Surface-Functionalized Hyperbranched Poly(Amido Acid) Magnetic Nanocarriers for Covalent Immobilization of a Bacterial γ-Glutamyltranspeptidase

Abstract
:1. Introduction
2. Results and Discussion
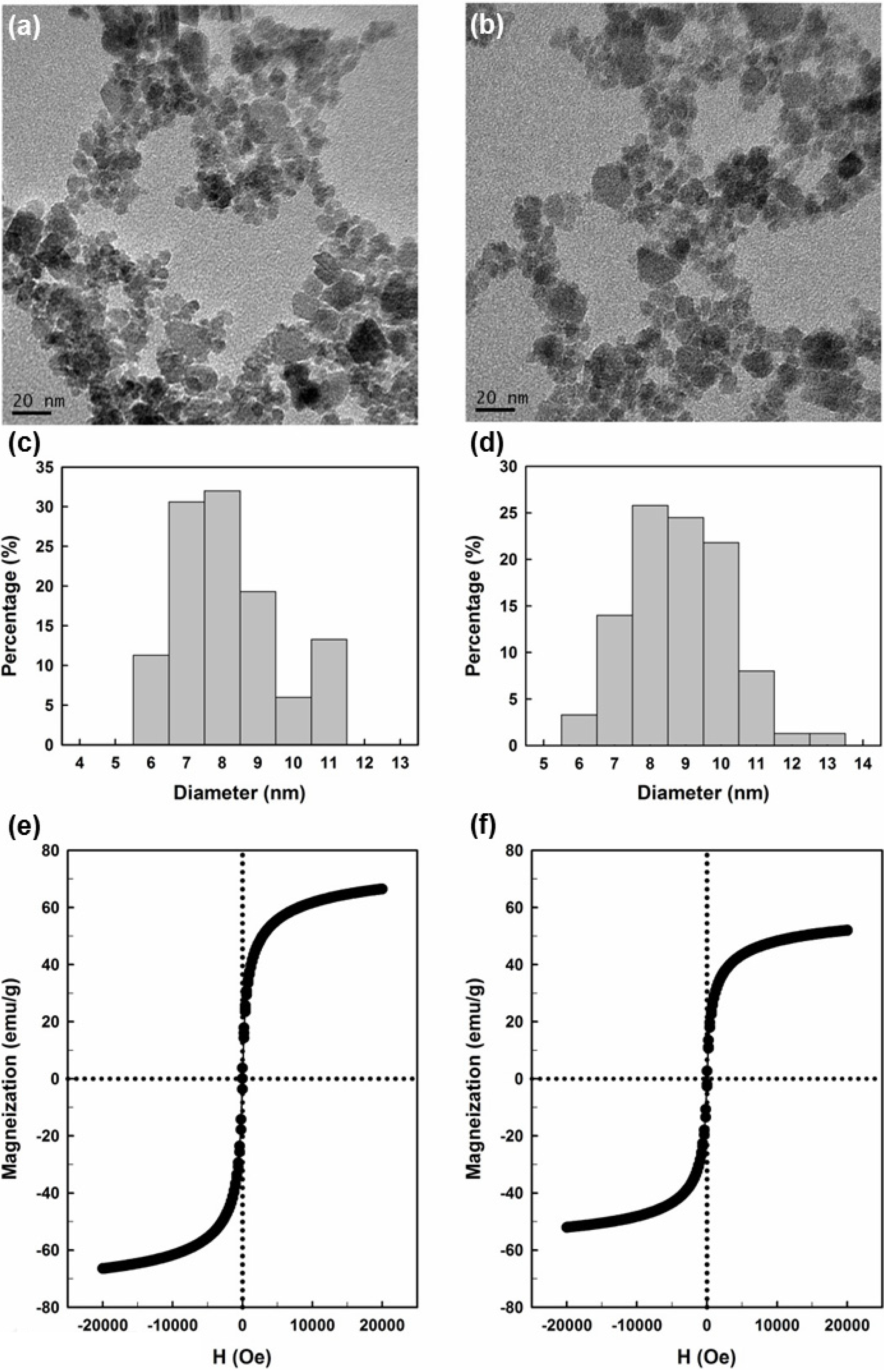
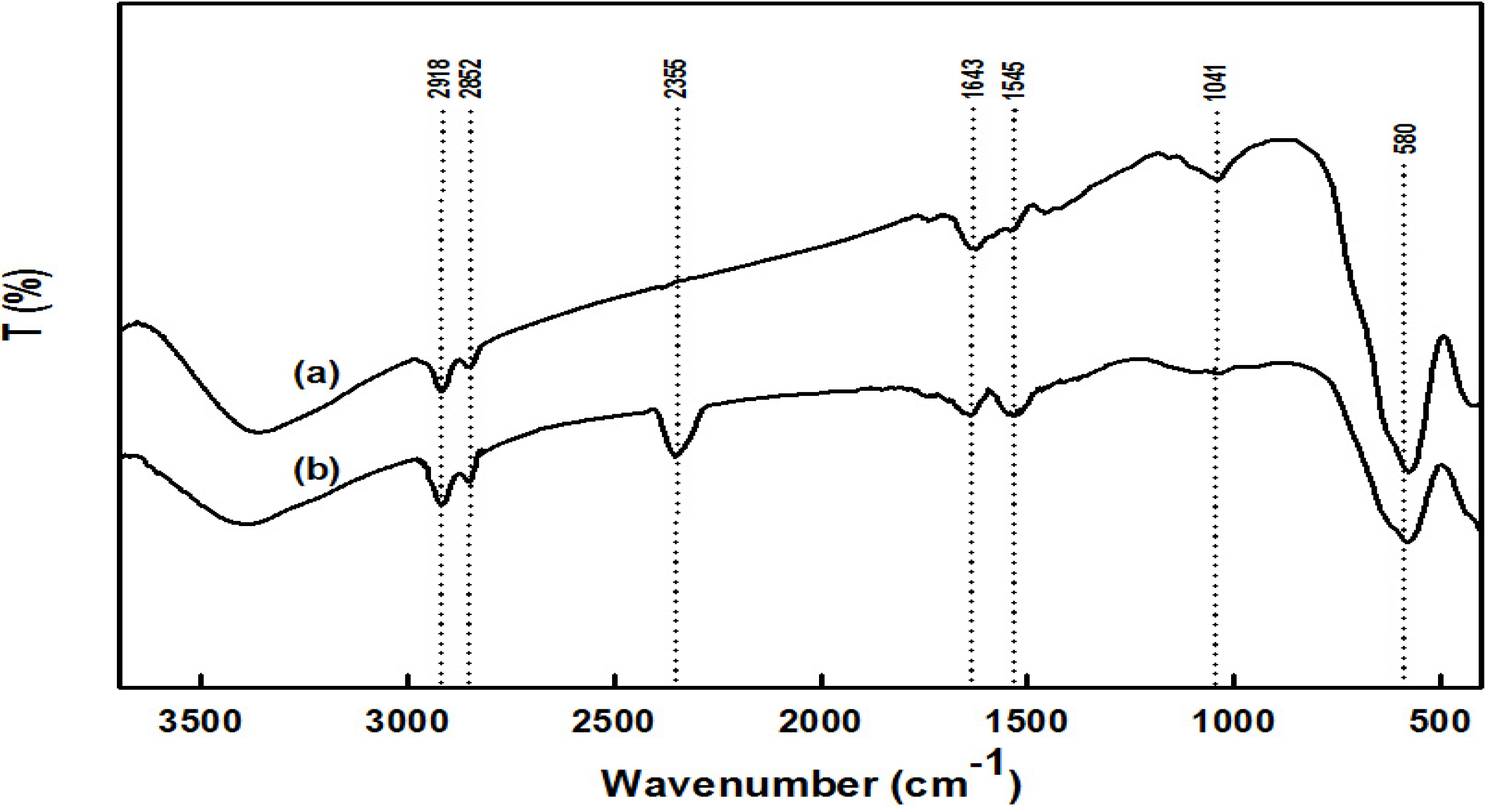
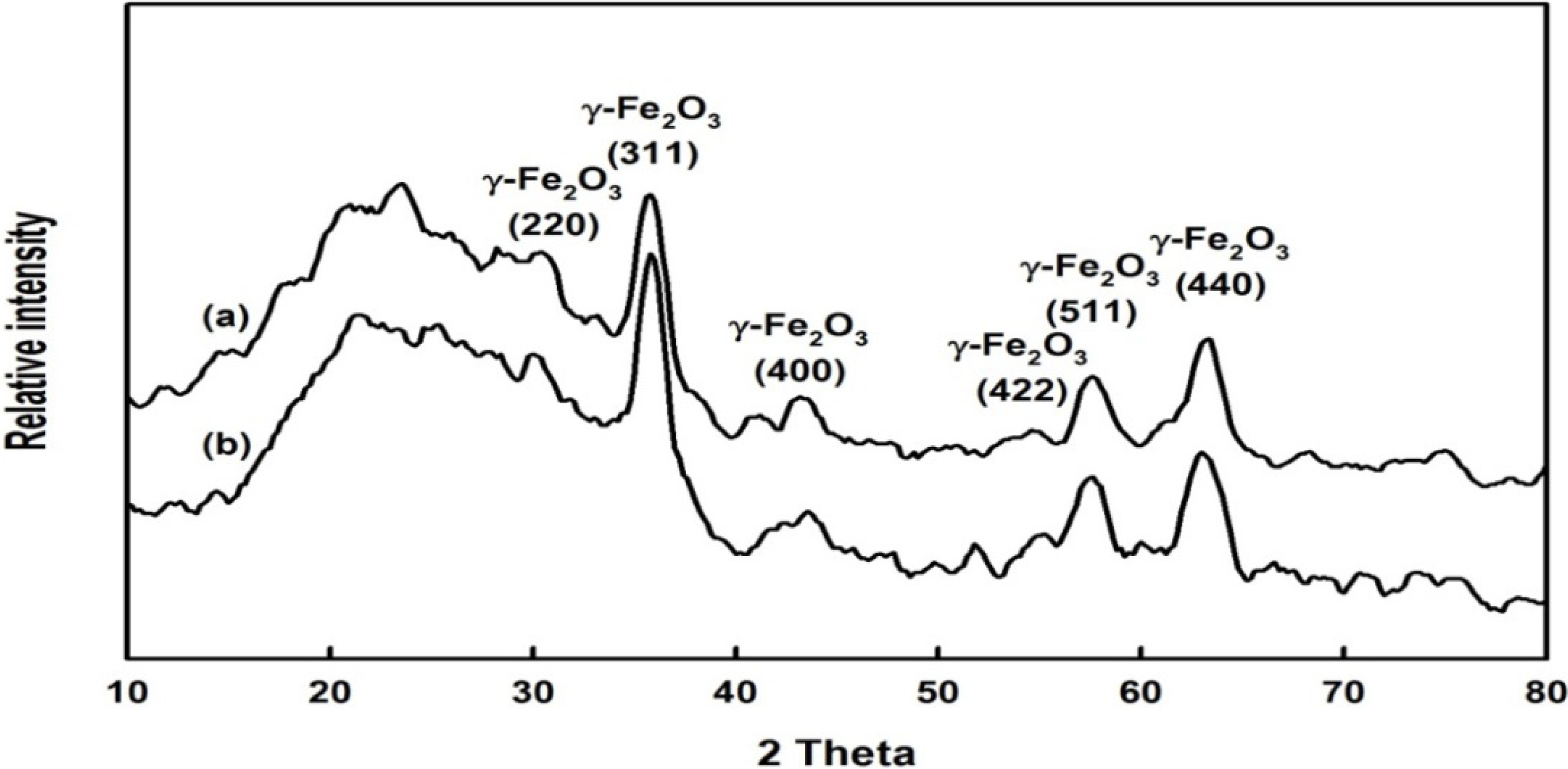
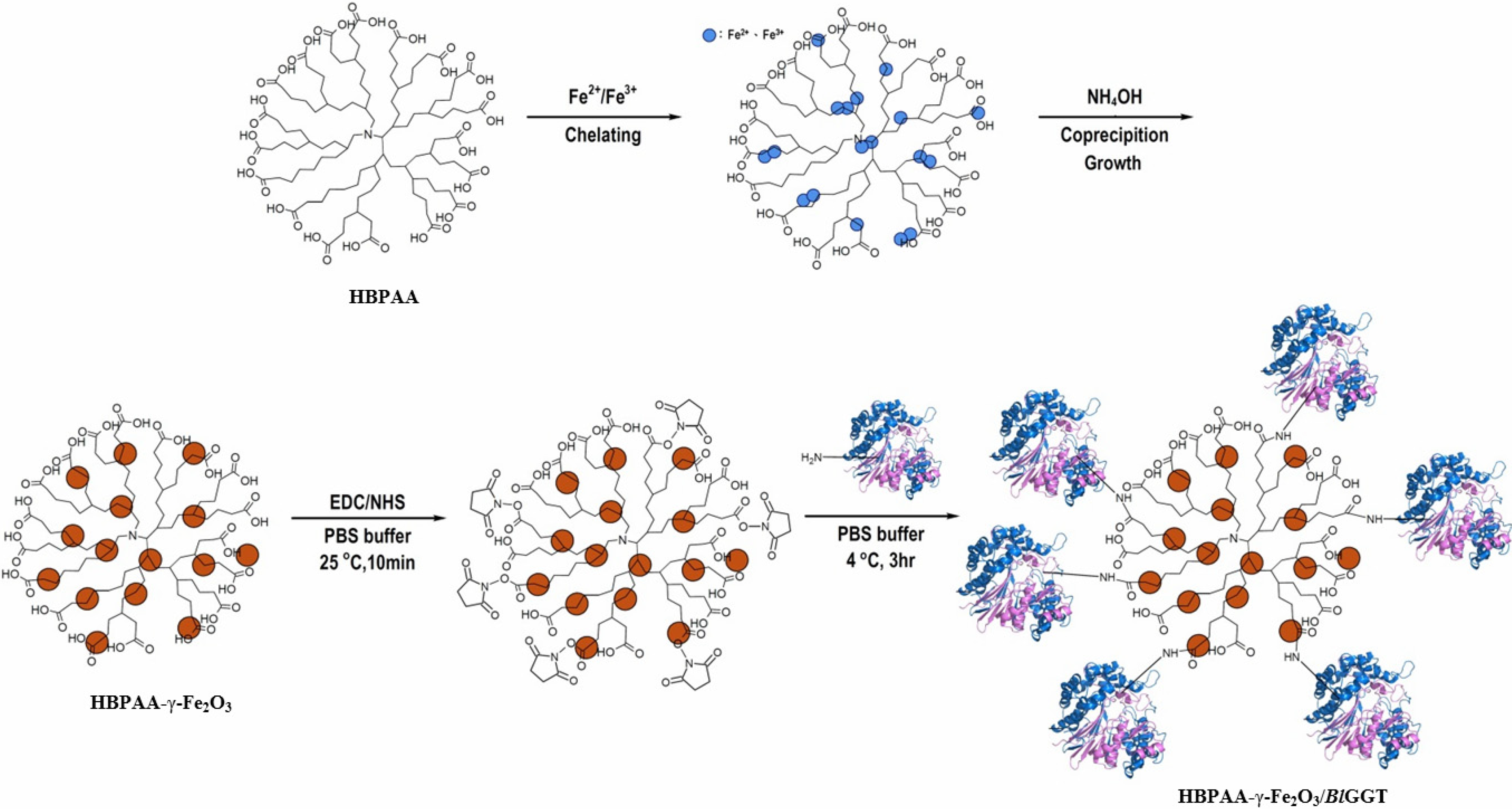
2.1. Preparation and Properties of the Synthesized Organic/Magnetic NPs
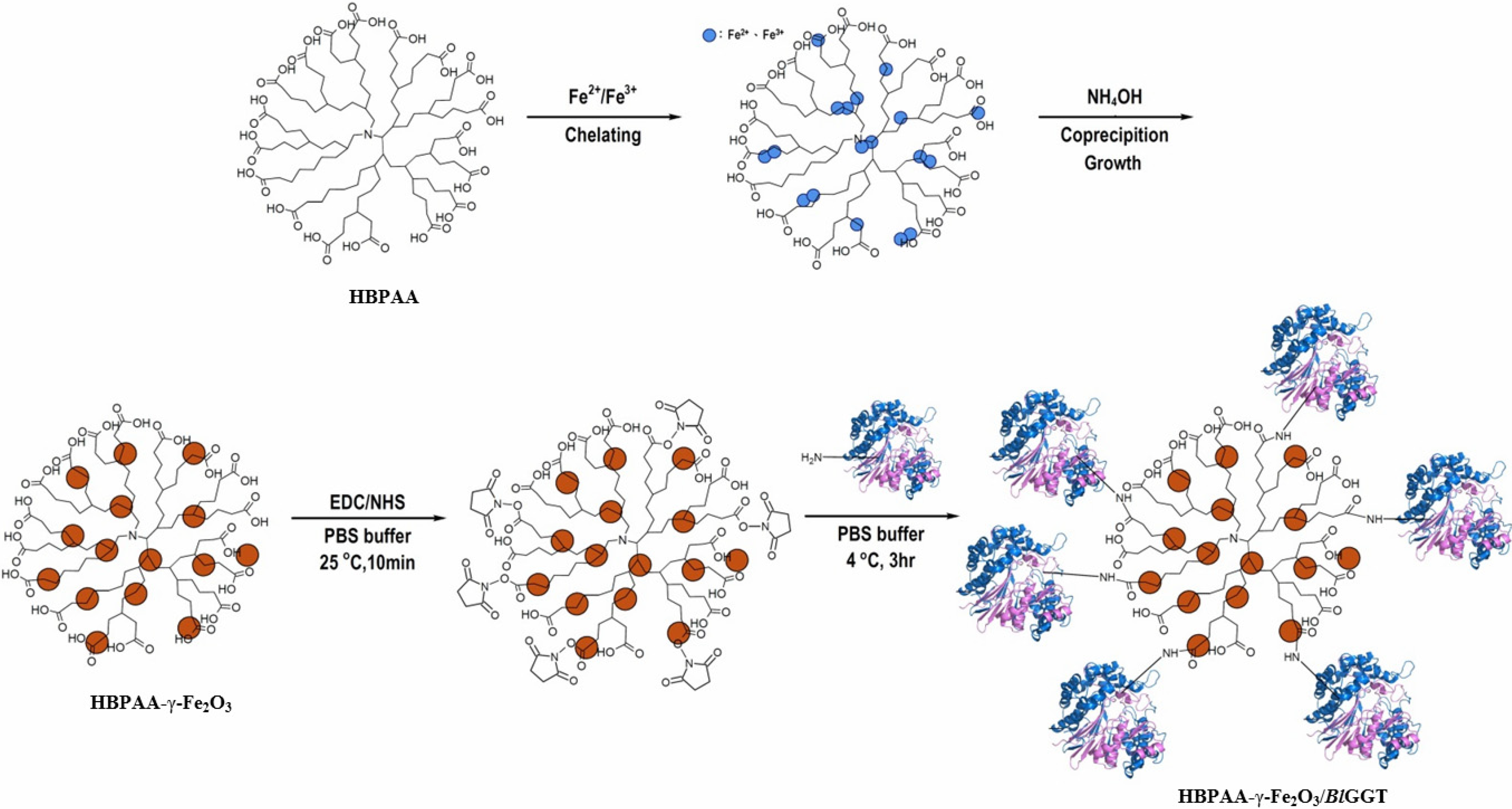
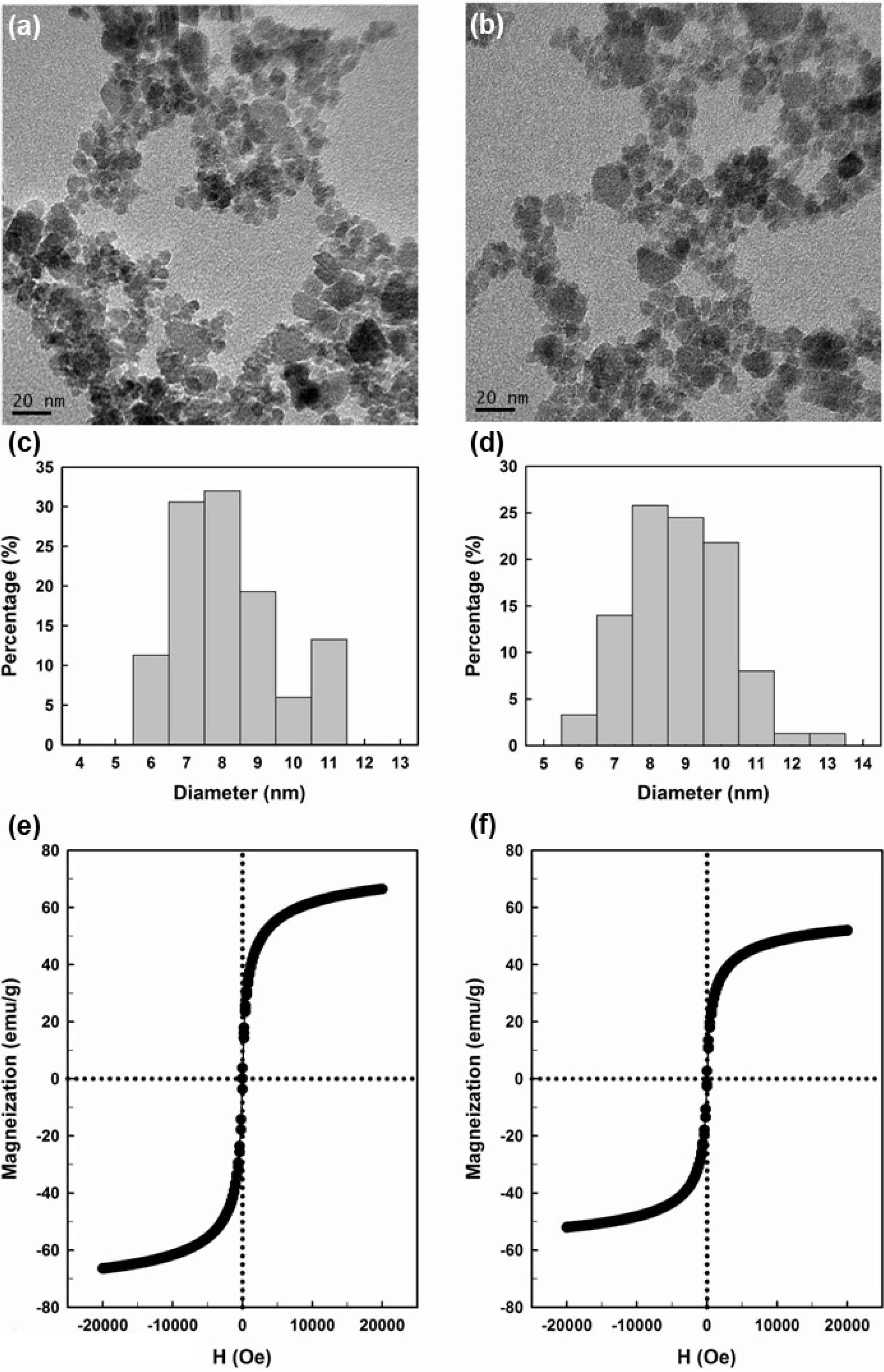

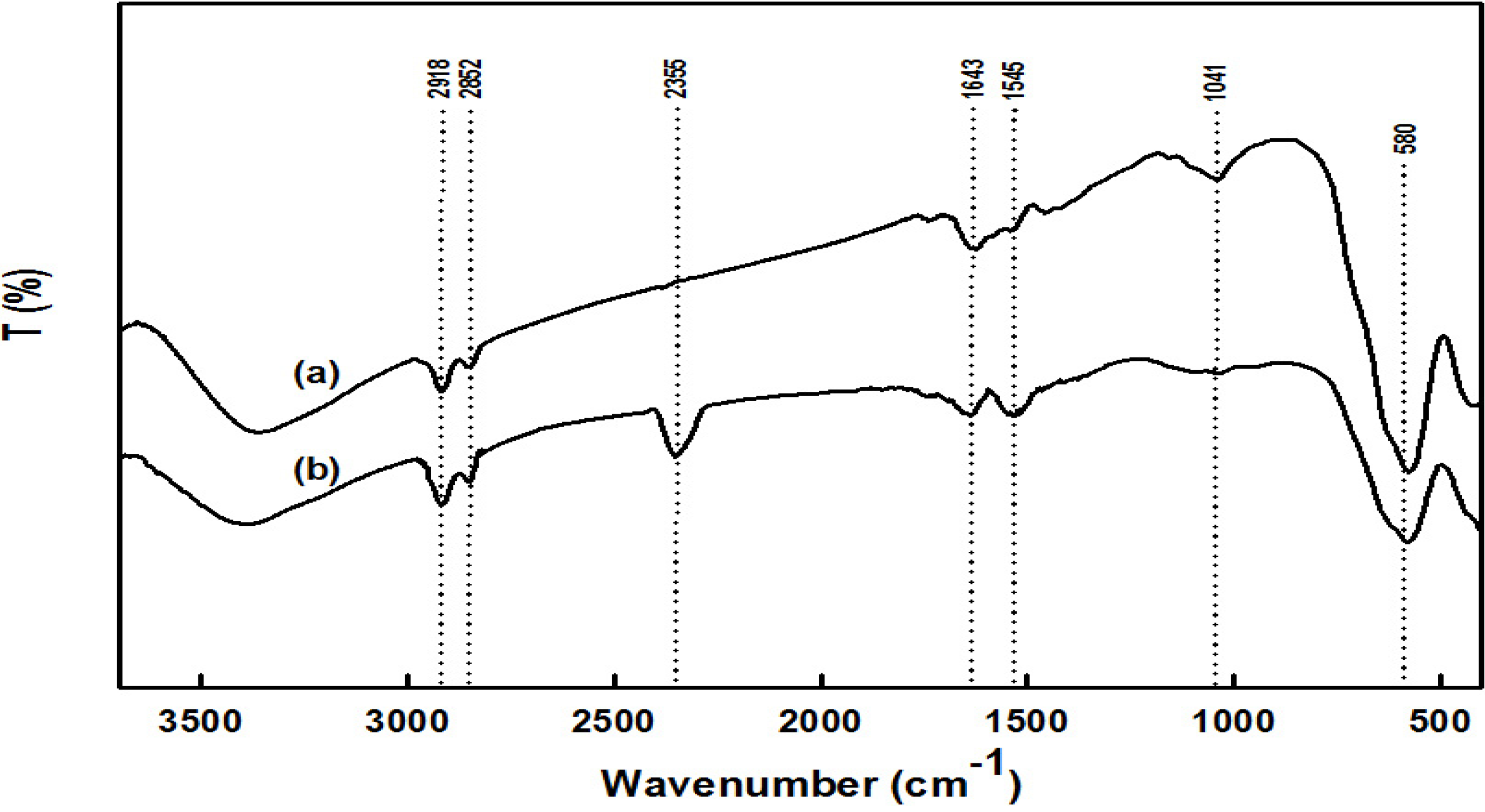
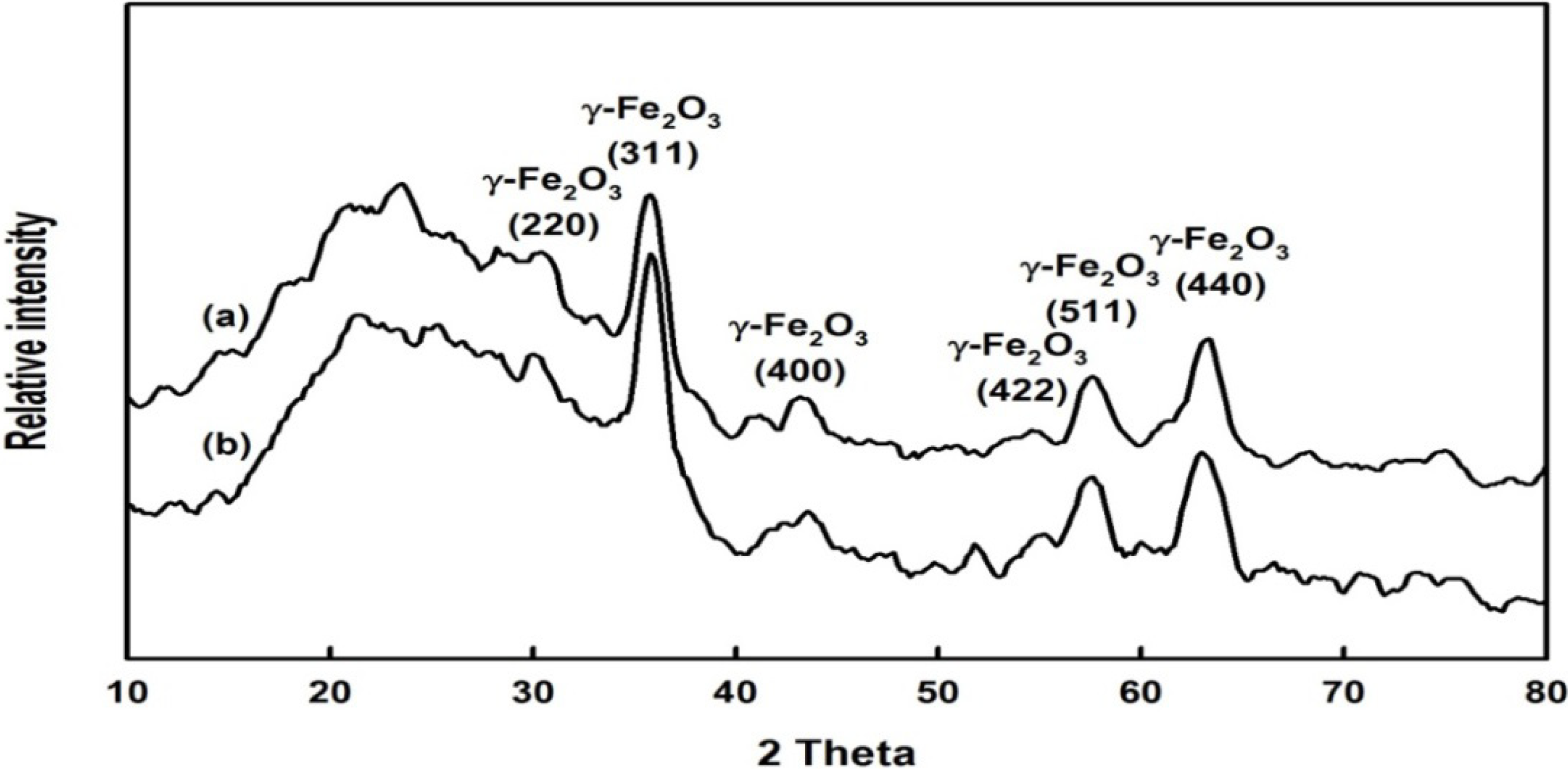
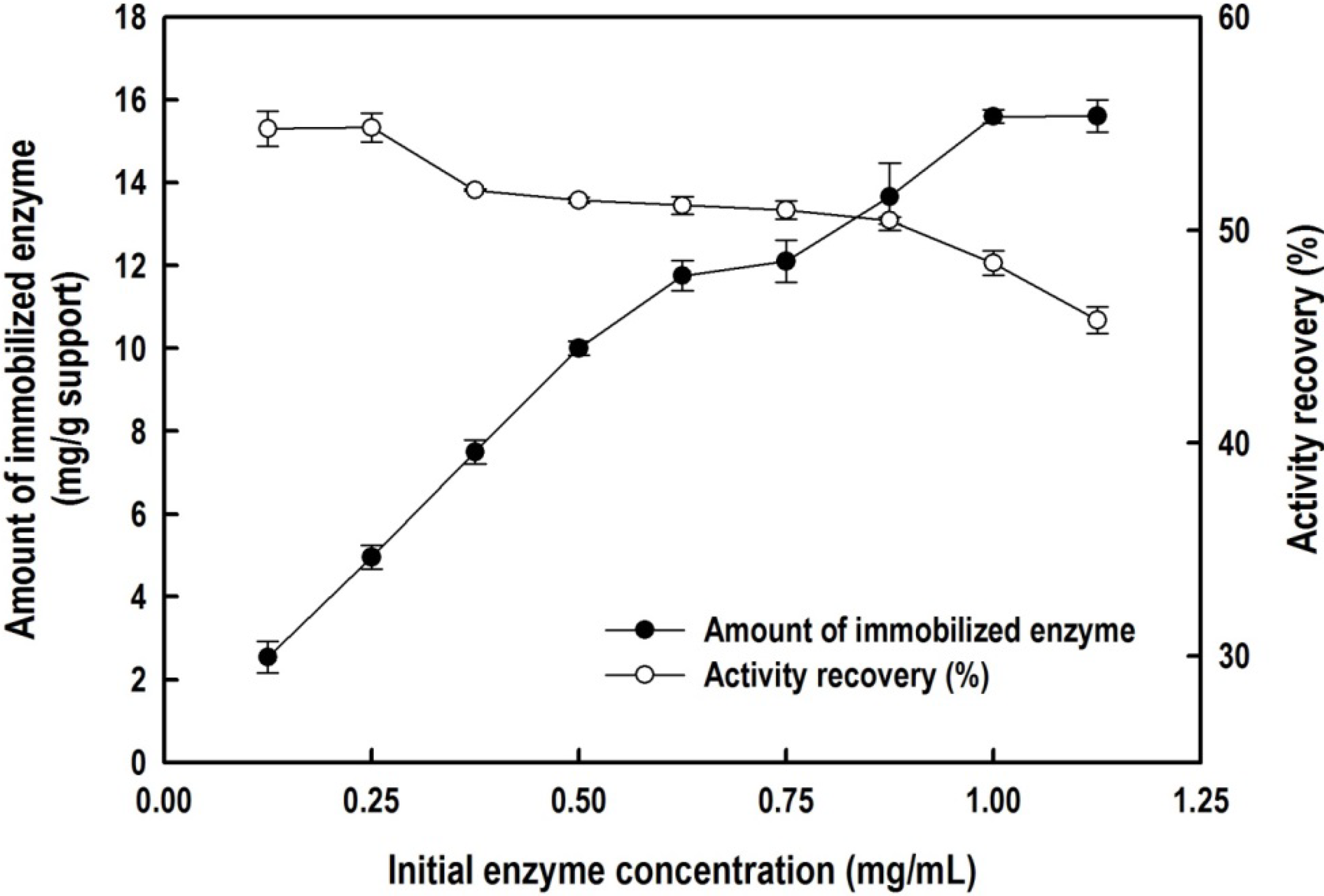
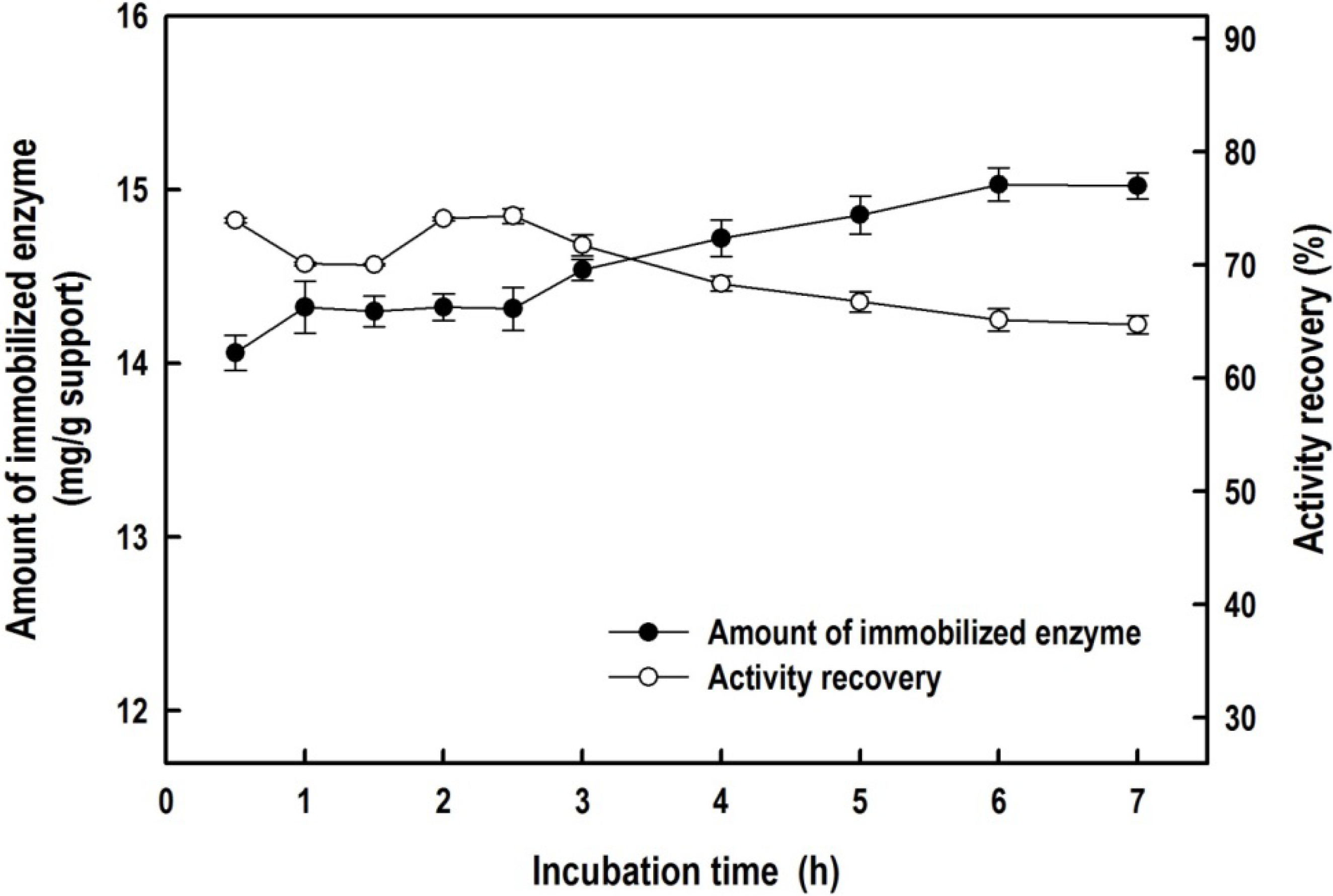
2.2. Immobilization of the Recombinant Enzyme


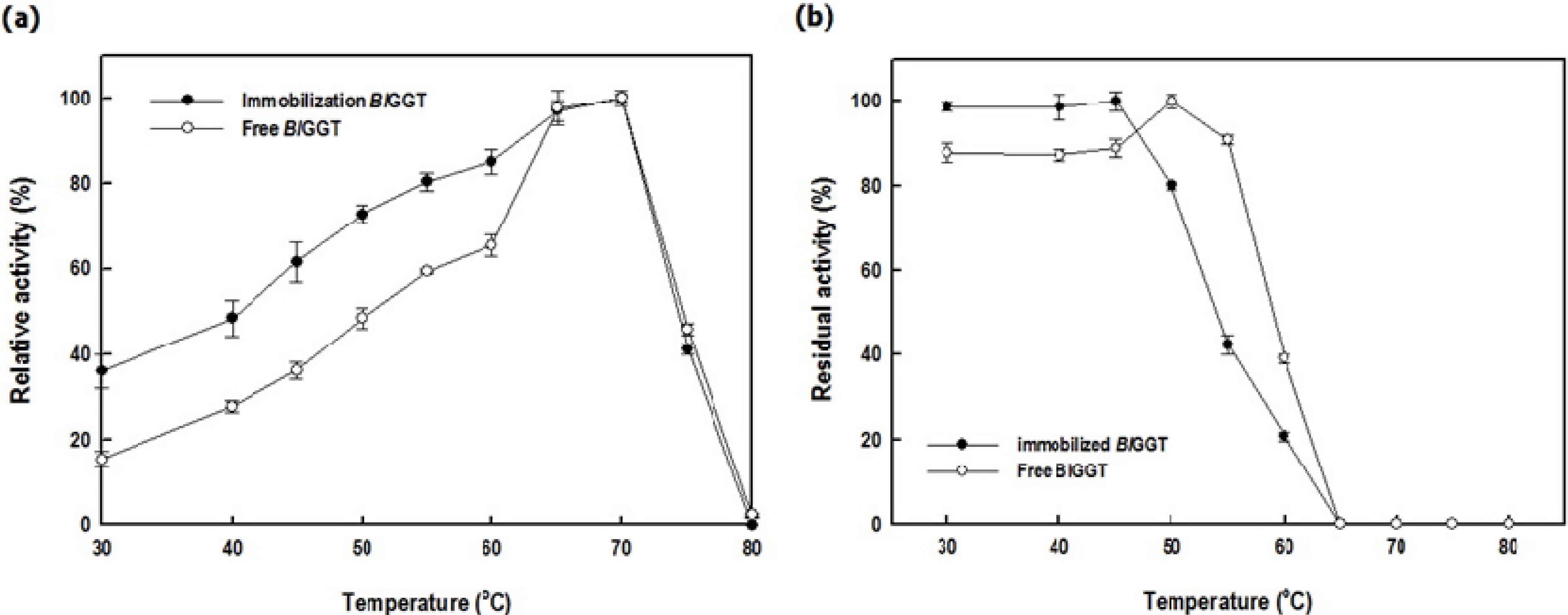
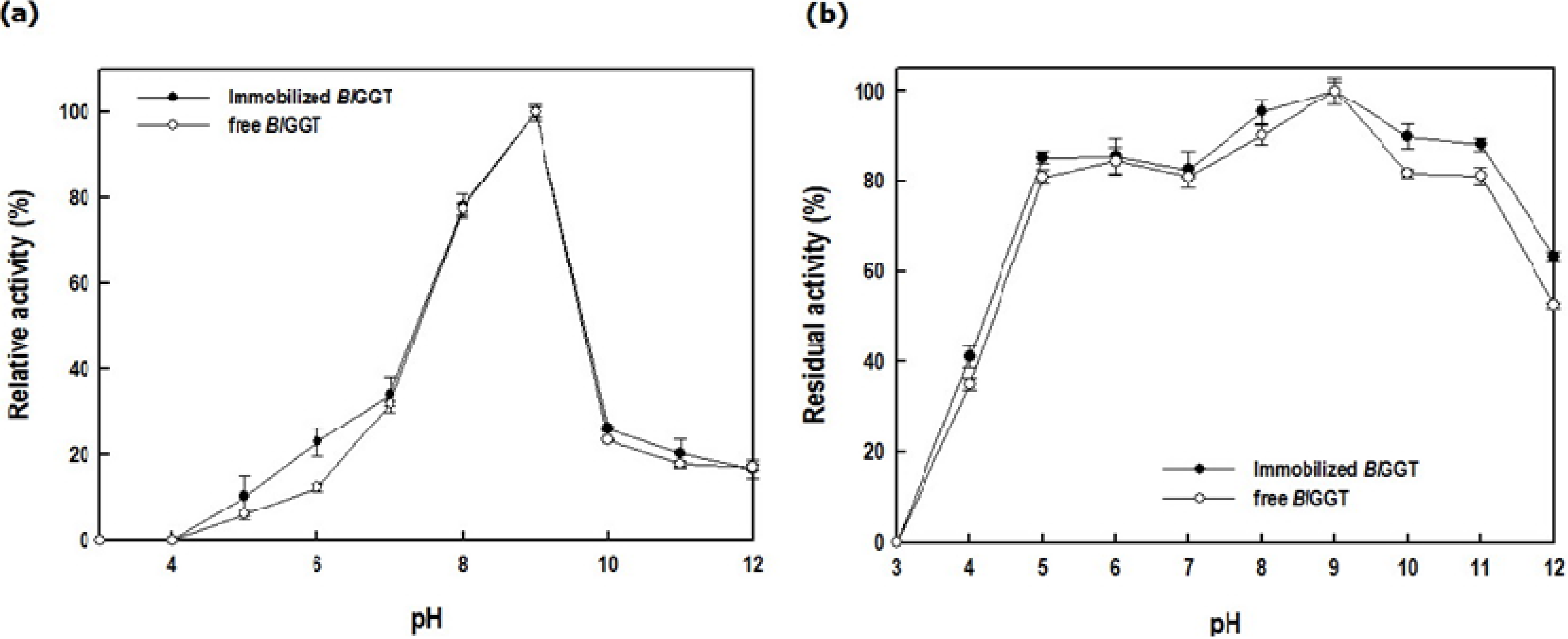
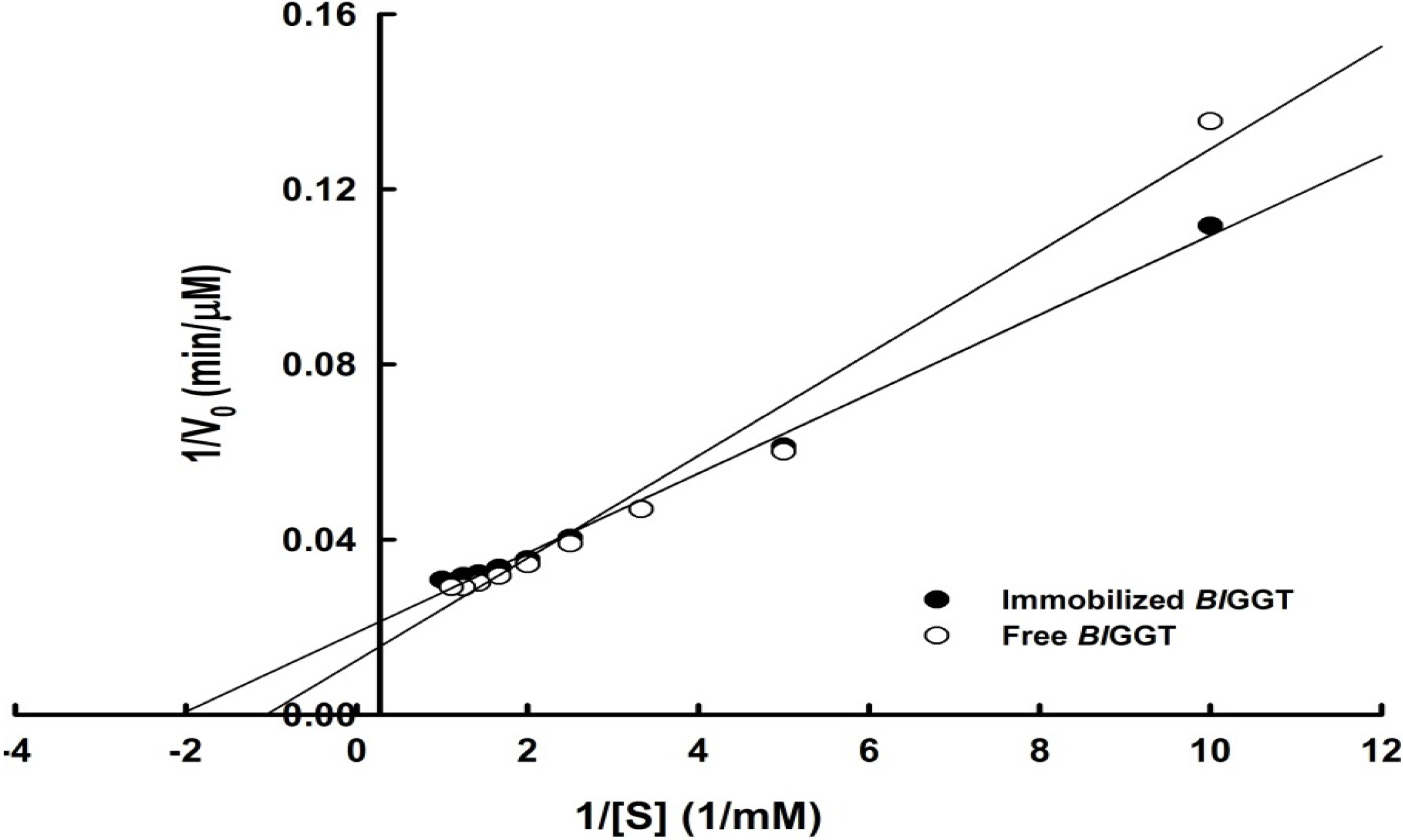
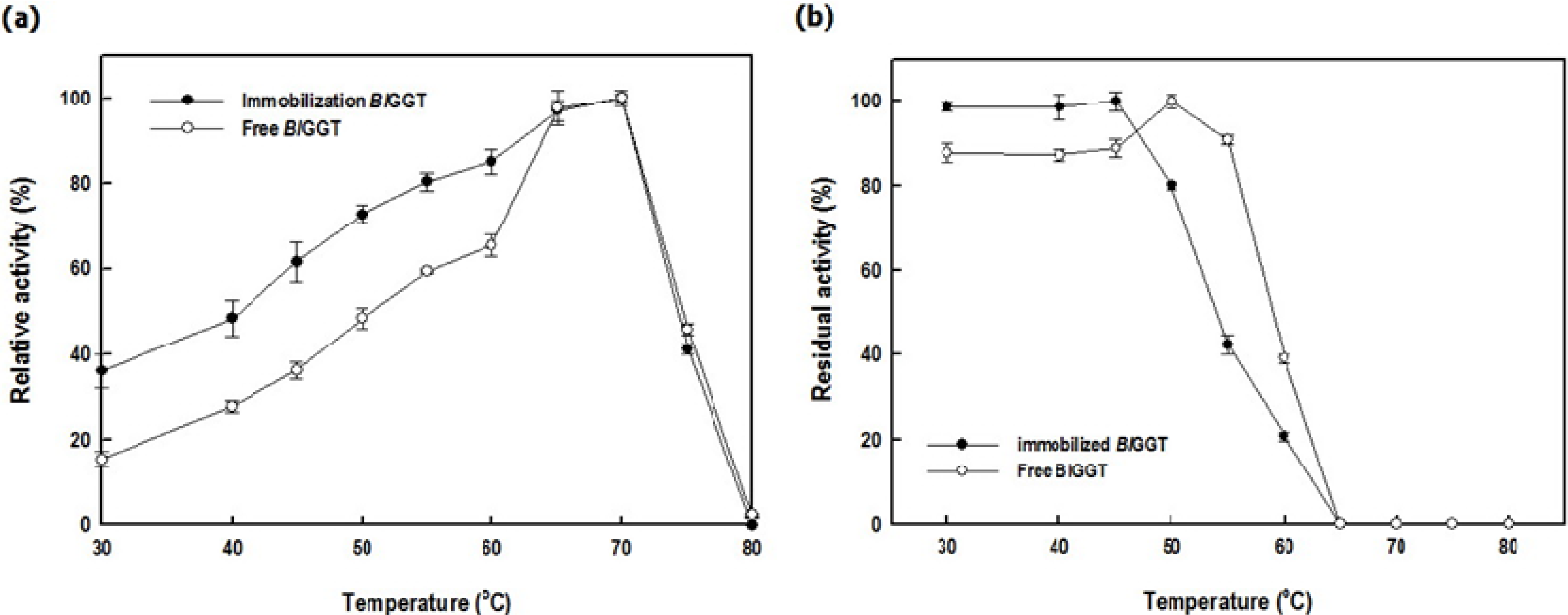
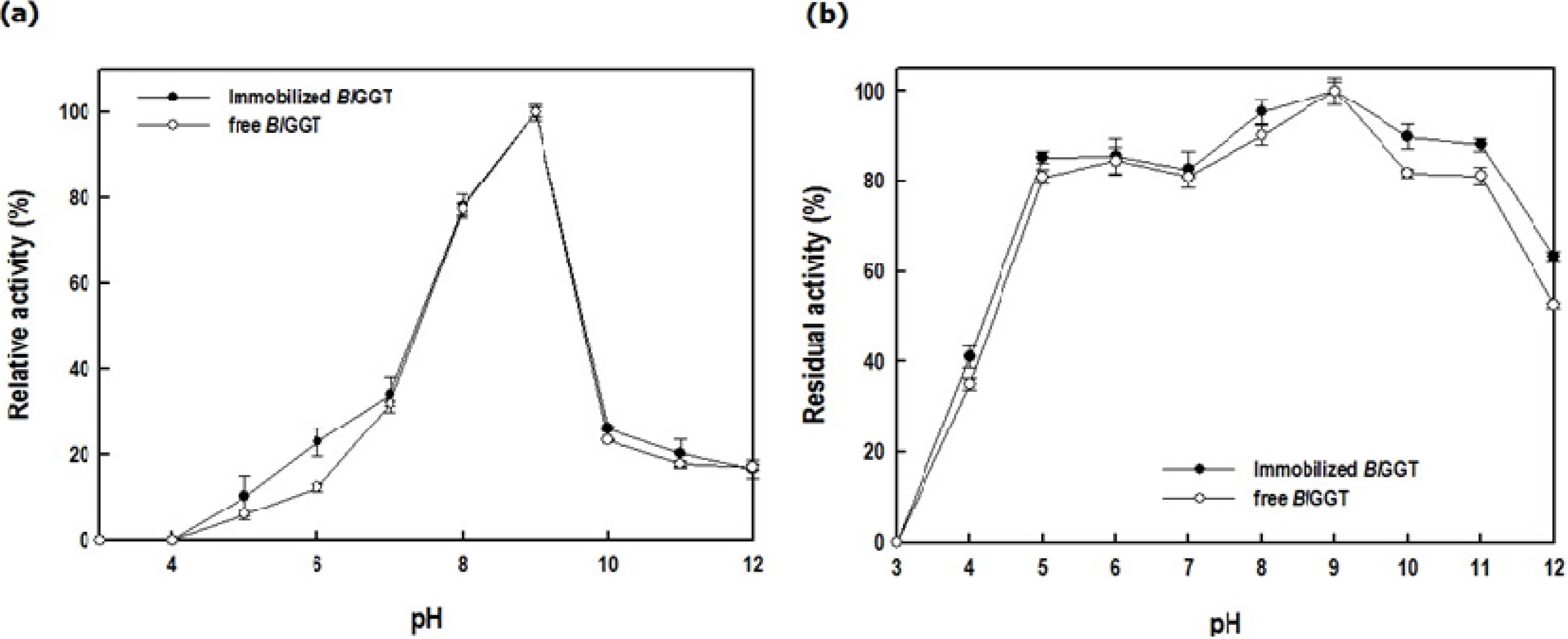
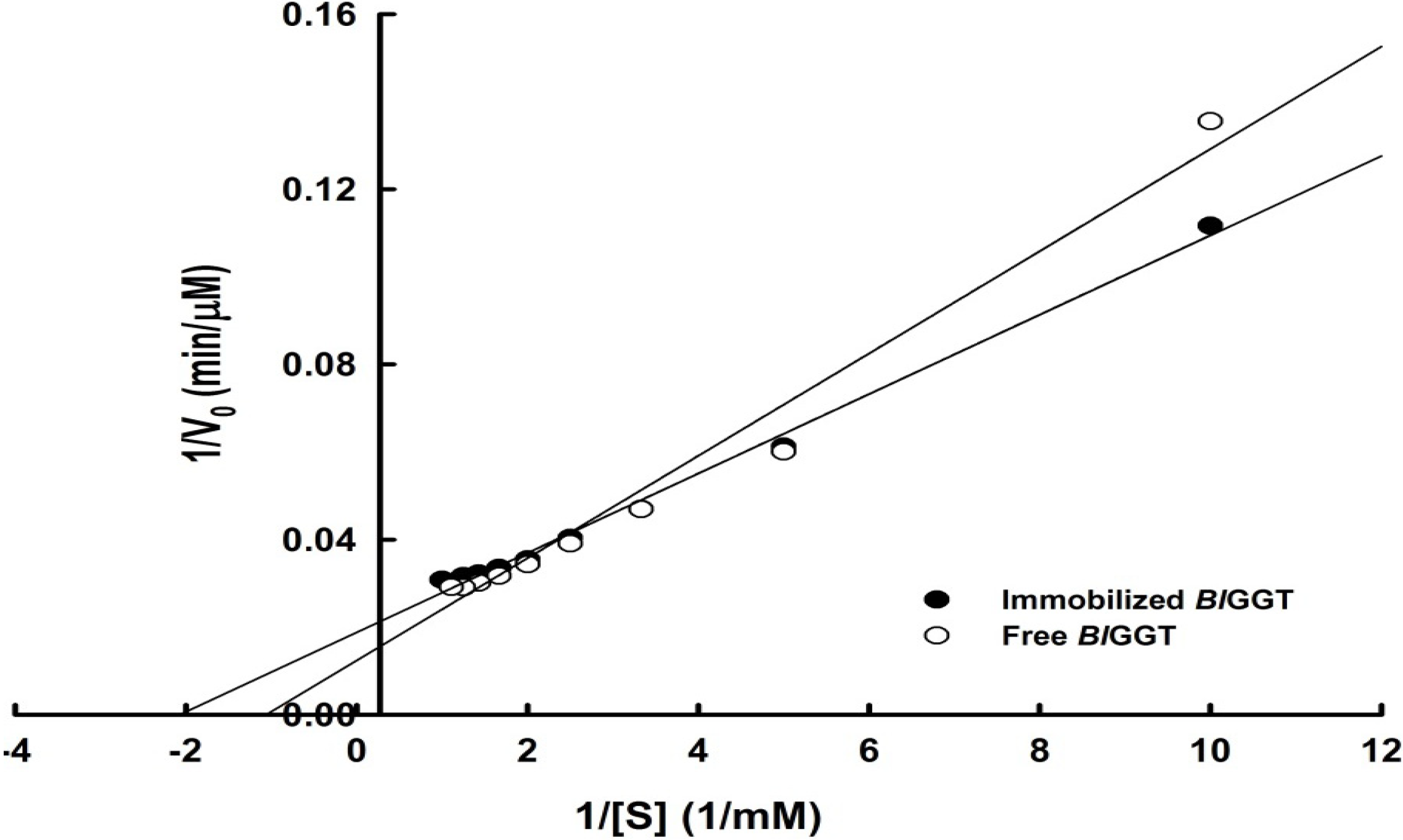
2.3. Characterization of Free and Immobilized Enzymes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Enzyme | Specific activity (U mg−1) | Vmax (µM min−1) | Km (mM) | kcat (s−1) | kcat/Km (mM−1 s−1) |
|---|---|---|---|---|---|
| Immobilized BlGGT | 4.24 ± 0.08 | 53.19 | 0.48 | 7.13 | 14.85 |
| Free BlGGT | 12.08 ± 0.09 | 80.64 | 0.94 | 31.68 | 33.70 |
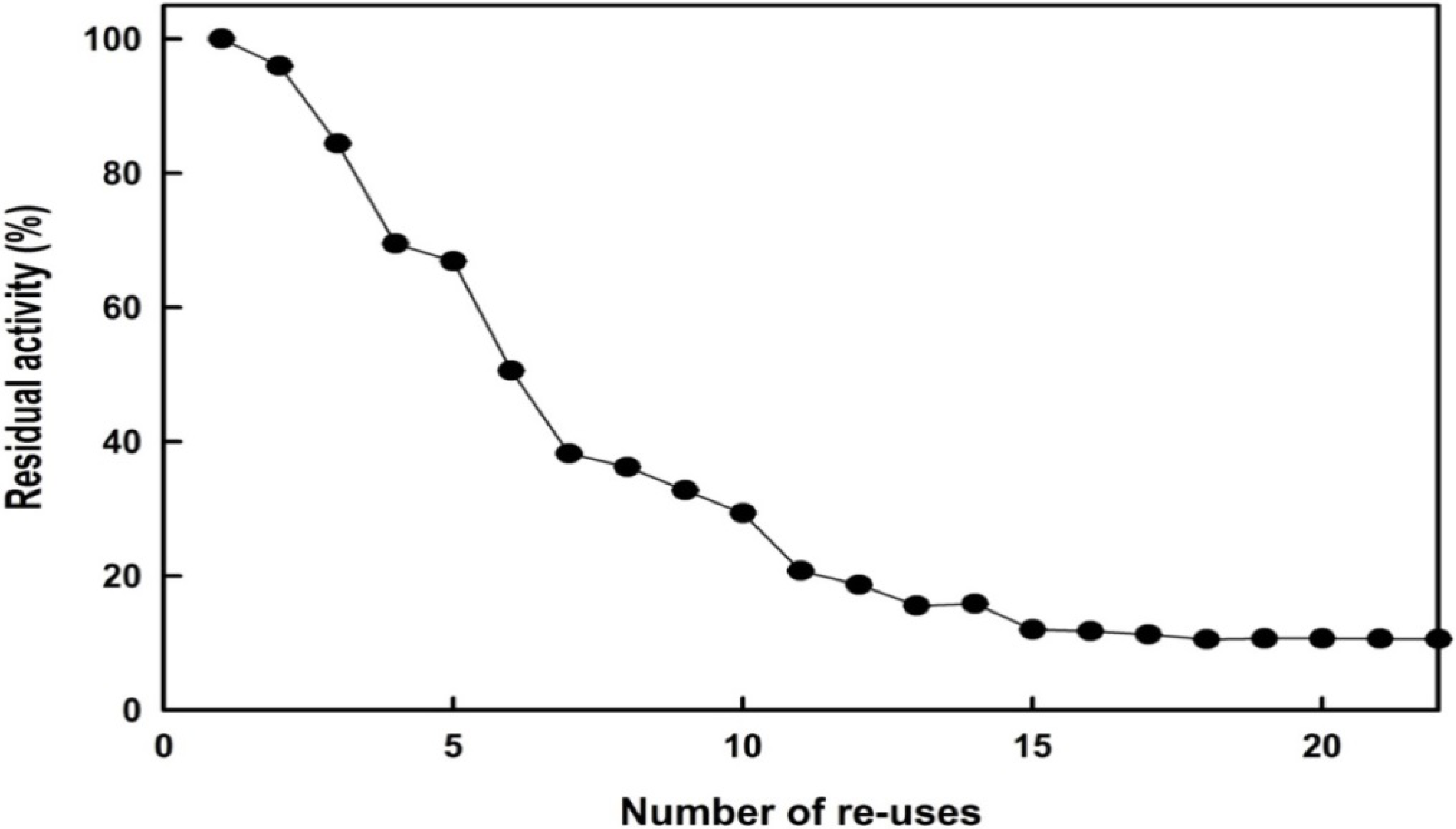
2.4. Reusability

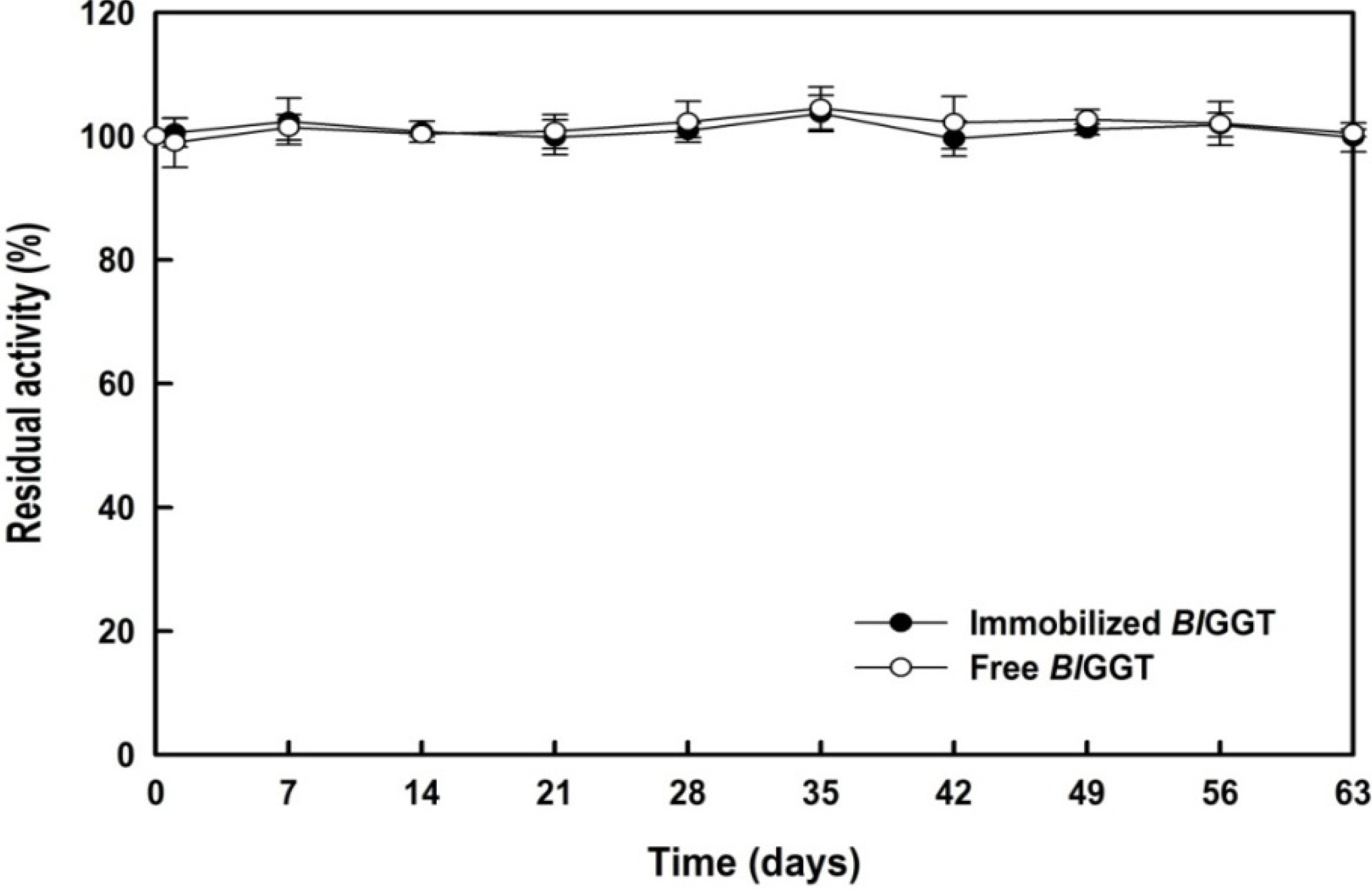
2.5. Storage Stability of Free and Immobilized Enzymes

3. Experimental
3.1. Materials
3.2. HBPAA-γ-Fe2O3 Magnetic Nanohybrids
3.3. HBPAA-γ-Fe2O3/BlGGT-Modified Magnetic Nanocarriers
3.4. Enzyme Activity
3.5. Effects of Temperature and pH
3.6. Reusability of Immobilized Enzyme
3.7. Storage Stability of the Immobilized Enzyme
3.8. Measurements
4. Conclusions
Supplementary Materials
Acknowledgments
Author Contributions
Conflicts of Interest
References
- Chen, L.; Wei, B.; Zhang, X.; Li, C. Bifunctional graphene/γ-Fe2O3 hybrid aerogels with double nanocrystalline networks for enzyme immobilization. Small 2013, 9, 2331–2340. [Google Scholar] [CrossRef]
- Swarnalatha, V.; Aluri Esther, R.; Dhamodharan, R. Immobilization of α-amylase on gum acacia stabilized magnetite nanoparticles, an easily recoverable and reusable support. J. Mol. Catal. B Enzym. 2013, 96, 6–13. [Google Scholar] [CrossRef]
- Chen, Y.-Y.; Tsai, M.-G.; Chi, M.-C.; Wang, T.-F.; Lin, L.-L. Covalent immobilization of Bacillus licheniformis γ-glutamyl transpeptidase on aldehyde-functionalized magnetic nanoparticles. Int. J. Mol. Sci. 2013, 14, 4613–4628. [Google Scholar] [CrossRef]
- Yu, C.-C.; Kuo, Y.-Y.; Liang, C.-F.; Chien, W.-T.; Wu, H.-T.; Chang, T.-C.; Jan, F.-D.; Lin, C.-C. Site-specific immobilization of enzymes on magnetic nanoparticles and their use in organic synthesis. Bioconjug. Chem. 2012, 23, 714–724. [Google Scholar] [CrossRef]
- Liao, M.-H.; Chen, D.-H. Immobilization of yeast alcohol dehydrogenase on magnetic nanoparticles for improving its stability. Biotechnol. Lett. 2001, 23, 1723–1727. [Google Scholar] [CrossRef]
- Schmid, A.; Dordick, J.; Hauer, B.; Kiener, A.; Wubbolts, M.; Witholt, B. Industrial biocatalysis today and tomorrow. Nature 2001, 409, 258–268. [Google Scholar] [CrossRef]
- Khan, A.A.; Alzohairy, M.A. Recent advances and applications of immobilized enzyme technologies: A review. Res. J. Biol. Sci. 2010, 5, 565–575. [Google Scholar] [CrossRef]
- Hanafeld, U.; Gardossib, L.; Magnerc, E. Understanding enzyme immobilization. Chem. Soc. Rev. 2009, 38, 453–468. [Google Scholar] [CrossRef]
- Yancey, D.F.; Carino, E.V.; Crooks, R.M. Electrochemical synthesis and electrocatalytic properties of Au@Pt dendrimer-encapsulated nanoparticles. J. Am. Chem. Soc. 2010, 132, 10988–10989. [Google Scholar] [CrossRef]
- Yang, W.; Pan, C.-Y.; Liu, X.-Q.; Wang, J. Multiple functional hyperbranched poly(amido amine)nanoparticles: Synthesis and application in cell imaging. Biomacromolecules 2011, 12, 1523–1531. [Google Scholar] [CrossRef]
- Shi, X.; Wang, S.H.; Swanson, S.D.; Ge, S.; Cao, Z.; Antwerp, M.E.V.; Landmark, K.J.; Baker, J.R., Jr. Dendrimer-functionalized shell-crosslinked iron oxide nanoparticles for in vivo magnetic resonance imaging of tumors. Adv. Mater. 2008, 20, 1671–1678. [Google Scholar] [CrossRef]
- Haba, Y.; Kojima, C.; Harada, A.; Ura, T.; Horinaka, H.; Kono, K. Preparation of poly(ethylene glycol)-modified poly(amido amine) dendrimers encapsulating gold nanoparticles and their heat-generating ability. Langmuir 2007, 23, 5243–5246. [Google Scholar] [CrossRef]
- Zhou, Y.; Yan, D. Supramolecular self-assembly of amphiphilic hyperbranched polymers at all scales and dimensions: Progress, characteristics and perspectives. Chem. Commun. 2009, 10, 1172–1188. [Google Scholar] [CrossRef]
- Shau, S.-M.; Juang, T.-Y.; Lin, H.-S.; Huang, C.-L.; Hsieh, C.-F.; Wu, J.-Y.; Jeng, R.-J. Individual graphene oxide platelets through direct molecular exfoliation with globular amphiphilic hyperbranched polymers. Polym. Chem. 2012, 3, 1249–1259. [Google Scholar] [CrossRef]
- Juang, T.Y.; Chen, Y.C.; Tsai, C.C.; Dai, S.A.; Wu, T.M.; Jeng, R.J. Nanoscale organic/inorganic hybrids based on self-organized dendritic macromolecules on montmorillonites. Appl. Clay Sci. 2010, 48, 103–110. [Google Scholar] [CrossRef]
- Chen, Y.-C.; Wu, W.-Y.; Juang, T.-Y.; Dai, S.A.; Su, W.-C.; Liu, Y.-L.; Lee, R.-H.; Jeng, R.-J. Poly(urethane/malonamide) dendritic structures featuring blocked/deblocked isocyanate units. Polym. Chem. 2011, 2, 1139–1145. [Google Scholar] [CrossRef]
- Bronstein, L.M.; Shifrina, Z.B. Dendrimers as encapsulating, stabilizing, or directing agents for inorganic nanoparticles. Chem. Rev. 2011, 111, 5301–5344. [Google Scholar] [CrossRef]
- Chang, Y.; Li, Y.; Meng, X.; Liu, N.; Sun, D.; Liu, H.; Wang, J. Dendrimer functionalized water soluble magnetic iron oxide conjugates as dual imaging probe for tumor targeting and drug delivery. Polym. Chem. 2012, 4, 789–794. [Google Scholar]
- Shau, S.-M.; Chang, C.-C.; Lo, C.-H.; Chen, Y.-C.; Juang, T.-Y.; Dai, S.A.; Lee, R.-H.; Jeng, R.-J. Organic/metallic nanohybrids based on amphiphilic dumbbell-Shaped dendrimers. ACS Appl. Mater. Interfaces 2012, 4, 1897–1908. [Google Scholar] [CrossRef]
- Feng, G.; Liang, J.; Liu, B. Hyperbranched conjugated polyelectrolytes for biological sensing and imaging. Macromol. Rapid Commun. 2013, 34, 705–715. [Google Scholar] [CrossRef]
- Li, D.; He, Q.; Cui, Y.; Duan, L.; Li, J. Immobilization of glucose oxidase onto gold nanoparticles with enhanced thermostability. Biochem. Biophys. Res. Commun. 2007, 355, 488–493. [Google Scholar] [CrossRef]
- Seleci, M.; Ag, D.; Yalcinkaya, E.E.; Demirkol, D.O.; Guler, C.; Timur, S. Amine-intercalated montmorillonite matrices for enzyme immobilization and biosensing applications. RSC Adv. 2012, 2, 2112–2118. [Google Scholar] [CrossRef]
- Zhang, J.; Zhang, F.; Yang, H.; Huang, X.; Liu, H.; Zhang, J.; Guo, S. Graphene oxide as a matrix for enzyme immobilization. Langmuir 2010, 26, 6083–6085. [Google Scholar]
- Lin, J.J.; Wei, J.C.; Juang, T.Y.; Tsai, W.C. Preparation of protein-silicate hybrids from polyamine intercalation of layered montmorillonite. Langmuir 2007, 23, 1995–1999. [Google Scholar] [CrossRef]
- Li, Y.; Wang, P.; Li, F.; Huang, X.; Wang, L.; Lin, X. Covalent immobilization of single-walled carbon nanotubes and single-stranded deoxyribonucleic acid nanocomposites on glassy carbon electrode: Preparation, characterization, and applications. Talanta 2008, 77, 833–838. [Google Scholar] [CrossRef]
- Lee, C.-H.; Lin, T.-S.; Mou, C.-Y. Mesoporous materials for encapsulating enzymes. Nano Today 2009, 4, 165–179. [Google Scholar] [CrossRef]
- Shiau, S.F.; Juang, T.Y.; Chou, H.W.; Liang, M. Synthesis and properties of new water-soluble aliphatic hyperbranched poly(amido acids) with high pH-dependent photoluminescence. Polymer 2013, 54, 623–630. [Google Scholar] [CrossRef]
- Huang, C.-L.; Cheng, W.-C.; Yang, J.-C.; Chi, M.-C.; Chen, J.-H.; Lin, H.-P.; Lin, L.-L. Preparation of carboxylated magnetic particles for the efficient immobilization of C-terminally lysine-tagged Bacillus stearothermophilus aminopeptidase II. J. Ind. Microbiol. Biotechnol. 2010, 37, 717–725. [Google Scholar] [CrossRef]
- Lian, X.; Zhu, W.; Wen, Y.; Li, L.; Zhao, X. Effects of soy protein hydrolysates on maize starch retrogradation studied by IR spectra and ESI-MS analysis. Int. J. Biol. Macromol. 2013, 59, 143–150. [Google Scholar] [CrossRef]
- Orlowski, M.; Meister, A. γ-Glutamyl transpeptidase: A new convenient substrate for determinationand study of l-and d-γ-glutamyltranspeptidase activities. Biochim. Biophys. Acta 1963, 73, 679–681. [Google Scholar]
- Sample Availability: Samples of the compounds hyperbranched poly(amido acid)s and γ-glutamyltranspeptidase are available from the authors.
© 2014 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Juang, T.-Y.; Kan, S.-J.; Chen, Y.-Y.; Tsai, Y.-L.; Lin, M.-G.; Lin, L.-L. Surface-Functionalized Hyperbranched Poly(Amido Acid) Magnetic Nanocarriers for Covalent Immobilization of a Bacterial γ-Glutamyltranspeptidase. Molecules 2014, 19, 4997-5012. https://doi.org/10.3390/molecules19044997
Juang T-Y, Kan S-J, Chen Y-Y, Tsai Y-L, Lin M-G, Lin L-L. Surface-Functionalized Hyperbranched Poly(Amido Acid) Magnetic Nanocarriers for Covalent Immobilization of a Bacterial γ-Glutamyltranspeptidase. Molecules. 2014; 19(4):4997-5012. https://doi.org/10.3390/molecules19044997
Chicago/Turabian StyleJuang, Tzong-Yuan, Shao-Ju Kan, Yi-Yu Chen, Yi-Lin Tsai, Min-Guan Lin, and Long-Liu Lin. 2014. "Surface-Functionalized Hyperbranched Poly(Amido Acid) Magnetic Nanocarriers for Covalent Immobilization of a Bacterial γ-Glutamyltranspeptidase" Molecules 19, no. 4: 4997-5012. https://doi.org/10.3390/molecules19044997