembling carbohydrate-recognition domains. J. Biol. Chem. 1990, 265, 12156–12162. [Google Scholar] [PubMed]
- Taylor, M.E.; Drickamer, K. Structural requirement for high affinity binding of complex ligands by the macrophage mannose receptor. J. Biol. Chem. 1993, 268, 399–404. [Google Scholar] [PubMed]
- Samad, A.; Sultana, Y.; Aqil, M. Liposomal Drug Delivery Systems: An Update Review. Curr. Drug Deliv. 2007, 4, 297–305. [Google Scholar] [CrossRef] [PubMed]
- Grit, M.; Crommelin, D.J. Chemical stability of liposomes: Implications for their physical stability. Chem. Phys. Lipids 1993, 64, 3–18. [Google Scholar] [CrossRef]
- Qiao, C.M.; Liu, J.D.; Yang, J.; Li, Y.; Weng, J.; Shao, Y.M.; Zhang, X. Enhanced non-inflammasome mediated immune response by mannosylated zwitterionic-based cationic liposomes for HIV DNA vaccines. Biomaterials 2016, 85, 1–17. [Google Scholar] [CrossRef] [PubMed]
- Barratt, G.; Tenu, J.P.; Yapo, A.; Pettit, J.F. Preparation and characterization of liposomes containing mannosylated phospholipids capable of targeting drugs to macrophages. Biochim. Biophys. Acta 1986, 862, 153–164. [Google Scholar] [CrossRef]
- Zhang, Y.Y.; Mintzer, E.; Uhrich, K.E. Synthesis and characterization of PEGylated bolaamphiphiles with enhanced retention in liposomes. J. Colloid Interf. Sci. 2016, 482, 19–26. [Google Scholar] [CrossRef] [PubMed]
- Sliedregt, L.A.J.M.; Rensen, P.C.N.; Rump, E.T.; van Santbrink, P.J.; Bijsterbosch, M.K.; Valentijn, A.R.P.M.; van der Marel, G.A.; van Boom, J.H.; van Berkel, T.J.C.; Biessen, E.A.L. Design and synthesis of novel amphiphilic dendritic galactosides for selective targeting of liposomes to the hepatic asialoglycoprotein receptor. J. Med. Chem. 1999, 42, 609–618. [Google Scholar] [CrossRef] [PubMed]
- Engel, A.; Chatterjee, S.K.; Al-arifi, A.; Riemann, D.; Langner, J.; Nuhn, P. Influence of spacer length on interaction of mannosylated liposomes with human phagocytic cells. Pharm. Res. 2003, 20, 51–57. [Google Scholar] [CrossRef] [PubMed]
- Jeong, H.S.; Na, K.S.; Hwang, H.; Oh, P.S.; Kim, D.H.; Lim, S.T.; Sohn, M.H.; Jeong, H.J. Effect of space length of mannose ligand on uptake of mannosylated liposome in RAW 264.7 cells: In vitro and in vivo studies. J. Biomed. Mater. Res. A 2014, 102, 4545–4553. [Google Scholar] [CrossRef] [PubMed]
- Crucianelli, E.; Bruni, P.; Frontini, A.; Massaccesi, L.; Pasani, M.; Smorlesi, A.; Mobbili, G. Liposomes containing mannose-6-phosphate-cholesteryl conjugates for lysosome-specific delivery. RSC Adv. 2014, 4, 58204–58207. [Google Scholar] [CrossRef]
- Wang, N.; Wang, T.; Zhang, M.L.; Chen, R.N.; Nu, R.W.; Deng, Y.H. Mannose derivative and lipid A dually decorated cationic liposomes as an effective cold chain free oral mucosal vaccine adjuvant-delivery system. Eur. J. Pharm. Biopharm. 2014, 88, 194–206. [Google Scholar] [CrossRef] [PubMed]
- Nguyen, H.; Katavic, P.; Bashah, N.A.H.; Ferro, V. Synthesis of mannose-cholesterol conjugates for targeted liposomal drug delivery. Chem. Sel. 2016, 1, 31–35. [Google Scholar] [CrossRef]
- Ferrer, M.; Cruces, M.A.; Plou, F.J.; Pastor, E.; Fuentes, G.; Bernabé, M.; Parra, J.; Ballesteros, A. Chemical versus enzymatic catalysis for the regioselective synthesis of sucrose esters of fatty acids. Stud. Surf. Sci. Catal. 2000, 130, 509–514. [Google Scholar] [CrossRef]
- Zaks, A.; Klibanov, A.M. Enzymatic catalysis in nonaqueous solvents. J. Biol. Chem. 1988, 263, 3194–3201. [Google Scholar] [PubMed]
- Klibanov, A.M. Improving enzymes by using them in organic solvents. Nature 2001, 409, 241–246. [Google Scholar] [CrossRef] [PubMed]
- Ferrer, M.; Plou, F.J.; Lopez-Cortes, N.; Reyes-Duarte, D.; Christensen, M.; Copa-Patino, J.L.; Ballesteros, A. Synthesis of sugar esters in solvent mixtures by lipases from Thermomyces lanuginosus and Candida antarctica B, and their antimicrobial properties. Enzym. Microb. Technol. 2005, 36, 391–398. [Google Scholar] [CrossRef]
- Ohvo-Rekila, H.; Ramstedt, B.; Leppimaki, P.; Slotte, J.P. Cholesterol interactions with phospholipids in membranes. Prog. Lipid Res. 2002, 41, 66–97. [Google Scholar] [CrossRef]
- Nie, H.; Zheng, P.J.; Luo, L.H.; Cheng, Y. Lipase-catalyzed synthesis of cholesterol vinyl hemi-sebacate for selective targeting of liposomes in organic media. Chin. Trad. Herb. Drugs 2013, 44, 3289–3295. [Google Scholar]
- Chen, X.Y.; Zong, M.H.; Lou, W.Y.; Wu, H. Highly efficient regioselective synthesis of 5′-O-lauroyl-5-azacytidine catalyzed by Candida antarctica lipase B. Biochem. Biotechnol. 2008, 151, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Miura, Y.; Ikeda, T.; Kobayashi, K. Chemoenzymatically synthesized glycoconjugate polymers. Biomacromolecules 2003, 4, 410–415. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Mori, A.; Huang, L. Role of liposome size and RES blockade in controlling biodistribution and tumor uptake of GM1-containing liposomes. BBA Biomenbranes 1992, 1104, 95–101. [Google Scholar] [CrossRef]
- Maruyama, K.; Yuda, T.; Okamoto, A.; Kojima, S.; Suginaka, A.; Iwatsuru, M. Prolonged circulation time in vivo of large unilamellar liposomes composed of distearoyl phosphatidylcholine and cholesterol containing amphipathic poly(ethylene glycol). Biochim. Biophys. Acta 1992, 1128, 44–49. [Google Scholar] [CrossRef]
- Maruyama, K. Intracellular targeting delivery of liposomal drugs to solid tumors based on EPR effects. Adv. Drug Deliver. Rev. 2011, 63, 161–169. [Google Scholar] [CrossRef] [PubMed]
- National Pharmacopoeia Committee. Pharmacopoeia of the People’s Republic of China; The Medicine Science and Technology Press of China: Beijing, China, 2015; Volume 4, p. 371. [Google Scholar]
- Guo, B.H.; Cheng, Y.; Lin, L.P.; Lin, D.H.; Wu, W. Preparation and characterization of galactose-modified liposomes by a nonaqueous enzymatic reaction. J. Liposome Res. 2011, 21, 255–260. [Google Scholar] [CrossRef] [PubMed]
- Singodia, D.; Verma, A.; Verma, R.K.; Mishra, P.R. Investigations into an alternate approach to target mannose receptors on macrophages using 4-sulfated N-acetyl galactosamine more efficiently in comparison with mannose-decorated liposomes: An application in drug delivery. Nanomedicine 2012, 8, 468–477. [Google Scholar] [CrossRef] [PubMed]
- Lian, H.; Sun, J.; Yu, Y.P.; Cao, W.; Wang, Y.J.; Sun, Y.H.; Wang, S.L.; He, Z.G. Supramolecular micellar nanoaggregates based on a novel chitosan/vitamin E succinate copolymer for paclitaxel selective delivery. Int. J. Nanomed. 2011, 6, 3323–3334. [Google Scholar] [CrossRef]
- Cai, L.L.; Wang, X.H.; Wang, W.W.; Qiu, N.; Wen, J.L.; Duan, X.M.; Li, X.; Chen, X.; Yang, L.; Qian, Z.Y.; et al. Peptide ligand and PEG-mediated long-circulating liposome targeted to FGFR overexpressing tumor in vivo. Int. J. Nanomed. 2012, 7, 4499–4510. [Google Scholar] [CrossRef]
- Chen, L.Y.; Liu, Y.B.; Wang, W.Y.; Lin, K. Effect of integrin receptor-targeted liposomal paclitaxel for hepatocellular carcinoma targeting and therapy. Int. J. Nanomed. 2015, 10, 77–84. [Google Scholar] [CrossRef] [PubMed]
Sample Availability: Samples of the compounds mannose-diester lauric diacid-cholesterol and glycyrrhetinic acid are available from the authors. |

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Sample | Particlesize(nm) | Zeta Potential (mV) | PDI | EE (%) |
|---|---|---|---|---|
| GA-Lp | 124.37 ± 1.43 | −32.47 ± 1.78 | 0.13 ± 0.00 | 86.34 ± 2.34 |
| Man-DLD-Chol-GA-Lp | 148.10 ± 0.95 | −38.63 ± 1.07 | 0.14 ± 0.01 | 88.18 ± 1.31 |
| Sample | Zero-Order Formula Model | First-Order Formula Model | Higuchi Formula Model |
|---|---|---|---|
| GA-S | 0.753 | −0.992 | 0.889 |
| GA-Lp | 0.941 | −0.984 | 0.990 |
| Man-DLD-Chol-GA-Lp | 0.964 | −0.990 | 0.993 |
| Parameters | GA-S | GA-Lp | Man-DLD-Chol-GA-Lp |
|---|---|---|---|
| AUC0–∞ (μg/L·h) | 1562.72 ± 50.45 | 1053.55 ± 65.44 * | 906.37 ± 48.99 * |
| MRT0-∞ (h) | 2.68 ± 0.12 | 1.77 ± 0.06 * | 1.35 ± 0.05 * |
| t1/2z (h) | 2.65 ± 0.25 | 2.51 ± 0.44 | 1.78 ± 0.08 * |
| CLz (L/(h∙kg)) | 3.36 ± 0.11 | 5.00 ± 0.30 * | 5.81 ± 0.30 * |
| Vd (L/kg) | 12.83 ± 0.88 | 18.15 ± 3.42 * | 14.90 ± 0.55 |
| Biosample | Linear Curve | Linear Coefficient (r2) | Linear Range (ng/mL) |
|---|---|---|---|
| Plasma | Y = 0.0063X + 0.1950 | 0.9991 | 5–5000 |
| Heart | Y = 0.0054X + 0.2093 | 0.9989 | 5–5000 |
| Liver | Y = 0.0056X + 0.4971 | 0.9993 | 5–12,000 |
| Spleen | Y = 0.0059X + 0.1126 | 0.9994 | 5–5000 |
| Lung | Y = 0.0067X + 0.3202 | 0.9991 | 5–5000 |
| Kidney | Y = 0.0050X + 0.2621 | 0.9990 | 5–5000 |
| Parameters | Tissues | GA-S | GA-Lp | Man-DLD-Chol-GA-Lp |
|---|---|---|---|---|
| AUC0–t | Heart | 2.5973 ± 0.1435 | 1.4244 ± 0.0936 * | 1.4482 ± 0.1024 * |
| (mg/L·h) | Liver | 4.0944 ± 0.2211 | 5.8582 ± 0.2376 | 19.5541 ± 0.6356 * |
| Spleen | 0.9401 ± 0.0744 | 1.0280 ± 0.068 | 1.3993 ± 0.1322 * | |
| Lung | 2.6747 ± 0.1262 | 3.7460 ± 0.1737 | 3.0815 ± 0.1363 | |
| Kidney | 7.3979 ± 0.2813 | 3.0836 ± 0.1424 * | 3.053 ± 0.2258 * | |
| Plasma | 7.6867 ± 0.1673 | 7.5861 ± 0.5270 | 7.2320 ± 0.1950 | |
| Cmax (mg/L) | Heart | 2.1362 ± 0.2284 | 1.2913 ± 0.1100 * | 1.1061 ± 0.1705 * |
| Liver | 3.1618 ± 0.355 | 4.8747 ± 0.4704 | 10.9554 ± 0.9288 * | |
| Spleen | 0.9382 ± 0.0954 | 1.0545 ± 0.1058 | 1.5679 ± 0.1045 * | |
| Lung | 2.4842 ± 0.1903 | 3.8845 ± 0.3562 * | 3.0607 ± 0.2937 | |
| Kidney | 3.1537 ± 0.1802 | 2.6341 ± 0.2846 | 2.8216 ± 0.3714 | |
| Plasma | 3.6551 ± 0.3398 | 3.8124 ± 0.3767 | 3.4521 ± 0.2840 |
| Parameter | Heart | Liver | Spleen | Lung | Kidney | Plasma |
|---|---|---|---|---|---|---|
| Te (%) | 10.23 | 16.13 | 3.70 | 10.53 | 29.14 | 30.27 |
| Parameter | Formulation | Heart | Liver | Spleen | Lung | Kidney | Plasma |
|---|---|---|---|---|---|---|---|
| Te (%) | GA-Lp | 6.27 | 25.78 | 4.52 | 16.48 | 13.57 | 33.38 |
| Man-DLD-Chol-GA-Lp | 4.05 | 54.67 | 3.91 | 8.62 | 8.54 | 20.22 | |
| RTe | GA-Lp | 0.61 | 1.60 | 1.22 | 1.56 | 0.47 | 1.10 |
| Man-DLD-Chol-GA-Lp | 0.40 | 3.39 | 1.06 | 0.82 | 0.29 | 0.67 | |
| Re | GA-Lp | 0.55 | 1.43 | 1.09 | 1.40 | 0.42 | 0.99 |
| Man-DLD-Chol-GA-Lp | 0.56 | 4.78 | 1.49 | 1.15 | 0.41 | 0.94 | |
| Ce | GA-Lp | 0.60 | 1.54 | 1.12 | 1.56 | 0.84 | 1.04 |
| Man-DLD-Chol-GA-Lp | 0.52 | 3.46 | 1.67 | 1.23 | 0.89 | 0.94 |
© 2017 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chen, J.; Chen, Y.; Cheng, Y.; Gao, Y. Glycyrrhetinic Acid Liposomes Containing Mannose-Diester Lauric Diacid-Cholesterol Conjugate Synthesized by Lipase-Catalytic Acylation for Liver-Specific Delivery. Molecules 2017, 22, 1598. https://doi.org/10.3390/molecules22101598
Chen J, Chen Y, Cheng Y, Gao Y. Glycyrrhetinic Acid Liposomes Containing Mannose-Diester Lauric Diacid-Cholesterol Conjugate Synthesized by Lipase-Catalytic Acylation for Liver-Specific Delivery. Molecules. 2017; 22(10):1598. https://doi.org/10.3390/molecules22101598
Chicago/Turabian StyleChen, Jing, Yuchao Chen, Yi Cheng, and Youheng Gao. 2017. "Glycyrrhetinic Acid Liposomes Containing Mannose-Diester Lauric Diacid-Cholesterol Conjugate Synthesized by Lipase-Catalytic Acylation for Liver-Specific Delivery" Molecules 22, no. 10: 1598. https://doi.org/10.3390/molecules22101598