
Statistical Correlations between HPLC Activity-Based Profiling Results and NMR/MS Microfraction Data to Deconvolute Bioactive Compounds in Mixtures

 ,
,  , , and
, , and
Abstract
:
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Results and Discussion
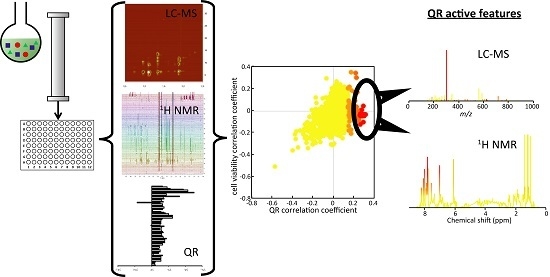
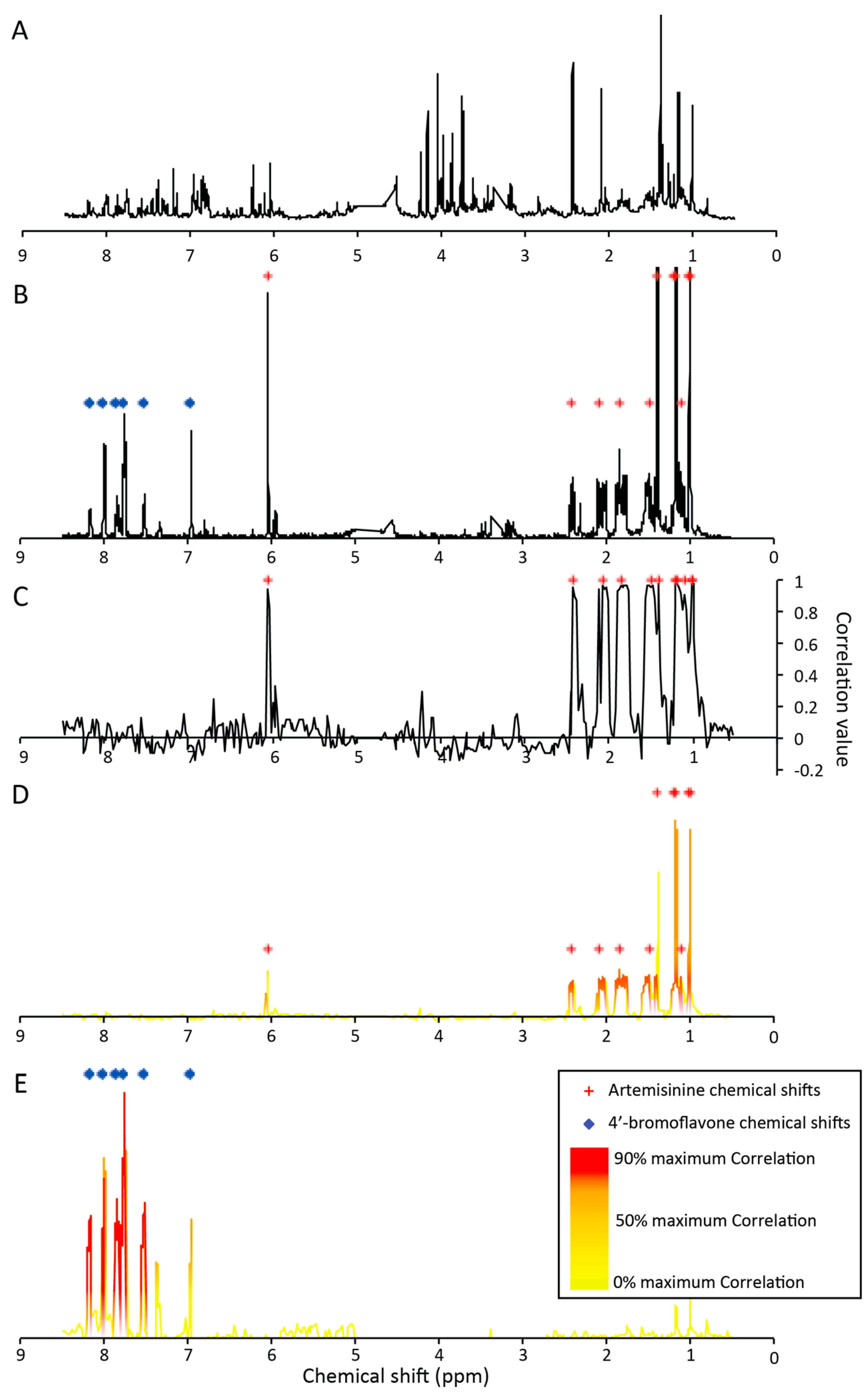
2.1. Microfractionation Combined with Statistical HeterospectroscopY to Improve Compound Identification
2.2. Microfractionation Combined with Biological Profiling to Highlight Relevant Biologically Active Compounds
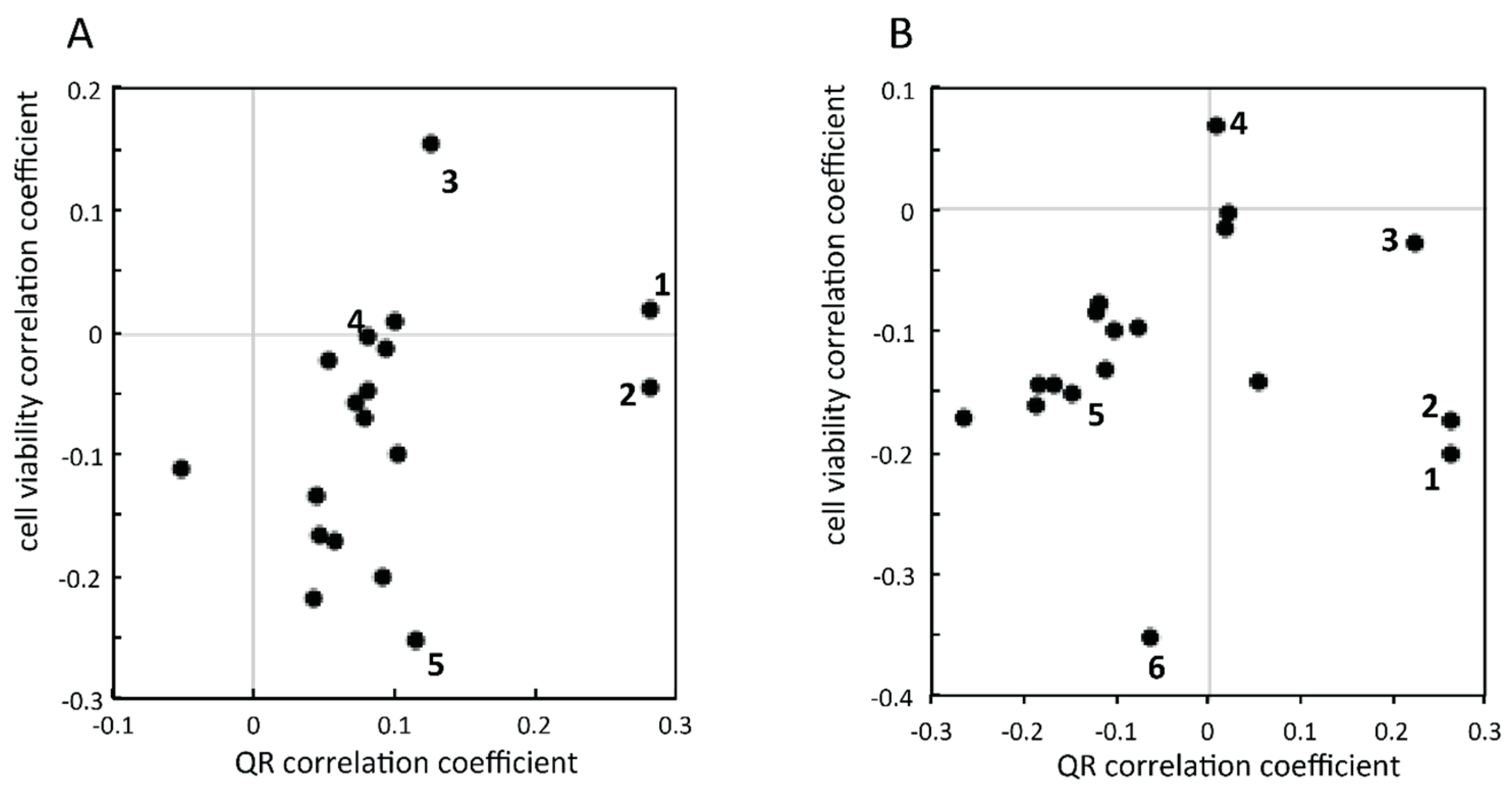
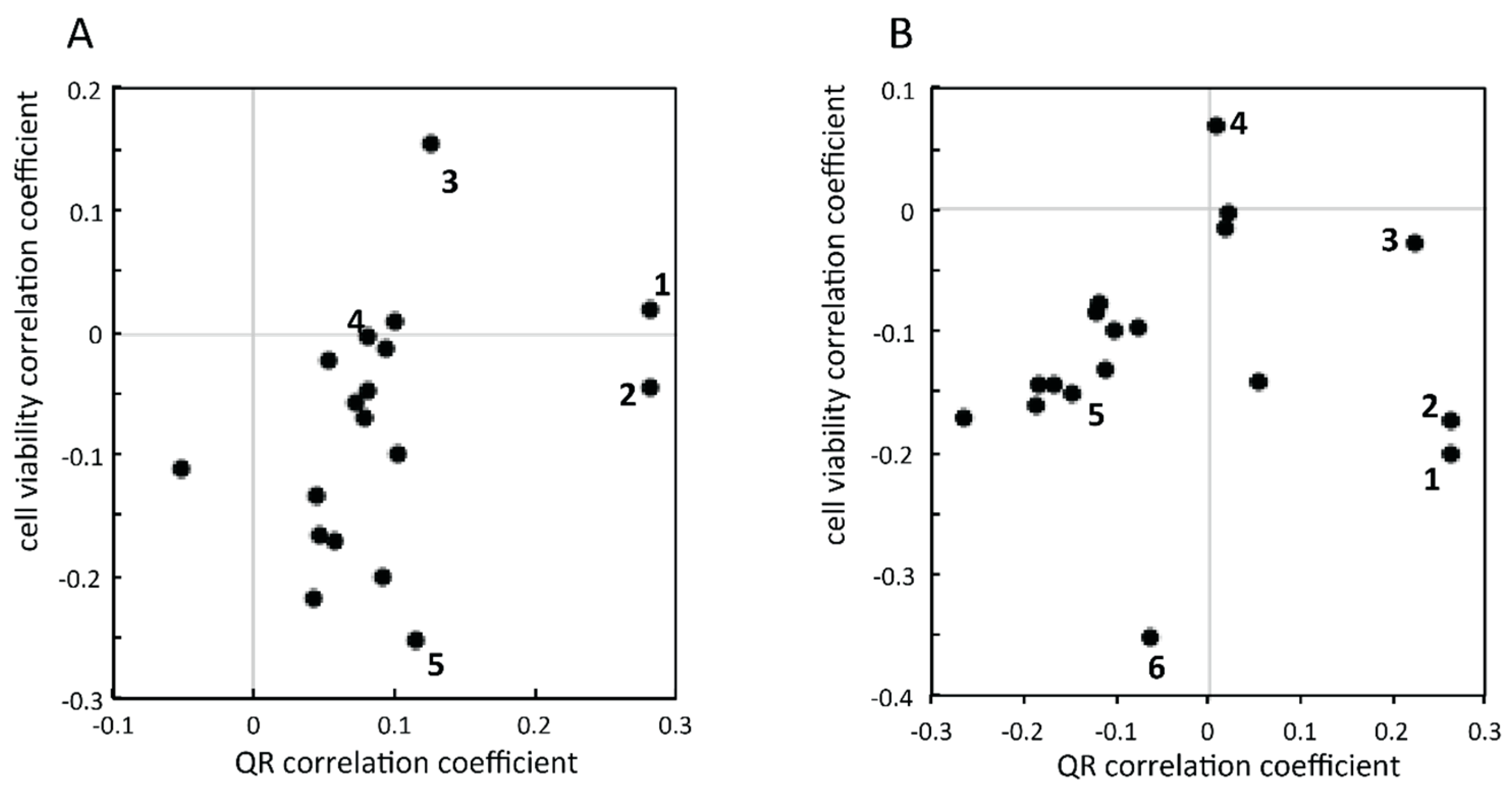
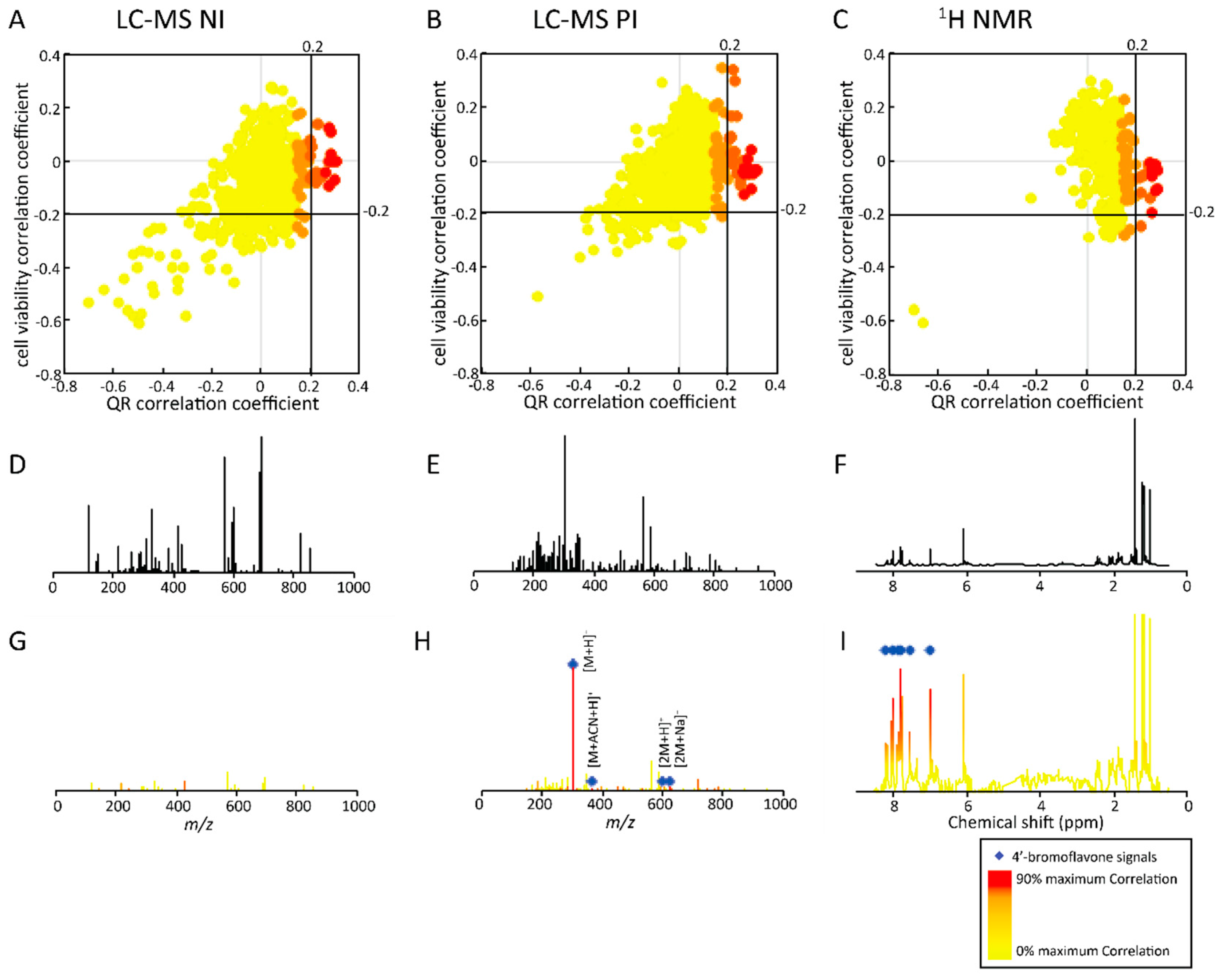
2.2.1. Targeted Deconvolution of Biologically Active Compounds Using LC-MS Data
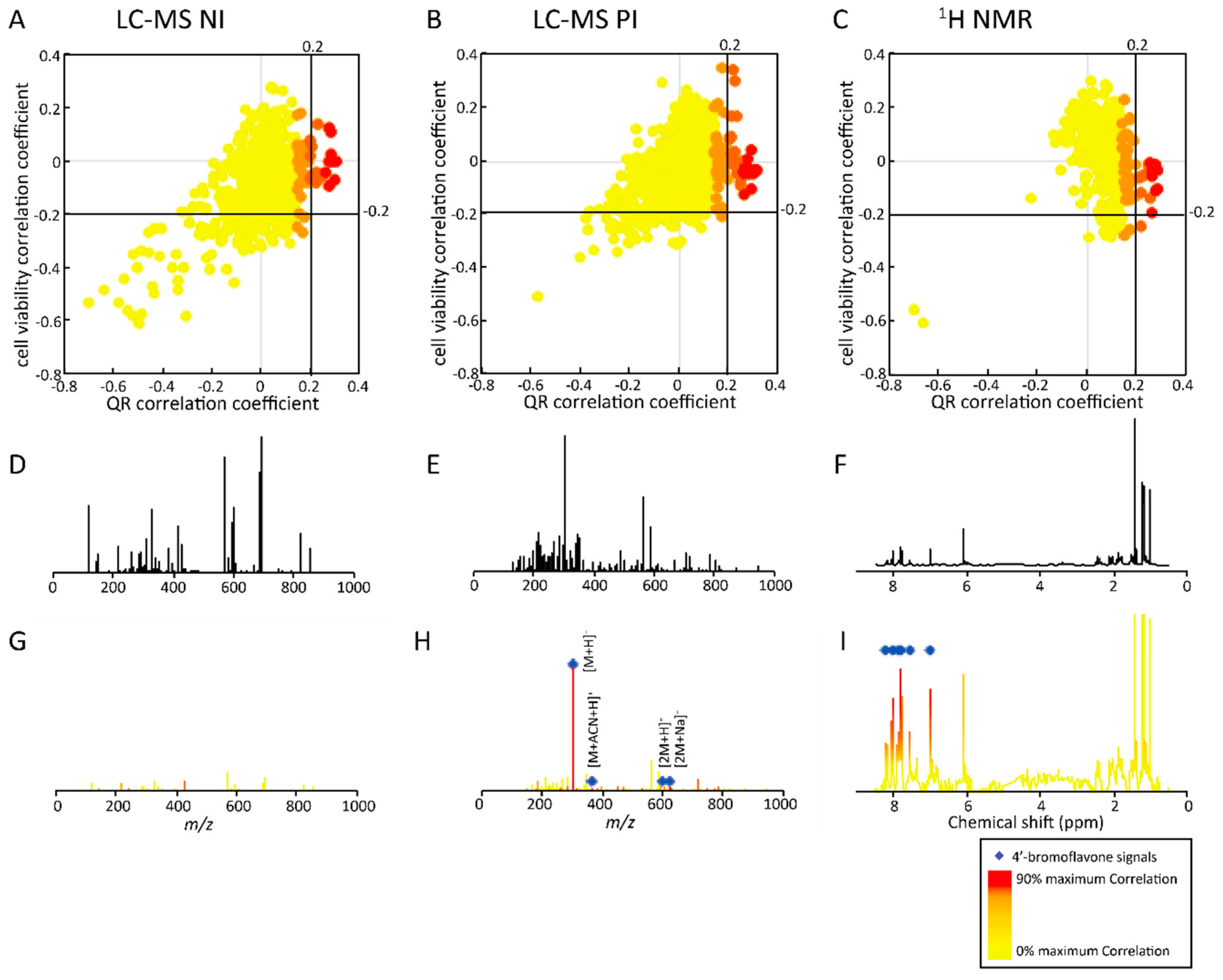
2.2.2. Untargeted Deconvolution of Biologically Active Compounds Using LC-MS Data
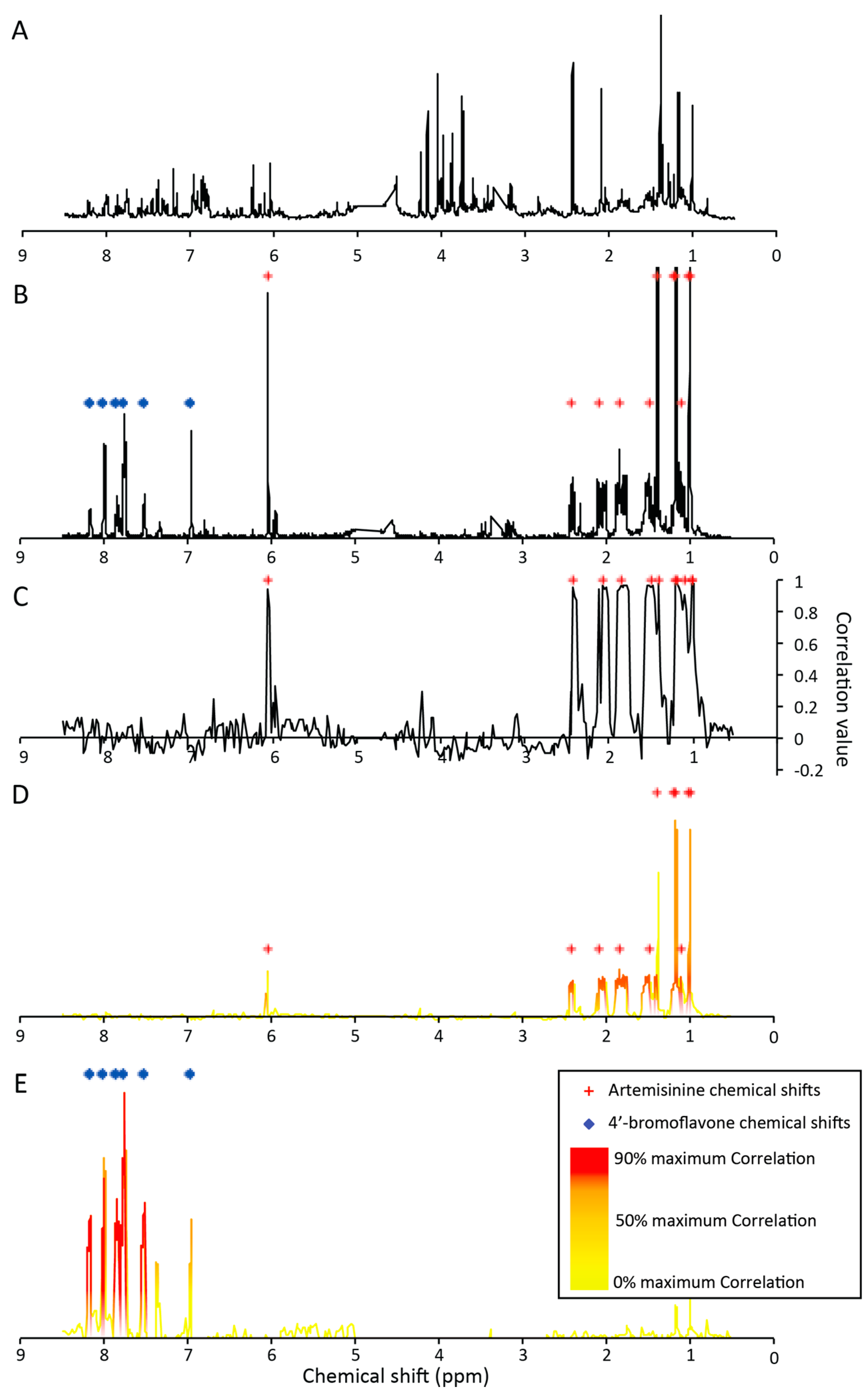
2.2.3. Untargeted Deconvolution of Biologically Active Compounds Using 1H-NMR Data
3. Experimental Section
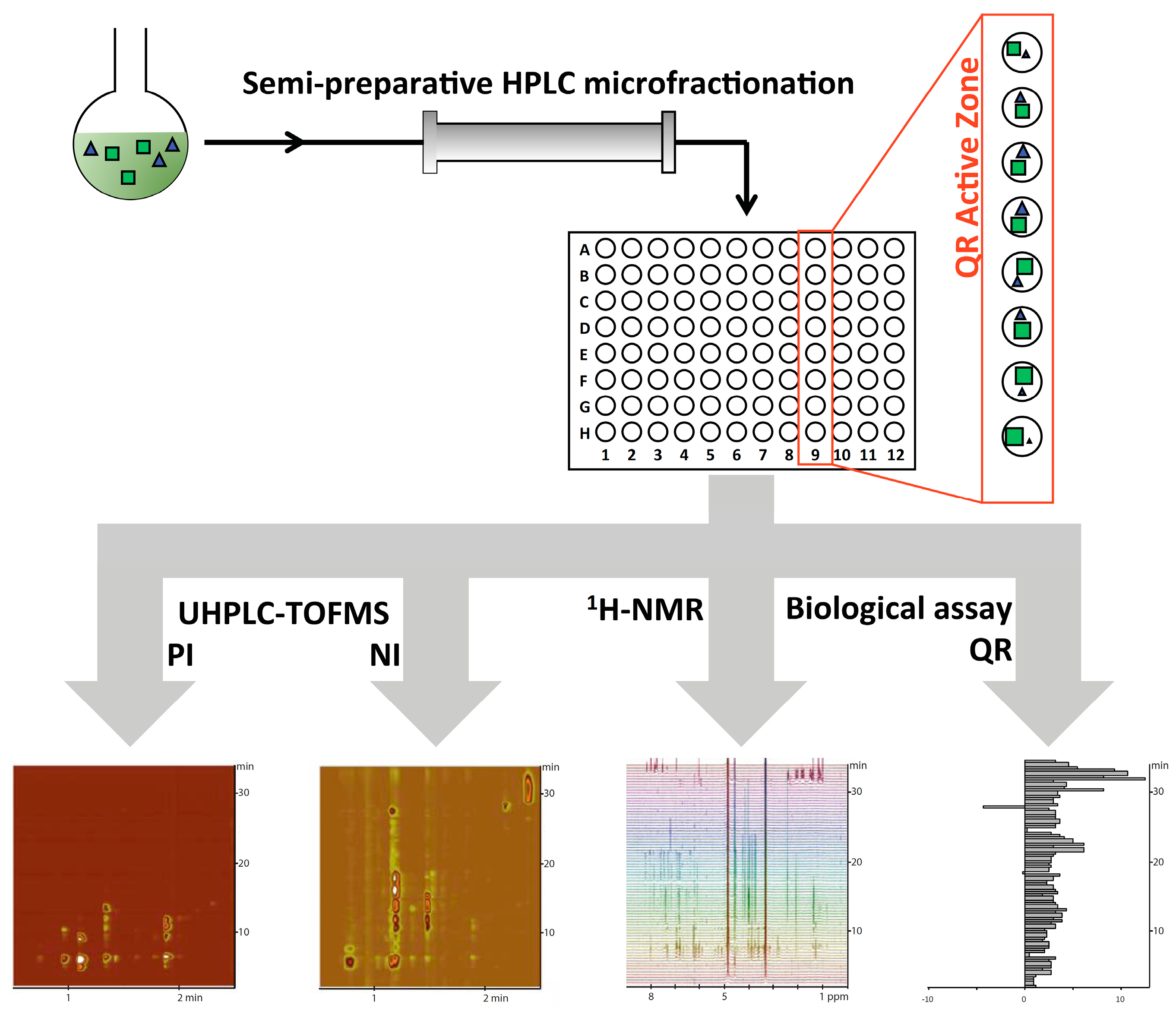
3.1. Creation of the Artificial Extract
3.2. Semi-Preparative Microfractionation of the Synthetic Extract by Reversed Phase HPLC
3.3. UHPLC-TOFMS Profiling of the Microfractions
3.4. Targeted and Untargeted Peak Picking of LC-MS Data
3.5. 1H-NMR Profiling of the Microfractions and NMR Data Matrix Creation
3.6. Quinone Reductase Induction Activity
3.7. Statistical Correlation
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Newman, D.J.; Cragg, G.M. Natural products as sources of new drugs over the 30 years from 1981 to 2010. J. Nat. Prod. 2012, 75, 311–335. [Google Scholar] [CrossRef] [PubMed]
- Rosén, J.; Gottfries, J.; Muresan, S.; Backlund, A.; Oprea, T.I. Novel chemical space exploration via natural products. J. Med. Chem. 2009, 52, 1953–1962. [Google Scholar] [CrossRef] [PubMed]
- Zhu, F.; Qin, C.; Tao, L.; Liu, X.; Shi, Z.; Ma, X.; Jia, J.; Tan, Y.; Cui, C.; Lin, J.; et al. Clustered patterns of species origins of nature-derived drugs and clues for future bioprospecting. Proc. Natl. Acad. Sci. USA 2011, 108, 12943–12948. [Google Scholar] [CrossRef] [PubMed]
- David, B.; Wolfender, J.-L.; Dias, D.A. The pharmaceutical industry and natural products: Historical status and new trends. Phytochem. Rev. 2014, 14, 299–315. [Google Scholar] [CrossRef]
- Koehn, F.E.; Carter, G.T. The evolving role of natural products in drug discovery. Nat. Rev. Drug Discov. 2005, 4, 206–220. [Google Scholar] [CrossRef] [PubMed]
- Lam, K.S. New aspects of natural products in drug discovery. Trends Microbiol. 2007, 15, 279–289. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-Sabin, J. Natural products: Back to the future in drug discovery. Biochem. Pharmacol. 2012, 1, e112. [Google Scholar]
- Roemer, T.; Xu, D.; Singh, S.B.; Parish, C.A.; Harris, G.; Wang, H.; Davies, J.E.; Bills, G.F. Confronting the challenges of natural product-based antifungal discovery. Chem. Biol. 2011, 18, 148–164. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, S.; Schumpp, O.; Bohni, N.; Monod, M.; Gindro, K.; Wolfender, J.-L. De novo production of metabolites by fungal co-culture of Trichophyton rubrum and Bionectria ochroleuca. J. Nat. Prod. 2013, 76, 1157–1165. [Google Scholar] [CrossRef] [PubMed]
- Glauser, G.; Guillarme, D.; Grata, E.; Boccard, J.; Thiocone, A.; Carrupt, P.-A.; Veuthey, J.-L.; Rudaz, S.; Wolfender, J.-L. Optimized liquid chromatography-mass spectrometry approach for the isolation of minor stress biomarkers in plant extracts and their identification by capillary nuclear magnetic resonance. J. Chromatogr. A 2008, 1180, 90–98. [Google Scholar] [CrossRef] [PubMed]
- Potterat, O.; Hamburger, M. Combined use of extract libraries and HPLC-based activity profiling for lead discovery: potential, challenges, and practical considerations. Planta Med. 2014, 80, 1171–1181. [Google Scholar] [CrossRef] [PubMed]
- Potterat, O.; Hamburger, M. Concepts and technologies for tracking bioactive compounds in natural product extracts: Generation of libraries, and hyphenation of analytical processes with bioassays. Nat. Prod. Rep. 2013, 30, 546–564. [Google Scholar] [CrossRef] [PubMed]
- Kool, J.; Giera, M.; Irth, H.; Niessen, W.A. Advances in mass spectrometry-based post-column bioaffinity profiling of mixtures. Anal. Bioanal. Chem. 2011, 399, 2655–2668. [Google Scholar] [CrossRef] [PubMed]
- Simões-Pires, C.A.; Queiroz, E.F.; Henriques, A.T.; Hostettmann, K. Isolation and on-line identification of anti-oxidant compounds from three Baccharis species by HPLC-UV-MS/MS with post-column derivatisation. Phytochem. Anal. 2005, 16, 307–314. [Google Scholar] [CrossRef] [PubMed]
- Rhee, I.K.; Appels, N.; Luijendijk, T.; Irth, H.; Verpoorte, R. Determining acetylcholinesterase inhibitory activity in plant extracts using a fluorimetric flow assay. Phytochem. Anal. 2003, 14, 145–149. [Google Scholar] [CrossRef] [PubMed]
- De Boer, A.R.; Alcaide-Hidalgo, J.M.; Krabbe, J.G.; Kolkman, J.; van Emde Boas, C.N.; Niessen, W.M.A.; Lingeman, H.; Irth, H. High-temperature liquid chromatography coupled on-line to a continuous-flow biochemical screening assay with electrospray ionization mass spectrometric detection. Anal. Chem. 2005, 77, 7894–7900. [Google Scholar] [CrossRef] [PubMed]
- Wubshet, S.G.; Nyberg, N.T.; Tejesvi, M.V.; Pirttilä, A.M.; Kajula, M.; Mattila, S.; Staerk, D. Targeting high-performance liquid chromatography-high-resolution mass spectrometry-solid-phase extraction−nuclear magnetic resonance analysis with high-resolution radical scavenging profiles-bioactive secondary metabolites from the endophytic fungus Penicillium namyslowskii. J. Chromatogr. A 2013, 1302, 34–39. [Google Scholar] [PubMed]
- Wolfender, J.-L.; Marti, G.; Thomas, A.; Bertrand, S. Current approaches and challenges for the metabolite profiling of complex natural extracts. J. Chromatogr. A 2015, 1382, 136–164. [Google Scholar] [CrossRef] [PubMed]
- Bohni, N.; Cordero-Maldonado, M.L.; Maes, J.; Siverio-Mota, D.; Marcourt, L.; Munck, S.; Kamuhabwa, A.R.; Moshi, M.J.; Esguerra, C.V.; de Witte, P.A.M.; et al. Integration of microfractionation, QNMR and zebrafish screening for the in vivo bioassay-guided isolation and quantitative bioactivity analysis of natural products. PLoS ONE 2013, 8, e64006. [Google Scholar] [CrossRef] [PubMed]
- Challal, S.; Buenafe, O.E.M.; Queiroz, E.F.; Maljevic, S.; Marcourt, L.; Bock, M.; Kloeti, W.; Dayrit, F.M.; Harvey, A.L.; Lerche, H.; et al. Zebrafish-bioassay guided microfractionation identifies anticonvulsant steroid glycosides from the Philippine medicinal plant Solanum torvum. ACS Chem. Neurosci. 2014, 5, 993–1004. [Google Scholar] [CrossRef] [PubMed]
- El-Elimat, T.; Figueroa, M.; Ehrmann, B.M.; Cech, N.B.; Pearce, C.J.; Oberlies, N.H. High-resolution MS, MS/MS, and UV database of fungal secondary metabolites as a dereplication protocol for bioactive natural products. J. Nat. Prod. 2013, 76, 1709–1716. [Google Scholar] [CrossRef] [PubMed]
- Smyth, W.F.; Smyth, T.J.P.; Ramachandran, V.N.; O’Donnell, F.; Brooks, P. Dereplication of phytochemicals in plants by LC-ESI-MS and ESI-MSn. Trends Anal. Chem. 2012, 33, 46–54. [Google Scholar] [CrossRef]
- Grosso, C.; Jäger, A.K.; Staerk, D. Coupling of a high-resolution monoamine oxidase-A inhibitor assay and HPLC–SPE–NMR for advanced bioactivity profiling of plant extracts. Phytochem. Anal. 2012, 24, 141–147. [Google Scholar] [CrossRef] [PubMed]
- Queiroz, E.F.; Wolfender, J.L.; Atindehou, K.K.; Traore, D.; Hostettmann, K. On-line identification of the antifungal constituents of Erythrina vogelii by liquid chromatography with tandem mass spectrometry, ultraviolet absorbance detection and nuclear magnetic resonance spectrometry combined with liquid chromatographic micro-fractionation. J. Chromatogr. A 2002, 974, 123–134. [Google Scholar] [PubMed]
- Yuliana, N.; Jahangir, M.; Verpoorte, R.; Choi, Y. Metabolomics for the rapid dereplication of bioactive compounds from natural sources. Phytochem. Rev. 2013, 12, 293–304. [Google Scholar] [CrossRef]
- Bertrand, S.; Petit, C.; Marcourt, L.; Ho, R.; Gindro, K.; Monod, M.; Wolfender, J.-L. HPLC profiling with at-line microdilution assay for the early identification of antifungal compounds in plants from French Polynesia. Phytochem. Anal. 2014, 25, 106–112. [Google Scholar] [CrossRef] [PubMed]
- Landreau, A.; Bertrand, S.; Simoes-Pires, C.; Marcourt, L.; dan Bach, T.; Litaudon, M.; Guilet, D.; Richomme, P.; Carrupt, P.-A.; Wolfender, J.-L. Guttiferones as acetylcholinesterase inhibitors retrieved by normal phase HPLC profiling of apolar plant extract. Nat. Prod. Res. 2016. [Google Scholar] [CrossRef]
- Crockford, D.J.; Holmes, E.; Lindon, J.C.; Plumb, R.S.; Zirah, S.; Bruce, S.J.; Rainville, P.; Stumpf, C.L.; Nicholson, J.K. Statistical heterospectroscopy, an approach to the integrated analysis of NMR and UPLC-MS data sets: Application in metabonomic toxicology studies. Anal. Chem. 2006, 78, 363–371. [Google Scholar] [CrossRef] [PubMed]
- Crockford, D.J.; Maher, A.D.; Ahmadi, K.R.; Barrett, A.; Plumb, R.S.; Wilson, I.D.; Nicholson, J.K. 1H-NMR and UPLC-MS(E) statistical heterospectroscopy: Characterization of drug metabolites (xenometabolome) in epidemiological studies. Anal. Chem. 2008, 80, 6835–6844. [Google Scholar] [CrossRef] [PubMed]
- Marti, G.; Boccard, J.; Mehl, F.; Debrus, B.; Marcourt, L.; Merle, P.; Delort, E.; Baroux, L.; Sommer, H.; Rudaz, S.; et al. Comprehensive profiling and marker identification in non-volatile citrus oil residues by mass spectrometry and nuclear magnetic resonance. Food Chem. 2014, 150, 235–245. [Google Scholar] [CrossRef] [PubMed]
- Yuliana, N.; Khatib, A.; Choi, Y.; Verpoorte, R. Metabolomics for bioactivity assessment of natural products. Phytother. Res. 2011, 25, 157–169. [Google Scholar] [CrossRef] [PubMed]
- Wiegandt, A.; Meyer, B. Unambiguous characterization of N-glycans of monoclonal antibody cetuximab by integration of LC-MS/MS and 1H-NMR Spectroscopy. Anal. Chem. 2014, 86, 4807–4814. [Google Scholar] [CrossRef] [PubMed]
- Behnken, H.; Fellenberg, M.; Koetzler, M.; Jirmann, R.; Nagel, T.; Meyer, B. Resolving the problem of chromatographic overlap by 3D cross correlation (3DCC) processing of LC, MS and NMR data for characterization of complex glycan mixtures. Anal. Bioanal. Chem. 2012, 404, 1427–1437. [Google Scholar] [CrossRef] [PubMed]
- Inui, T.; Wang, Y.; Pro, S.M.; Franzblau, S.G.; Pauli, G.F. Unbiased evaluation of bioactive secondary metabolites in complex matrices. Fitoterapia 2012, 83, 1218–1225. [Google Scholar] [CrossRef] [PubMed]
- Guillarme, D.; Nguyen, D.T.T.; Rudaz, S.; Veuthey, J.-L. HPLC Calculator 3.0. Available online: http://www.unige.ch/sciences/pharm/fanal/lcap/telechargement.htm (accessed on 19 February 2016).
- Guillarme, D.; Nguyen, D.T.T.; Rudaz, S.; Veuthey, J.-L. Method transfer for fast liquid chromatography in pharmaceutical analysis: Application to short columns packed with small particle. Part II: Gradient experiments. Eur. J. Pharm. Biopharm. 2008, 68, 430–440. [Google Scholar] [CrossRef] [PubMed]
- Pluskal, T.; Castillo, S.; Villar-Briones, A.; Oresic, M. MZmine 2: Modular framework for processing, visualizing, and analyzing mass spectrometry-based molecular profile data. BMC Bioinf. 2010, 11, 395. [Google Scholar] [CrossRef] [PubMed]
- Van den Berg, R.A.; Hoefsloot, H.C.; Westerhuis, J.A.; Smilde, A.K.; van der Werf, M.J. Centering, scaling, and transformations: Improving the biological information content of metabolomics data. BMC Genom. 2006, 7, 142. [Google Scholar] [CrossRef] [PubMed]
- Bewick, V.; Cheek, L.; Ball, J. Statistics Review 14: Logistic regression. Crit. Care 2005, 9, 112–118. [Google Scholar] [CrossRef] [PubMed]
- Al-Massarani, S.M.; Bertrand, S.; Nievergelt, A.; Mohammed El-Shafae, A.; Abdullah Al-Howiriny, T.; Musayeib Al-Musayeib, N.; Cuendet, M.; Wolfender, J.-L. Acylated pregnane glycosides from Caralluma sinaica. Phytochemistry 2012, 79, 129–140. [Google Scholar] [CrossRef] [PubMed]
- Giacomelli, E.; Bertrand, S.; Nievergelt, A.; Zwick, V.; Simoes-Pires, C.; Marcourt, L.; Rivara-Minten, E.; Cuendet, M.; Bisio, A.; Wolfender, J.-L. Cancer chemopreventive diterpenes from Salvia corrugata. Phytochemistry 2013, 96, 257–264. [Google Scholar] [CrossRef] [PubMed]
- Misico, R.I.; Song, L.L.; Veleiro, A.S.; Cirigliano, A.M.; Tettamanzi, M.C.; Burton, G.; Bonetto, G.M.; Nicotra, V.E.; Silva, G.L.; Gil, R.R.; et al. Induction of quinone reductase by withanolides. J. Nat. Prod. 2002, 65, 677–680. [Google Scholar] [CrossRef] [PubMed]
- Kind, T.; Fiehn, O. Seven golden rules for heuristic filtering of molecular formulas obtained by accurate mass spectrometry. BMC Bioinf. 2007, 8, 105. [Google Scholar] [CrossRef] [PubMed]
- Bertrand, S.; Schumpp, O.; Bohni, N.; Bujard, A.; Azzollini, A.; Monod, M.; Gindro, K.; Wolfender, J.-L. Detection of metabolite induction in fungal co-cultures on solid media by high-throughput differential ultra-high pressure liquid chromatography-time-of-flight mass spectrometry fingerprinting. J. Chromatogr. A 2013, 1292, 219–228. [Google Scholar] [CrossRef] [PubMed]
- Kang, Y.-H.; Pezzuto, J.M. Induction of quinone reductase as a primary screen for natural product anticarcinogens. Methods Enzymol. 2004, 382, 380–414. [Google Scholar] [PubMed]

© 2016 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons by Attribution (CC-BY) license ( http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bertrand, S.; Azzollini, A.; Nievergelt, A.; Boccard, J.; Rudaz, S.; Cuendet, M.; Wolfender, J.-L. Statistical Correlations between HPLC Activity-Based Profiling Results and NMR/MS Microfraction Data to Deconvolute Bioactive Compounds in Mixtures. Molecules 2016, 21, 259. https://doi.org/10.3390/molecules21030259
Bertrand S, Azzollini A, Nievergelt A, Boccard J, Rudaz S, Cuendet M, Wolfender J-L. Statistical Correlations between HPLC Activity-Based Profiling Results and NMR/MS Microfraction Data to Deconvolute Bioactive Compounds in Mixtures. Molecules. 2016; 21(3):259. https://doi.org/10.3390/molecules21030259
Chicago/Turabian StyleBertrand, Samuel, Antonio Azzollini, Andreas Nievergelt, Julien Boccard, Serge Rudaz, Muriel Cuendet, and Jean-Luc Wolfender. 2016. "Statistical Correlations between HPLC Activity-Based Profiling Results and NMR/MS Microfraction Data to Deconvolute Bioactive Compounds in Mixtures" Molecules 21, no. 3: 259. https://doi.org/10.3390/molecules21030259
APA StyleBertrand, S., Azzollini, A., Nievergelt, A., Boccard, J., Rudaz, S., Cuendet, M., & Wolfender, J.-L. (2016). Statistical Correlations between HPLC Activity-Based Profiling Results and NMR/MS Microfraction Data to Deconvolute Bioactive Compounds in Mixtures. Molecules, 21(3), 259. https://doi.org/10.3390/molecules21030259