Practical Aspects and Mechanism of Asymmetric Hydrogenation with Chiral Half-Sandwich Complexes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
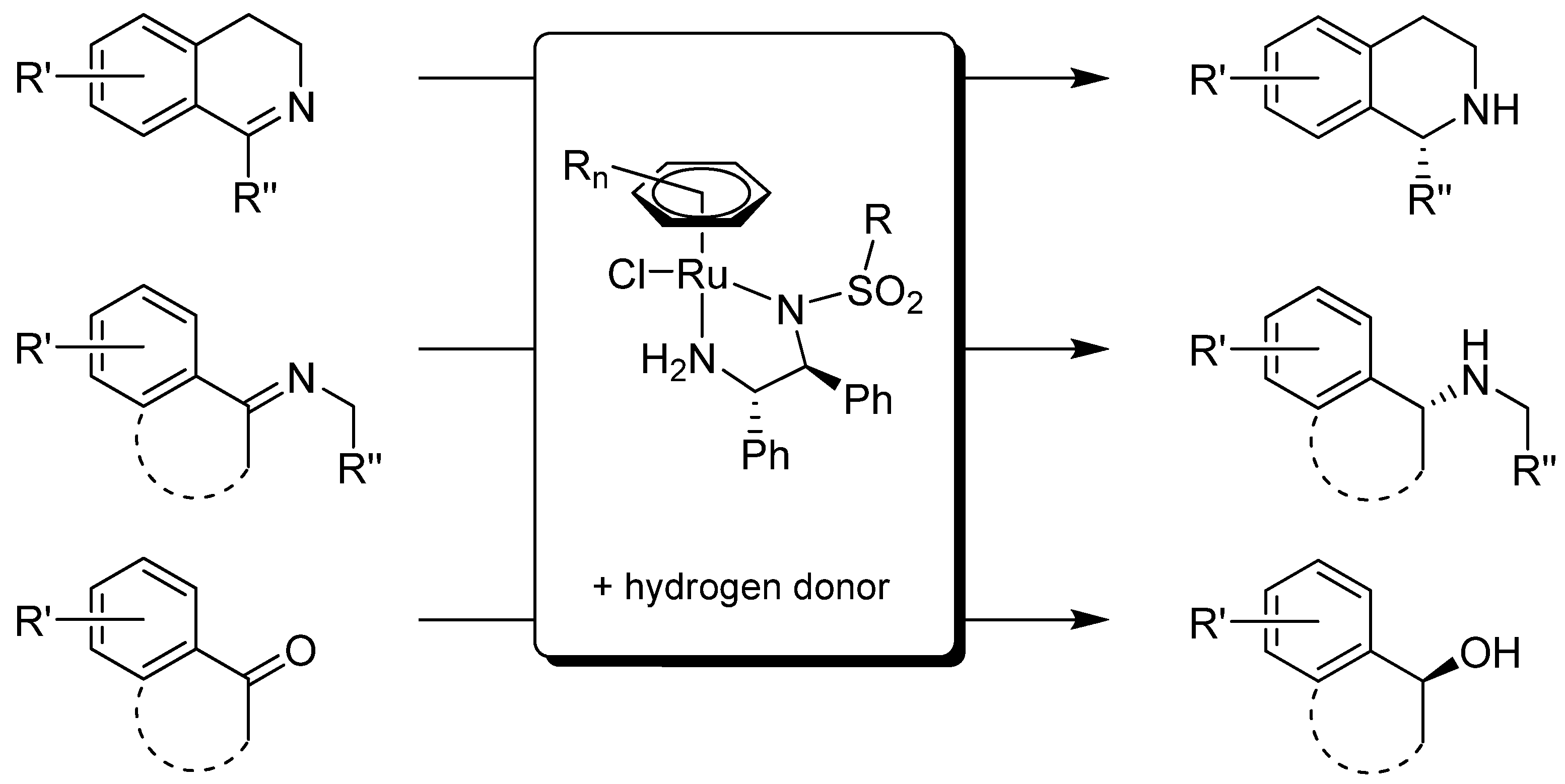
2. Analytical Methods Tailored for ATH
2.1. Monitoring of Reaction Kinetics
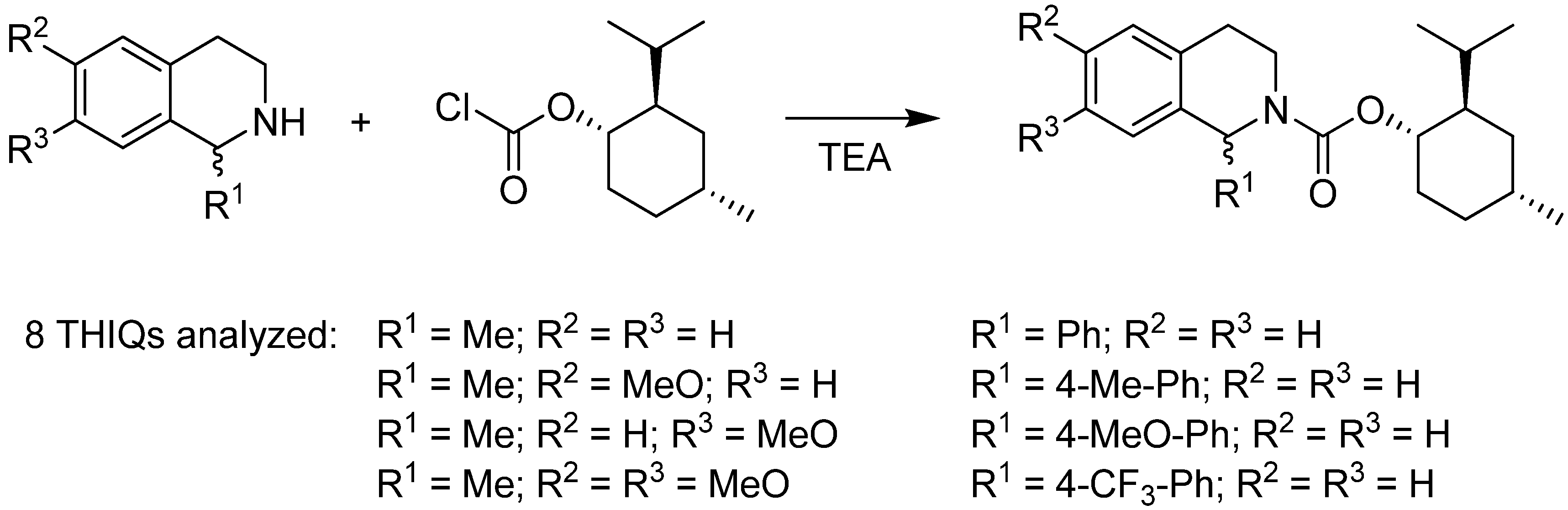
2.2. Determination of Enantioselectivity
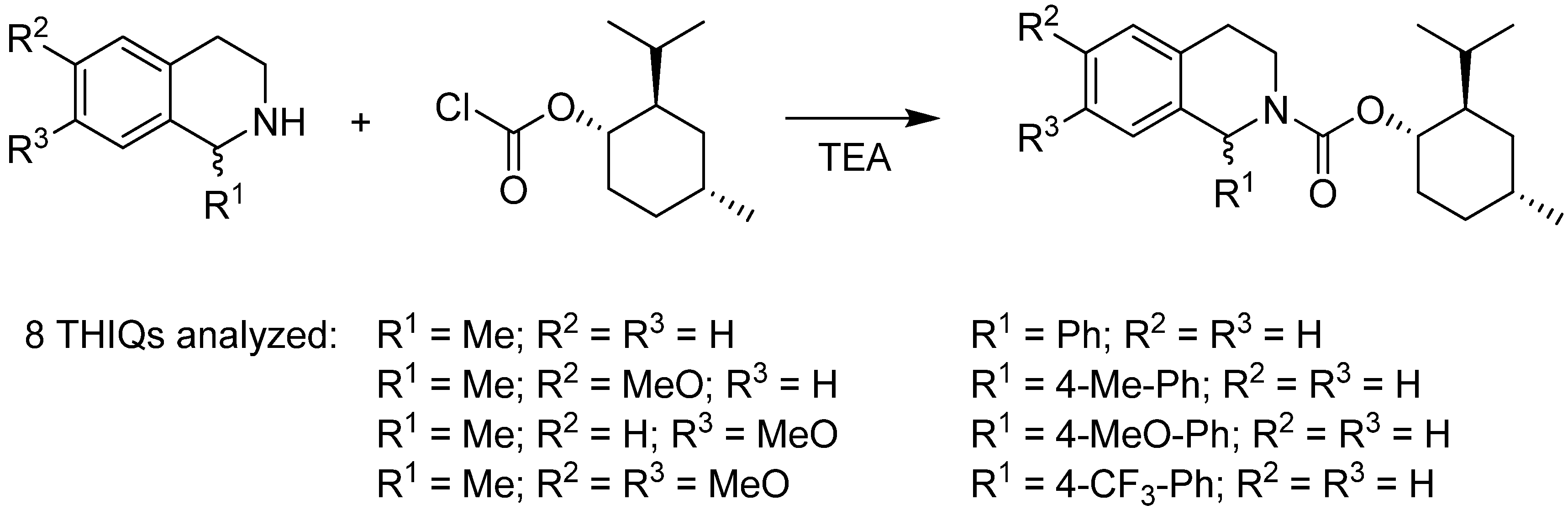
3. Parameters Governing the Reaction Performance
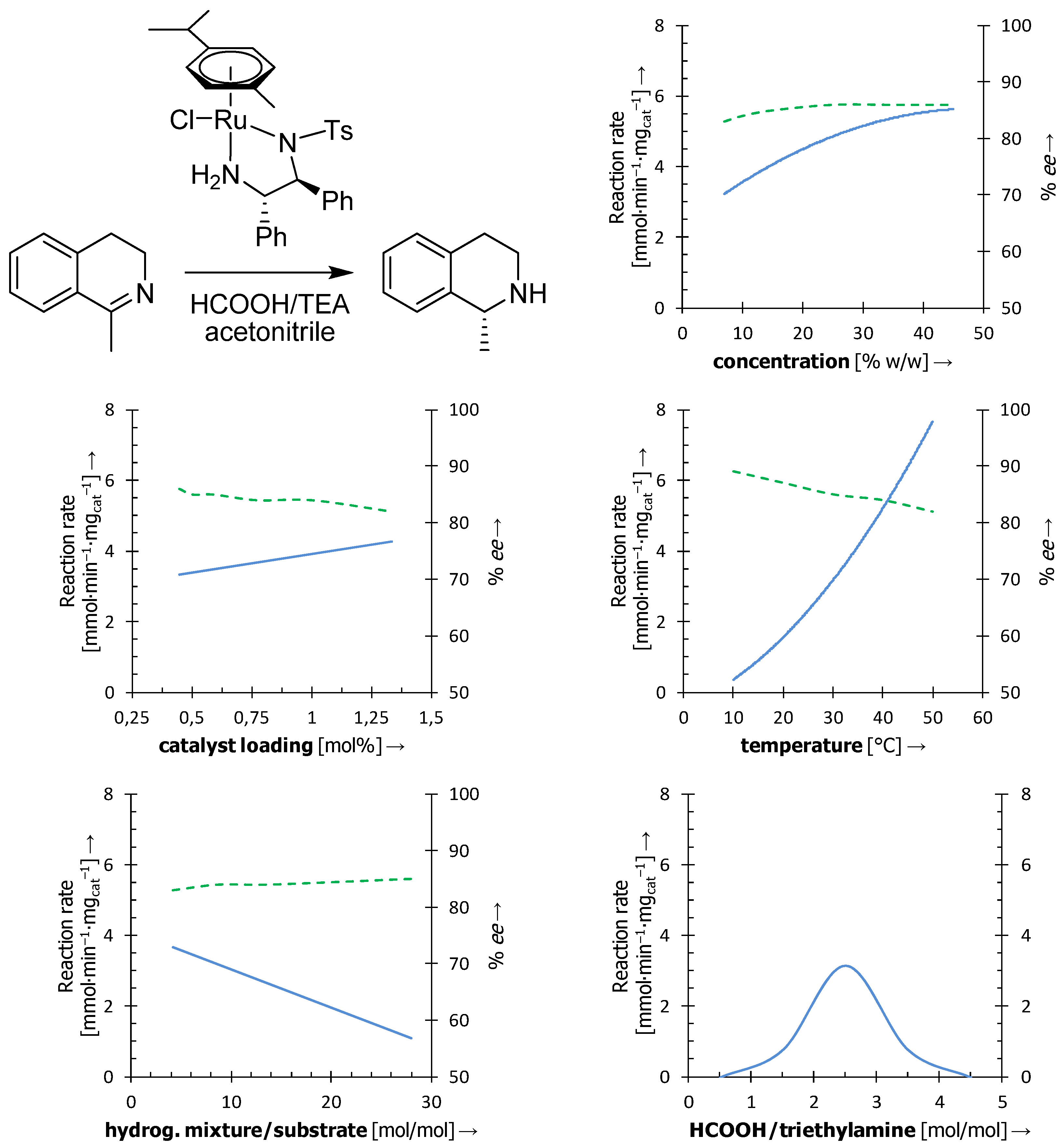
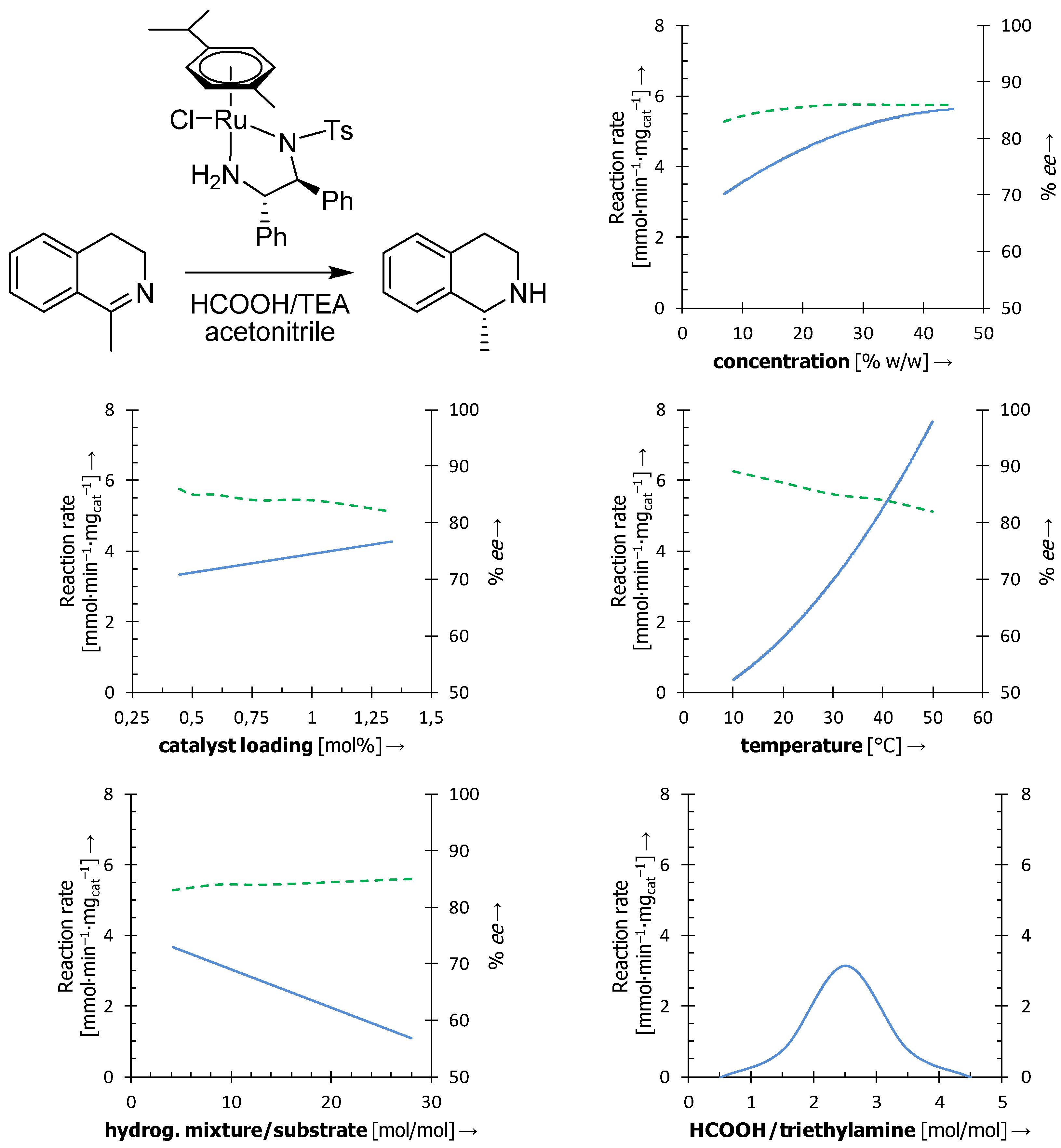
3.1. Influence of the Reaction Conditions
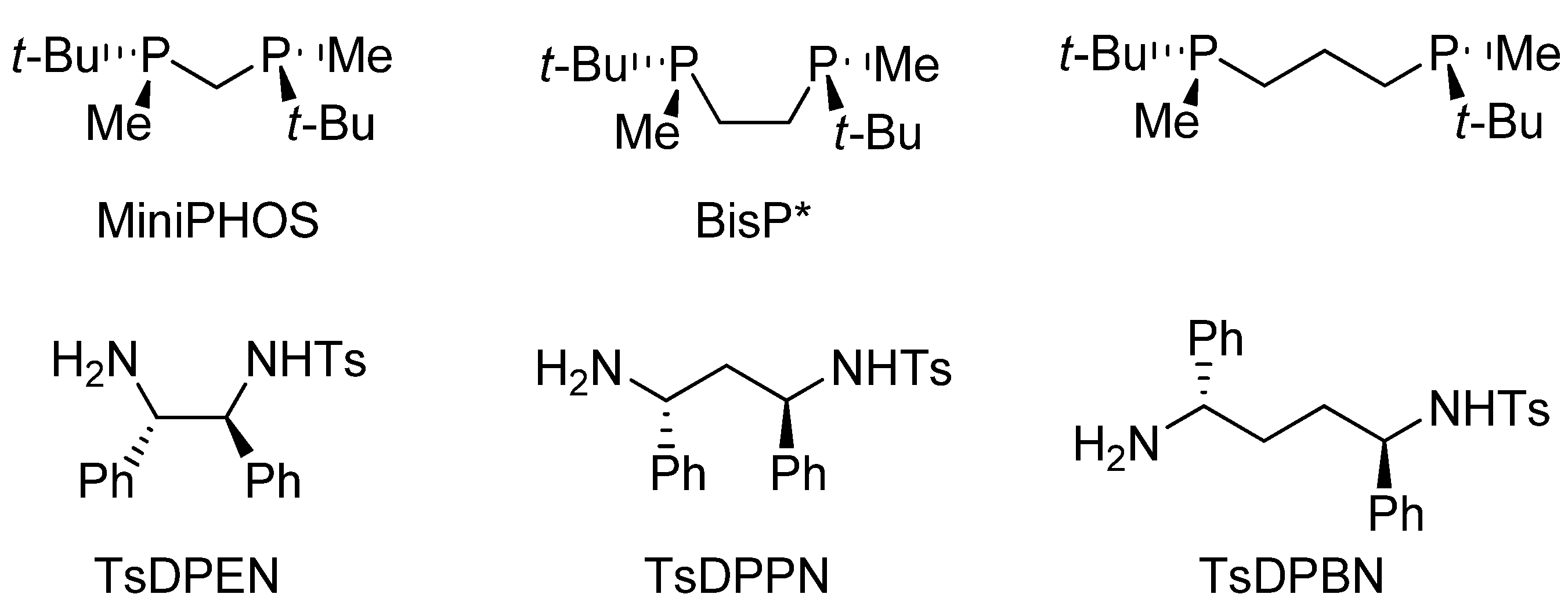
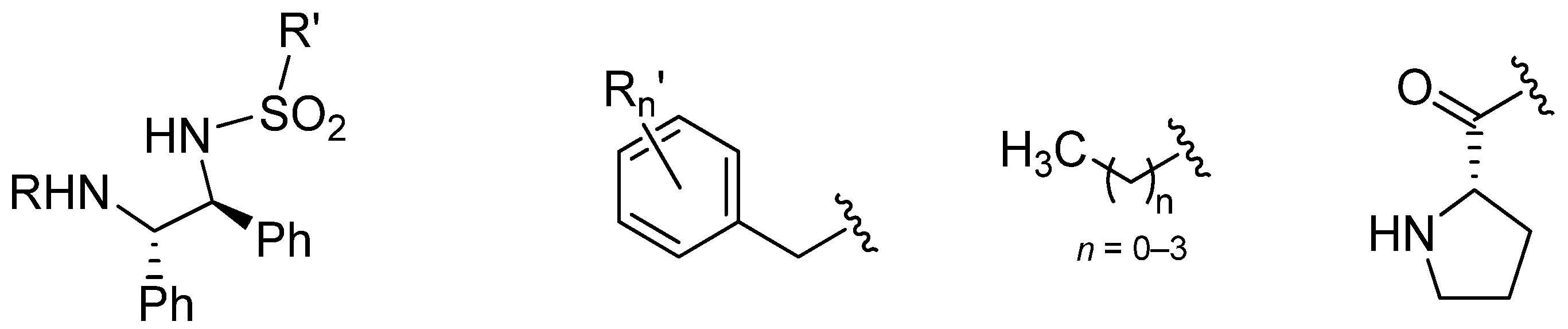
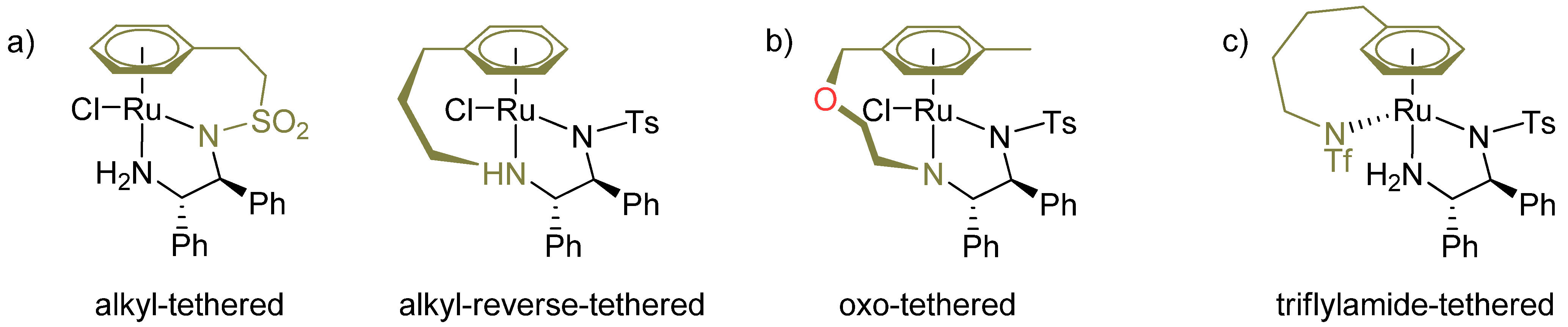
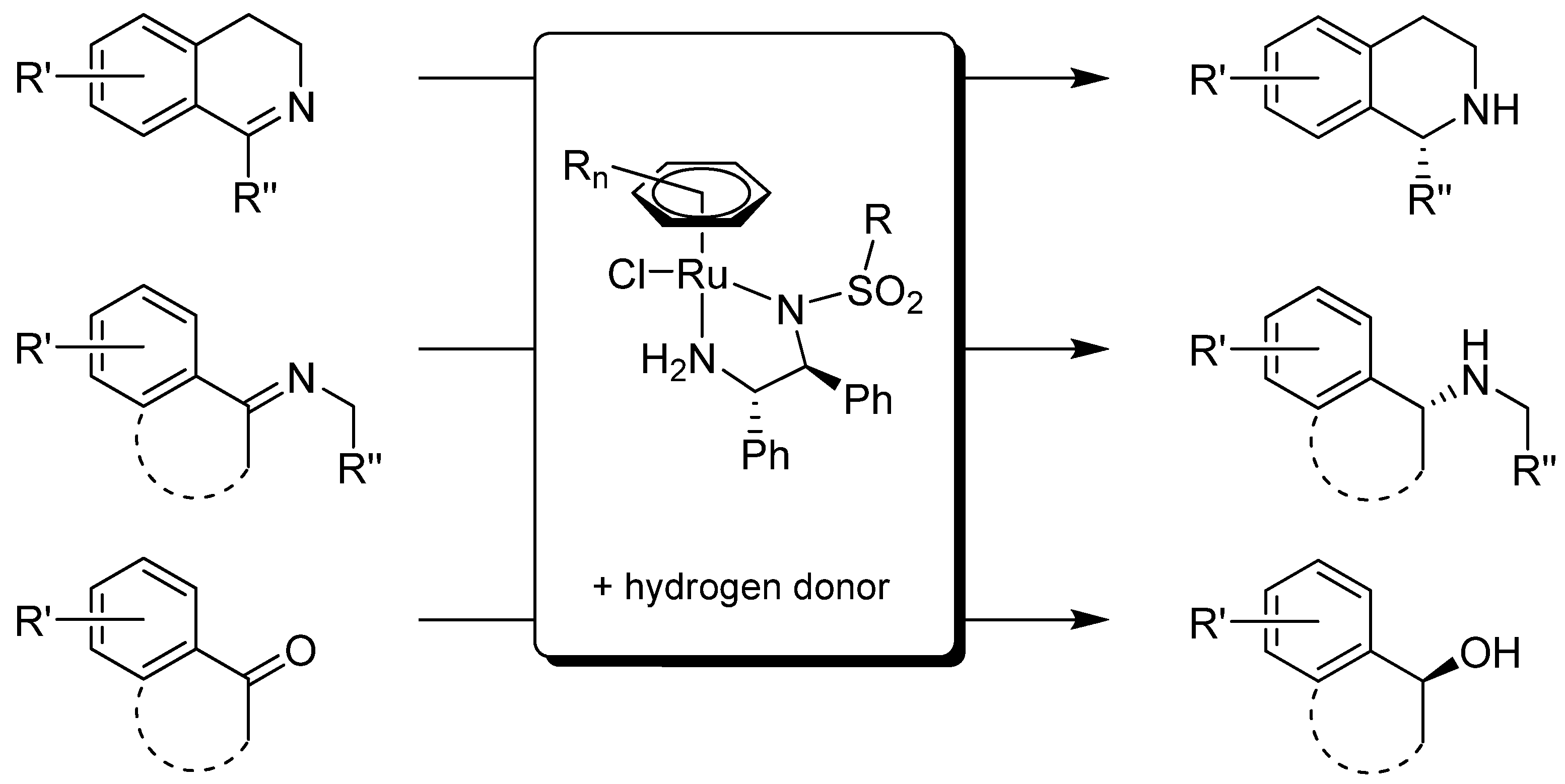
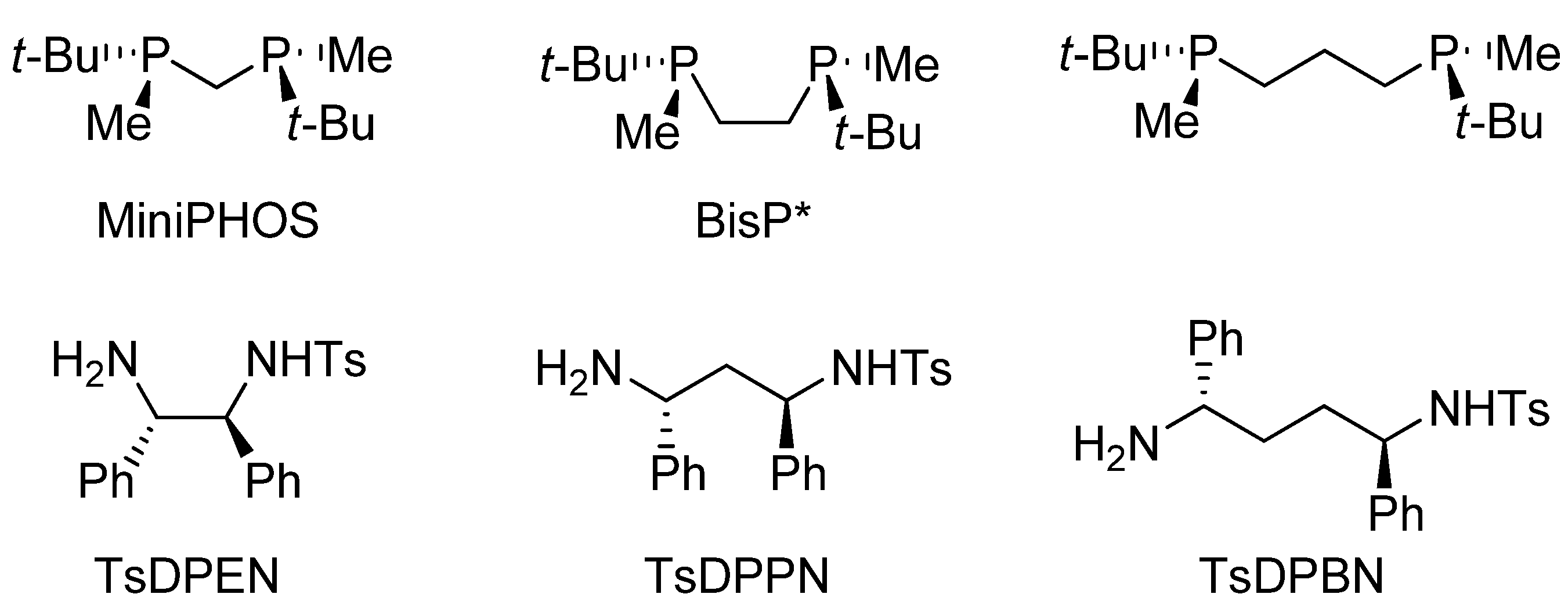
3.2. Structural Effects of the Catalyst


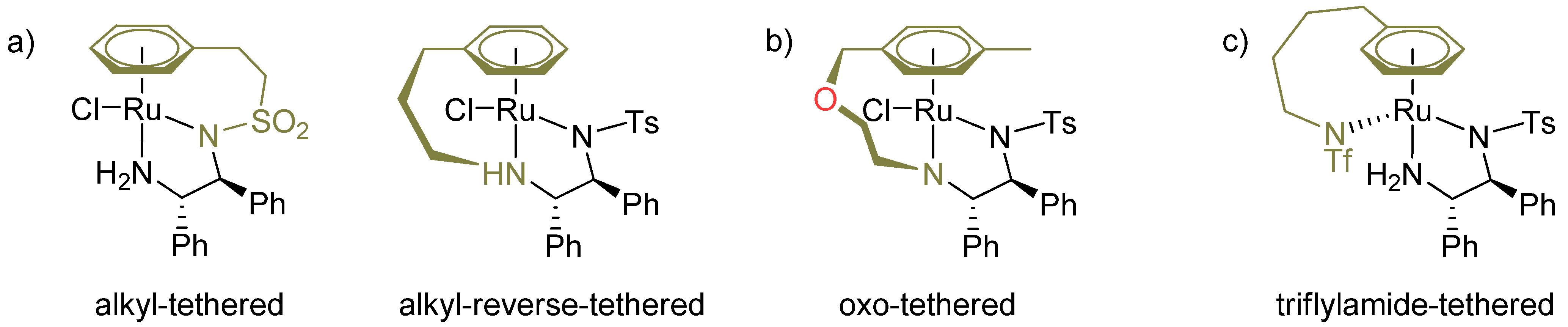
3.3. Structural Effects of the Substrate
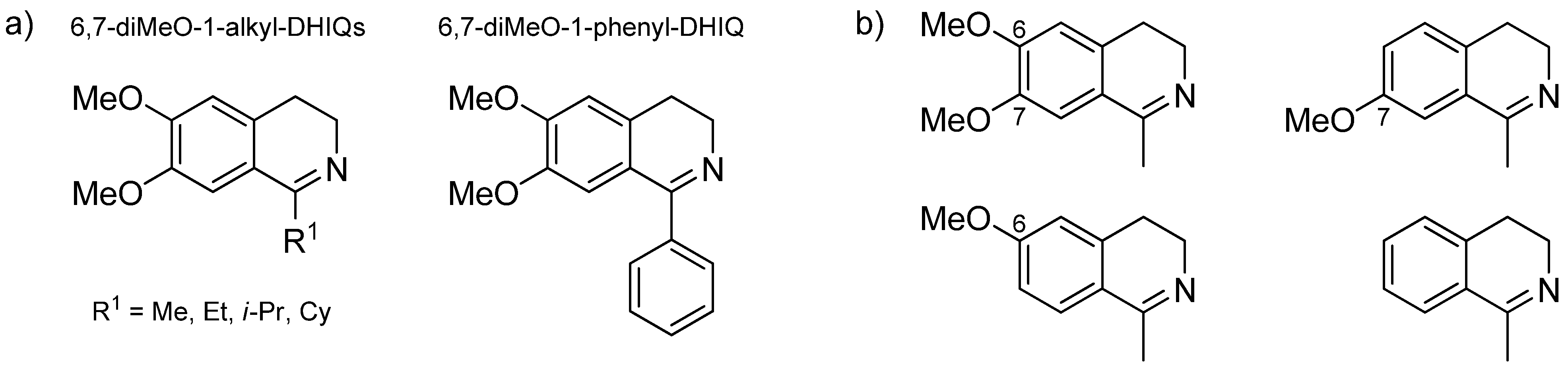
4. Reaction Mechanism
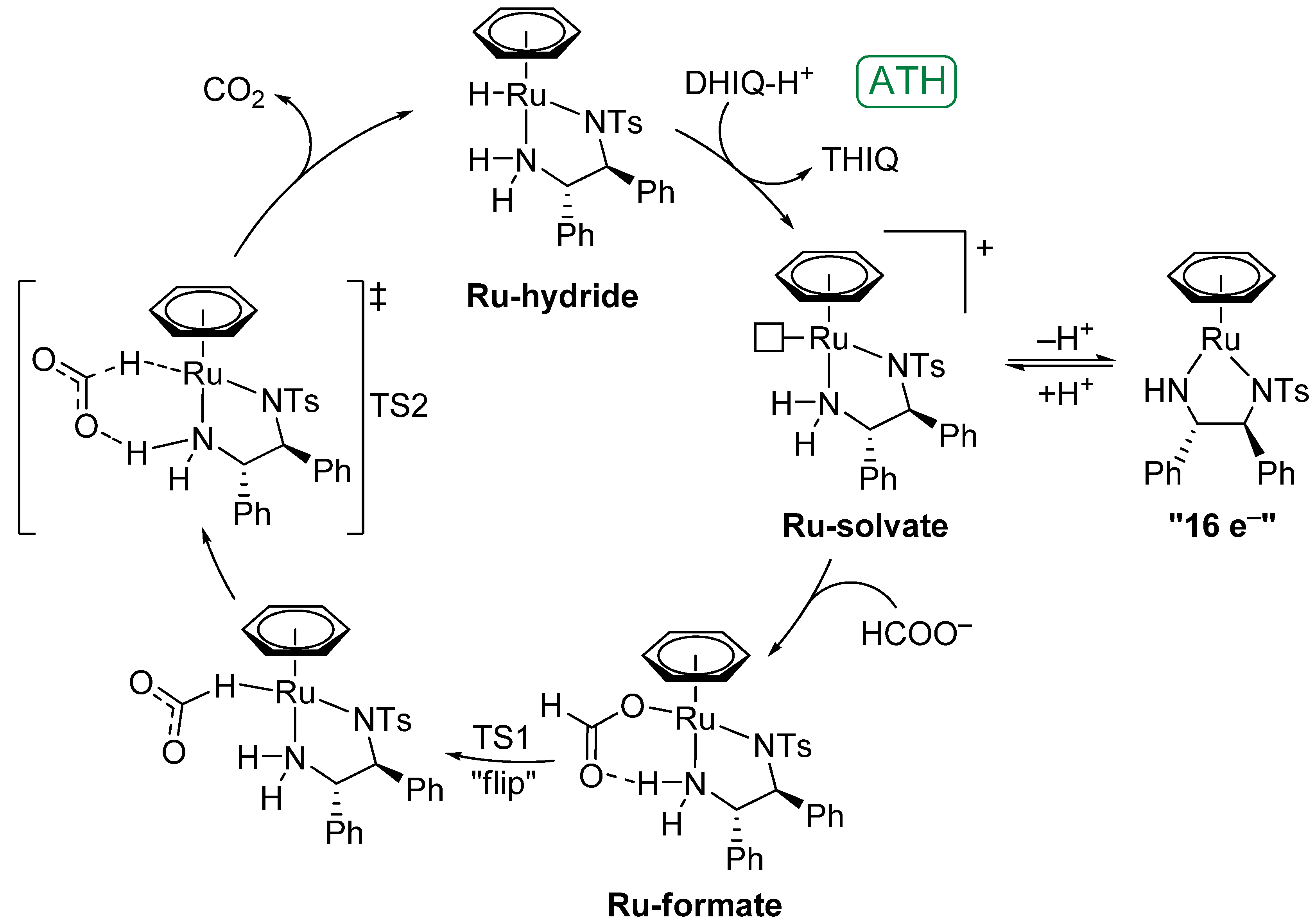
4.1. Formation of the Ruthenium-Hydride Species
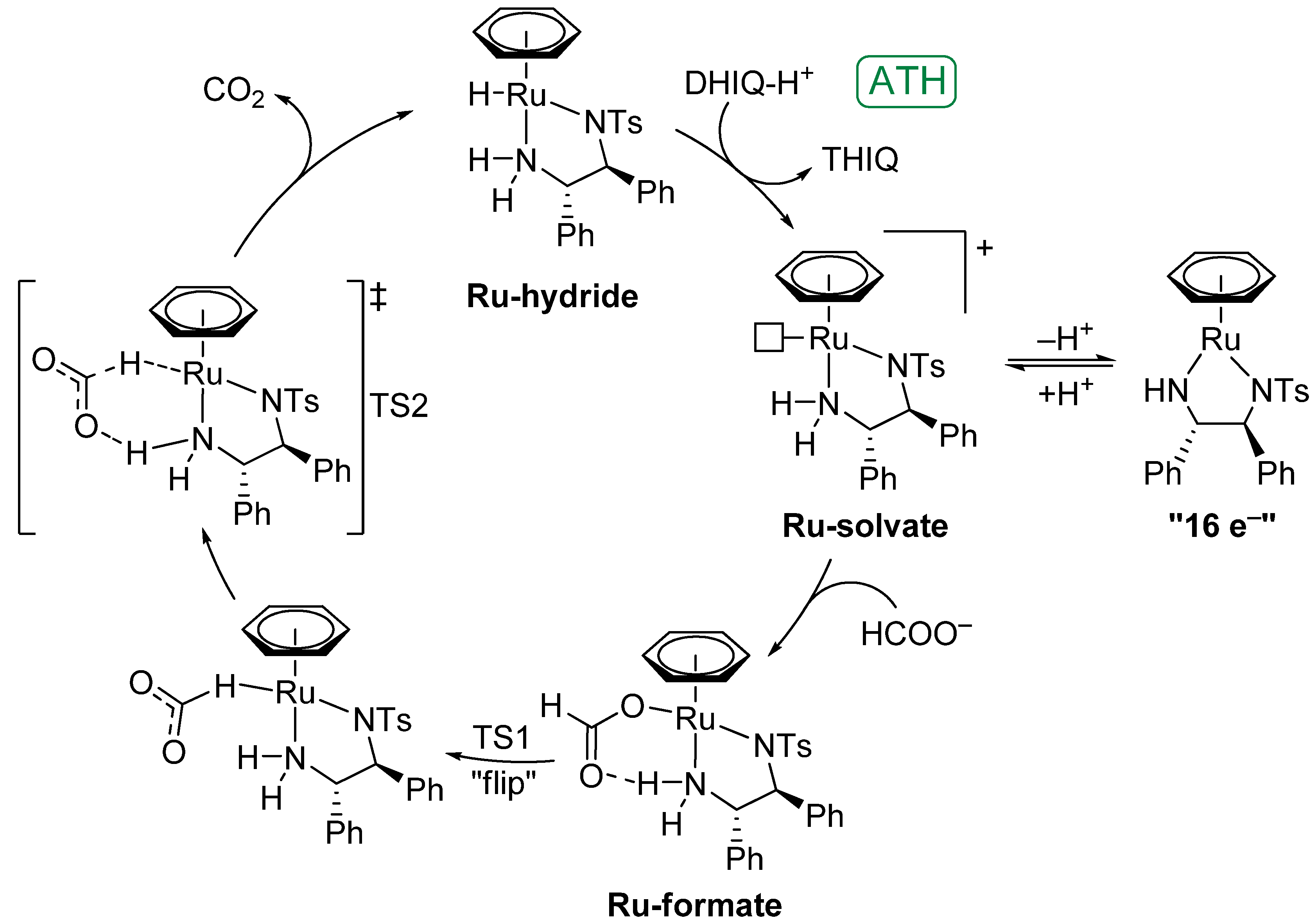

4.2. Mechanism of ATH of Imines and Catalyst Behaviour under Acidic Conditions
4.3. ATH of Activated Olefins
5. Asymmetric Hydrogenation using Gaseous Hydrogen
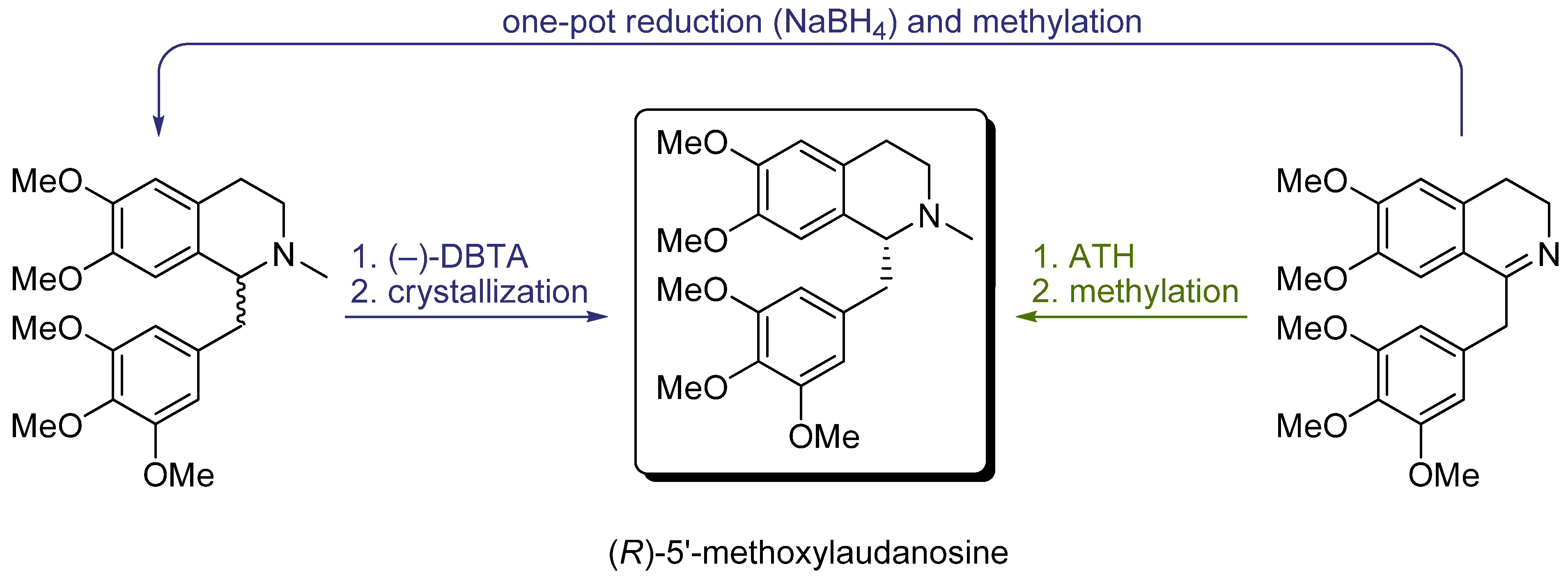
6. Examples of Practical Usage of ATH
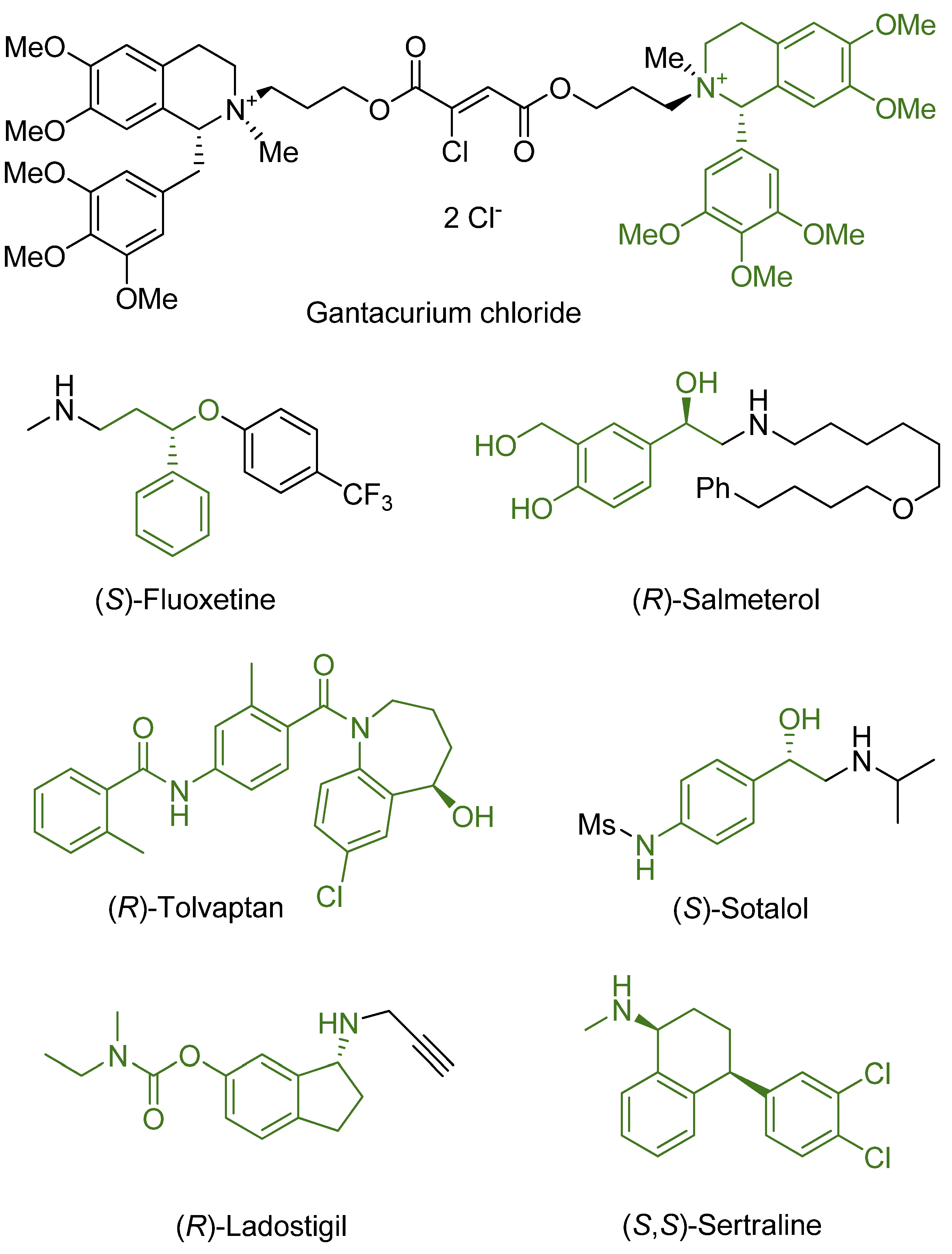
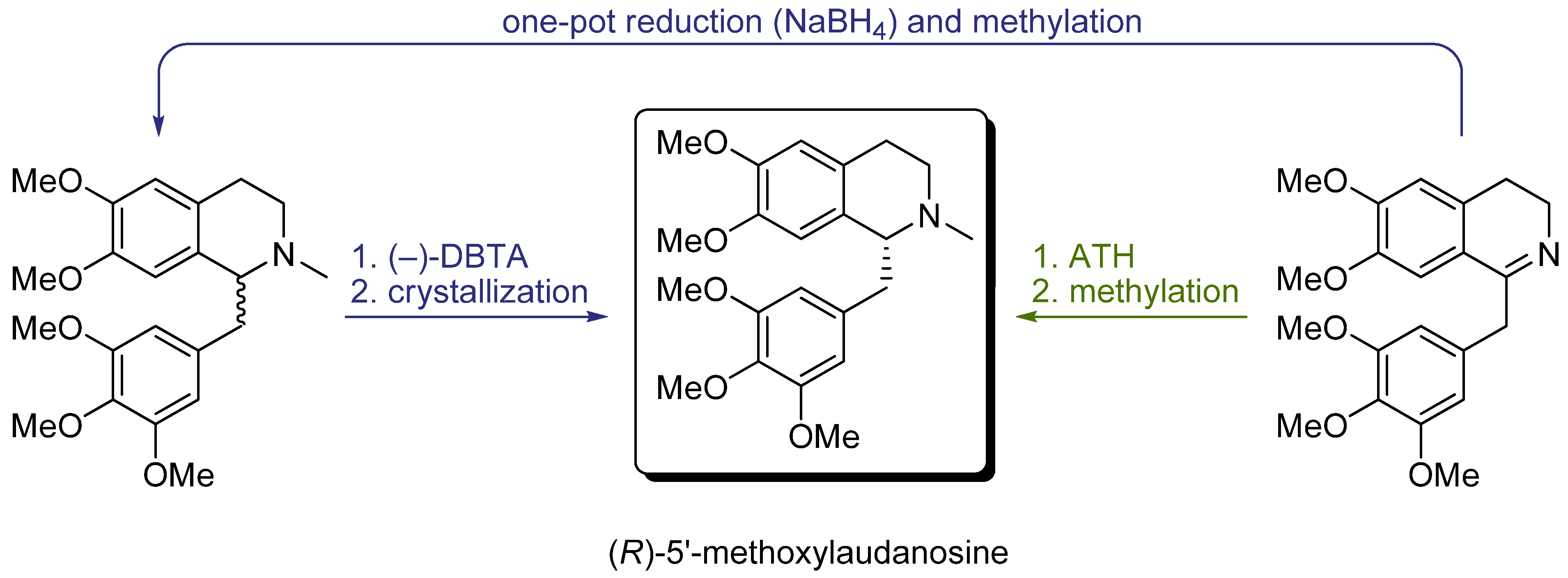
7. Conclusions
Acknowledgments
Conflicts of Interest
References
- Hashiguchi, S.; Fujii, A.; Takehara, J.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of aromatic ketones catalyzed by chiral ruthenium(II) complexes. J. Am. Chem. Soc. 1995, 117, 7562–7563. [Google Scholar] [CrossRef]
- Fujii, A.; Hashiguchi, S.; Uematsu, N.; Ikariya, T.; Noyori, R. Ruthenium(II)-catalyzed asymmetric transfer hydrogenation of ketones using a formic acid−triethylamine mixture. J. Am. Chem. Soc. 1996, 118, 2521–2522. [Google Scholar] [CrossRef]
- Uematsu, N.; Fujii, A.; Hashiguchi, S.; Ikariya, T.; Noyori, R. Asymmetric transfer hydrogenation of imines. J. Am. Chem. Soc. 1996, 118, 4916–4917. [Google Scholar] [CrossRef]
- Václavík, J.; Kačer, P.; Kuzma, M.; Červený, L. Opportunities offered by chiral η6-arene/N-arylsulfonyl-diamine-RuII catalysts in the asymmetric transfer hydrogenation of ketones and imines. Molecules 2011, 16, 5460–5495. [Google Scholar]
- Blackmond, D.G.; Ropic, M.; Stefinovic, M. Kinetic studies of the asymmetric transfer hydrogenation of imines with formic acid catalyzed by Rh-diamine catalysts. Org. Proc. Res. Dev. 2006, 10, 457–463. [Google Scholar] [CrossRef]
- Václavík, J.; Přech, J.; Kuzma, M.; Kačer, P.; Červený, L. In situ monitoring of asymmetric transfer hydrogenation of imines using NMR spectrometry. Chem. Listy 2012, 106, 206–210. [Google Scholar]
- Kajita, M.; Niwa, T.; Fujisaki, M.; Ueki, M.; Niimura, K.; Sato, M.; Egami, K.; Naoi, M.; Yoshida, M.; Nagatsu, T. Detection of 1-phenyl-N-methyl-1,2,3,4-tetrahydroisoquinoline and 1-phenyl-1,2,3,4-tetrahydroisoquinoline in human brain by gas chromatography-tandem mass spectrometry. J. Chromatogr. B 1995, 669, 345–351. [Google Scholar] [CrossRef]
- Makino, Y.; Tasaki, Y.; Ohta, S.; Hirobe, M. Confirmation of the enantiomers of 1-methyl-1,2,3,4-tetrahydroisoquinoline in the mouse brain and foods applying gas chromatography/mass spectrometry with negative ion chemical ionization. Biol. Mass Spectrom. 1990, 19, 415–419. [Google Scholar] [CrossRef]
- Kažoka, H.; Rotkaja, Q.; Varačeva, L. Enantioseparation of 1-phenyl-1,2,3,4-tetrahydroisoquinoline on polysaccharide-based chiral stationary phases. Chromatographia 2011, 73, 123–129. [Google Scholar]
- Lee, A.; Choi, H.J.; Jin, K.B.; Hyun, M.H. Liquid chromatographic resolution of 1-aryl-1,2,3,4-tetrahydroisoquinolines on a chiral stationary phase based on (+)-(18-crown-6)-2,3,11,12-tetracarboxylic acid. J. Chromatogr. A 2011, 1218, 4071–4076. [Google Scholar] [CrossRef]
- Inoue, H.; Matsubara, D.; Tsuruta, Y. Simultaneous analysis of 1,2,3,4-tetrahydroisoquinolines by high-performance liquid chromatography using 4-(5,6-dimethoxy-2-phthalimidinyl)-2-methoxyphenylsulfonyl chloride as a fluorescent labeling reagent. J. Chromatogr. B 2008, 867, 32–36. [Google Scholar] [CrossRef]
- Song, Y.; Xu, J.; Hamme, A.; Liu, Y.-M. Capillary liquid chromatography-tandem mass spectrometry of tetrahydroisoquinoline derived neurotoxins: a study on the blood-brain barrier of rat brain. J. Chromatogr. A 2006, 1103, 229–234. [Google Scholar] [CrossRef]
- Cai, M.; Liu, Y.-M. Quantification of salsolinol enantiomers by stable isotope dilution liquid chromatography with tandem mass spectrometric detection. Rapid Commun. Mass Spectrom. 2008, 22, 4171–4177. [Google Scholar] [CrossRef]
- Přech, J.; Matoušek, V.; Václavík, J.; Pecháček, J.; Syslová, K.; Šot, P.; Januščák, J.; Vilhanová, B.; Kuzma, M.; Kačer, P. Determination of enantiomeric composition of substituted tetrahydroisoquinolines based on derivatization with menthyl chloroformate. Am. J. Anal. Chem. 2013, 4, 125–133. [Google Scholar] [CrossRef]
- Dale, J.A.; Mosher, H.S. Nuclear magnetic resonance enantiomer reagents. Configurational correlations via nuclear magnetic resonance chemical shifts of diastereomeric mandelate, O-methylmandelate, and α-methoxy-α-trifluoromethylphenylacetate (MTPA) Esters. J. Am. Chem. Soc. 1973, 95, 512–519. [Google Scholar]
- Pirkle, W.H.; Burlingame, T.G.; Beare, S.D. Optically active NMR solvents VI. The determination of optical purity and absolute configuration of amines. Tetrahedron Lett. 1968, 9, 5849–5852. [Google Scholar] [CrossRef]
- Vilhanová, B.; Matoušek, V.; Václavík, J.; Syslová, K.; Přech, J.; Pecháček, J.; Šot, P.; Januščák, J.; Toman, J.; Zápal, J.; Kuzma, M.; Kačer, P. Two optimized synthetic pathways toward a chiral precursor of Mivacurium chloride and other skeletal muscle relaxants. Tetrahedron: Asymmetry 2013, 24, 50–55. [Google Scholar]
- Pecháček, J.; Václavík, J.; Přech, J.; Šot, P.; Januščák, J.; Vilhanová, B.; Vavřík, J.; Kuzma, M.; Kačer, P. Asymmetric transfer hydrogenation of imines catalyzed by a Noyori-type Ru(II) complex—a parametric study. Tetrahedron: Asymmetry 2013, 24, 233–239. [Google Scholar]
- Balcells, D.; Maseras, F. Computational approaches to asymmetric synthesis. New J. Chem. 2007, 31, 333–343. [Google Scholar] [CrossRef]
- Zhou, X.; Wu, X.; Yang, B.; Xiao, J. Varying the ratio of formic acid to triethylamine impacts on asymmetric transfer hydrogenation of ketones. J. Mol. Catal. A: Chem. 2012, 357, 133–140. [Google Scholar]
- Wu, X.; Li, X.; Zanotti-Gerosa, A.; Pettman, A.; Liu, J.; Mills, A. J.; Xiao, J. RhIII- and IrIII-catalyzed asymmetric transfer hydrogenation of ketones in water. Chem. Eur. J. 2008, 14, 2209–2222. [Google Scholar] [CrossRef]
- Zhang, J.; Blazecka, P.G.; Bruendl, M.M.; Huang, Y. Ru-TsDPEN with formic acid/Hünig’s base for asymmetric transfer hydrogenation, a practical synthesis of optically enriched N-propyl pantolactam. J. Org. Chem. 2009, 74, 1411–1414. [Google Scholar] [CrossRef]
- Kuzma, M.; Václavík, J.; Novák, P.; Přech, J.; Januščák, J.; Červený, J.; Pecháček, J.; Šot, P.; Vilhanová, B.; Matoušek, V.; Goncharova, I.I.; Urbanová, M.; Kačer, P. New insight into the role of a base in the mechanism of imine transfer hydrogenation on a Ru(II) half-sandwich complex. Dalton Trans. 2013, 42, 5174–5182. [Google Scholar]
- Yamakawa, M.; Yamada, I.; Noyori, R. CH/π attraction: The origin of enantioselectivity in transfer hydrogenation of aromatic carbonyl compounds catalyzed by chiral η6-arene-ruthenium(II) complexes. Angew. Chem. Int. Ed. 2001, 40, 2818–2821. [Google Scholar] [CrossRef]
- Takehara, J.; Hashiguchi, S.; Fujii, A.; Inoue, S.; Ikariya, T.; Noyori, R. Amino alcohol effects on the ruthenium (II)-catalysed asymmetric transfer hydrogenation of ketones in propan-2-ol. Chem. Commun. 1996, 233–234. [Google Scholar]
- Yamada, I.; Noyori, R. Asymmetric transfer hydrogenation of benzaldehydes. Org. Lett. 2000, 2, 3425–3427. [Google Scholar] [CrossRef]
- Vedejs, E.; Trapencieris, P.; Suna, E. Substituted isoquinolines by Noyori transfer hydrogenation: enantioselective synthesis of chiral diamines containing an aniline subunit. J. Org. Chem. 1999, 64, 6724–6729. [Google Scholar] [CrossRef]
- Samano, V.; Ray, J.A.; Thompson, J.B.; Mook, R.A.; Jung, D.K.; Koble, C.S.; Martin, M.T.; Bigham, E.C.; Regitz, C.S.; Feldman, P.L.; Boros, E.E. Synthesis of ultra-short-acting neuromuscular blocker GW 0430: A remarkably stereo- and regioselective tetrahydroisoquinolinium. Org. Lett. 1999, 1, 1993–1996. [Google Scholar] [CrossRef]
- Ohkuma, T.; Utsumi, N.; Watanabe, M.; Tsutsumi, K.; Arai, N.; Murata, K. Asymmetric hydrogenation of α-hydroxy ketones catalyzed by MsDPEN-Cp*Ir(III) complex. Org. Lett. 2007, 9, 2565–2567. [Google Scholar] [CrossRef]
- Shirai, S.; Nara, H.; Kayaki, Y.; Ikariya, T. Remarkable positive effect of silver salts on asymmetric hydrogenation of acyclic imines with Cp*Ir complexes bearing chiral N-sulfonylated diamine ligands. Organometallics 2009, 28, 802–809. [Google Scholar] [CrossRef]
- Chen, F.; Wang, T.; He, Y.; Ding, Z.; Li, Z.; Xu, L.; Fan, Q.-H. Asymmetric hydrogenation of N-alkyl ketimines with phosphine-free, chiral, cationic Ru-MsDPEN catalysts. Chem. Eur. J. 2011, 17, 1109–1113. [Google Scholar] [CrossRef]
- Ariger, M.A.; Carreira, E.M. pH-Independent transfer hydrogenation in water: Catalytic, enantioselective reduction of β-keto esters. Org. Lett. 2012, 14, 4522–4524. [Google Scholar] [CrossRef]
- Yin, L.; Zheng, Y.; Jia, X.; Li, X.; Chan, A. S. C. Efficient and promising asymmetric preparation of enantiopure tolvaptan via transfer hydrogenation with robust catalysts. Tetrahedron: Asymmetry 2010, 21, 2390–2393. [Google Scholar]
- Přech, J.; Václavík, J.; Šot, P.; Pecháček, J.; Vilhanová, B.; Januščák, J.; Syslová, K.; Pažout, R.; Maixner, J.; Zápal, J.; Kuzma, M.; Kačer, P. Asymmetric transfer hydrogenation of 1-phenyl dihydroisoquinolines using Ru(II) diamine catalysts. Catal. Commun. 2013, 36, 67–70. [Google Scholar]
- Li, X.; Blacker, J.; Houson, I.; Wu, X.; Xiao, J. An efficient Ir(III) catalyst for the asymmetric transfer hydrogenation of ketones in neat water. Synlett 2006, 2006, 1155–1160. [Google Scholar] [CrossRef]
- Lu, C.; Luo, Z.; Huang, L.; Li, X. The Ru-catalyzed enantioselective preparation of chiral halohydrins and their application in the synthesis of (R)-clorprenaline and (S)-sotalol. Tetrahedron: Asymmetry 2011, 22, 722–727. [Google Scholar]
- Luo, Z.; Qin, F.; Yan, S.; Li, X. An efficient and promising method to prepare Ladostigil (TV3326) via asymmetric transfer hydrogenation catalyzed by Ru–Cs-DPEN in an HCOONa–H2O–surfactant system. Tetrahedron: Asymmetry 2012, 23, 333–338. [Google Scholar]
- Crépy, K.V.L.; Imamoto, T. Recent developments in catalytic asymmetric hydrogenation. Adv. Synth. Catal. 2003, 345, 79–101. [Google Scholar] [CrossRef]
- Majdloch, R.; Januščák, J.; Kuzma, M.; Kačer, P. Institute of Chemical Technology in Prague. Manuscript in Preparation.
- Martins, J.E.D.; Clarkson, G.J.; Wills, M. Ru(II) complexes of N-alkylated TsDPEN ligands in asymmetric transfer hydrogenation of ketones and imines. Org. Lett. 2009, 11, 847–850. [Google Scholar] [CrossRef]
- Martins, J.E.D.; Contreras Redondo, M.A.; Wills, M. Applications of N′-alkylated derivatives of TsDPEN in the asymmetric transfer hydrogenation of CO and CN bonds. Tetrahedron: Asymmetry 2010, 21, 2258–2264. [Google Scholar]
- Manville, C.V.; Docherty, G.; Padda, R.; Wills, M. Application of proline-functionalised 1,2-diphenylethane-1,2-diamine (DPEN) in asymmetric transfer hydrogenation of ketones. Eur. J. Org. Chem. 2011, 2011, 6893–6901. [Google Scholar] [CrossRef]
- Johnson, T.C.; Totty, W.G.; Wills, M. Application of ruthenium complexes of triazole-containing tridentate ligands to asymmetric transfer hydrogenation of ketones. Org. Lett. 2012, 14, 5230–5233. [Google Scholar] [CrossRef]
- Hannedouche, J.; Clarkson, G.J.; Wills, M. A new class of “tethered” ruthenium(II) catalyst for asymmetric transfer hydrogenation reactions. J. Am. Chem. Soc. 2004, 126, 986–987. [Google Scholar] [CrossRef]
- Hayes, A.M.; Morris, D.J.; Clarkson, G.J.; Wills, M. A class of ruthenium(II) catalyst for asymmetric transfer hydrogenations of ketones. J. Am. Chem. Soc. 2005, 127, 7318–7319. [Google Scholar]
- Kei, F.; Cheung, K.; Hayes, A.M.; Hannedouche, J.; Yim, A.S.Y.; Wills, M. “Tethered” Ru(II) catalysts for asymmetric transfer hydrogenation of ketones. J. Org. Chem. 2005, 70, 3188–3197. [Google Scholar] [CrossRef]
- Morris, D.J.; Hayes, A.M.; Wills, M. The “reverse-tethered” ruthenium (II) catalyst for asymmetric transfer hydrogenation: further applications. J. Org. Chem. 2006, 71, 7035–7044. [Google Scholar] [CrossRef]
- Cheung, F.K.; Lin, C.; Minissi, F.; Crivillé, A.L.; Graham, M.A.; Fox, D.J.; Wills, M. An investigation into the tether length and substitution pattern of arene-substituted complexes for asymmetric transfer hydrogenation of ketones. Org. Lett. 2007, 9, 4659–4662. [Google Scholar] [CrossRef]
- Matharu, D. S.; Martins, J. E. D.; Wills, M. Asymmetric transfer hydrogenation of C=O and C=N bonds by tethered Rh(III) catalysts. Chem. Asian J. 2008, 3, 1374–1383. [Google Scholar] [CrossRef]
- Cheung, F.K.K.; Clarke, A.J.; Clarkson, G.J.; Fox, D.J.; Graham, M.A.; Lin, C.; Crivillé, A.L.; Wills, M. Kinetic and structural studies on “tethered” Ru(II) arene ketone reduction catalysts. Dalton Trans. 2010, 39, 1395–1402. [Google Scholar] [CrossRef]
- Soni, R.; Cheung, F.K.; Clarkson, G.C.; Martins, J.E.D.; Graham, M.A.; Wills, M. The importance of the N-H bond in Ru/TsDPEN complexes for asymmetric transfer hydrogenation of ketones and imines. Org. Biomol. Chem. 2011, 9, 3290–3294. [Google Scholar] [CrossRef]
- Kačer, P.; Kuzma, M.; Leitmannová, E.; Červený, L. Ruthenium complexes for asymmetric transfer hydrogenation. In Organometallic Compounds: Preparation, Structure and Properties; Chin, H.F., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2010; pp. 373–401. [Google Scholar]
- Touge, T.; Hakamata, T.; Hideki, N.; Kobayashi, T.; Sayo, N.; Saito, T.; Kayaki, Y.; Ikariya, T. Oxo-tethered ruthenium(II) complex as a bifunctional catalyst for asymmetric transfer hydrogenation and H2 hydrogenation. J. Am. Chem. Soc. 2011, 133, 14960–14963. [Google Scholar]
- Parekh, V.; Ramsden, A.; Wills, M. Ether-tethered Ru(II)/TsDPEN complexes; synthesis and applications to asymmetric transfer hydrogenation. Catal. Sci. Technol. 2012, 2, 406–414. [Google Scholar] [CrossRef]
- Sandoval, C.A.; Bie, F.; Matsuoka, A.; Yamaguchi, Y.; Naka, H.; Li, Y.; Kato, K.; Utsumi, N.; Tsutsumi, K.; Ohkuma, T.; Noyori, R. Chiral η6-arene/N-tosylethylenediamine–ruthenium (II) complexes: Solution behavior and catalytic activity for asymmetric hydrogenation. Chem. Asian J. 2010, 5, 806–816. [Google Scholar] [CrossRef]
- Ito, M.; Endo, Y.; Ikariya, T. Well-defined triflylamide-tethered arene-Ru(Tsdpen) complexes for catalytic asymmetric hydrogenation of ketones. Organometallics 2008, 27, 6053–6055. [Google Scholar] [CrossRef]
- Ito, M.; Endo, Y.; Tejima, N.; Ikariya, T. Bifunctional triflylamide-tethered Cp′Rh and Cp′Ir complexes: A new entry for asymmetric hydrogenation catalysts. Organometallics 2010, 29, 2397–2399. [Google Scholar] [CrossRef]
- Mao, J.; Baker, D. C. A chiral rhodium complex for rapid asymmetric transfer hydrogenation of imines with high enantioselectivity. Org. Lett. 1999, 1, 841–843. [Google Scholar] [CrossRef]
- Wu, J.; Wang, F.; Ma, Y.; Cui, X.; Cun, L.; Zhu, J.; Deng, J.; Yu, B. Asymmetric transfer hydrogenation of imines and iminiums catalyzed by a water-soluble catalyst in water. Chem. Commun. 2006, 1766–1768. [Google Scholar] [CrossRef]
- Evanno, L.; Ormala, J.; Pihko, P.M. A highly enantioselective access to tetrahydroisoquinoline and β-carboline alkaloids with simple Noyori-type catalysts in aqueous media. Chem. Eur. J. 2009, 15, 12963–12967. [Google Scholar] [CrossRef]
- Chang, M.; Li, W.; Zhang, X. A highly efficient and enantioselective access to tetrahydroisoquinoline alkaloids: asymmetric hydrogenation with an iridium catalyst. Angew. Chem. Int. Ed. 2011, 50, 10679–10681. [Google Scholar]
- Václavík, J.; Pecháček, J.; Vilhanová, B.; Šot, P.; Januščák, J.; Matoušek, V.; Přech, J.; Bártová, S.; Kuzma, M.; Kačer, P. Molecular structure effects in the asymmetric transfer hydrogenation of functionalized dihydroisoquinolines on (S,S)-[RuCl(η6-p-cymene)TsDPEN]. Catal. Lett. 2013, 143, 555–562. [Google Scholar]
- Dub, P. A.; Ikariya, T. Quantum chemical calculations with the inclusion of nonspecific and specific solvation: Asymmetric transfer hydrogenation with bifunctional ruthenium catalysts. J. Am. Chem. Soc. 2013, 135, 2604–2619. [Google Scholar] [CrossRef]
- Koike, T.; Ikariya, T. Mechanistic aspects of formation of chiral ruthenium hydride complexes from 16-electron ruthenium amide complexes and formic acid: Facile reversible decarboxylation and carboxylation. Adv. Synth. Catal. 2004, 346, 37–41. [Google Scholar] [CrossRef]
- Ogo, S.; Abura, T.; Watanabe, Y. pH-Dependent transfer hydrogenation of ketones with HCOONa as a hydrogen donor promoted by (η6-C6Me6)Ru complexes. Organometallics 2002, 21, 2964–2969. [Google Scholar] [CrossRef]
- Aliende, C.; Pérez-Manrique, M.; Jalón, F.A.; Manzano, B.R.; Rodíguez, A.M.; Espino, G. Arene ruthenium complexes as versatile catalysts in water in both transfer hydrogenation of ketones and oxidation of alcohols. Selective deuterium labeling of rac-1-phenylethanol. Organometallics 2012, 31, 6106–6123. [Google Scholar]
- Cao, L.; Sun, C.; Sun, N.; Meng, L.; Chen, D. Theoretical mechanism studies on the electrocatalytic reduction of CO2 to formate by water-stable iridium dihydride pincer complex. Dalton Trans. 2013, 42, 5755–5763. [Google Scholar] [CrossRef]
- Soldevila-Barreda, J.J.; Bruijnincx, P.C.A.; Habtemariam, A.; Clarkson, G.J.; Deeth, R.J.; Sadler, P.J. Improved catalytic activity of ruthenium–arene complexes in the reduction of NAD+. Organometallics 2012, 31, 5958–5967. [Google Scholar] [CrossRef]
- Letko, C.S.; Heiden, Z.M.; Rauchfuss, T.B.; Wilson, S.R. Coordination chemistry of the soft chiral Lewis acid [Cp*Ir(TsDPEN)]+. Inorg. Chem. 2011, 50, 5558–5566. [Google Scholar] [CrossRef]
- Jolley, K.E.; Zanotti-Gerosa, A.; Hancock, F.; Dyke, A.; Grainger, D.M.; Medlock, J.A.; Nedden, H.G.; Le Paih, J.J.M.; Roseblade, S.J.; Seger, A.; et al. Application of tethered ruthenium catalysts to asymmetric hydrogenation of ketones, and the selective hydrogenation of aldehydes. Adv. Synth. Catal. 2012, 354, 2545–2555. [Google Scholar]
- Noyori, R.; Hashiguchi, S. Asymmetric transfer hydrogenation catalyzed by chiral ruthenium complexes. Acc. Chem. Res. 1997, 30, 97–102. [Google Scholar] [CrossRef]
- Åberg, J.B.; Samec, J.S.M.; Bäckvall, J.-E. Mechanistic investigation on the hydrogenation of imines by [p-(Me2CH)C6H4Me]RuH(NH2CHPhCHPhNSO2C6H4-p-CH3). Experimental support for an ionic pathway. Chem. Commun. 2006, 2771–2773. [Google Scholar]
- Václavík, J.; Kuzma, M.; Přech, J.; Kačer, P. Asymmetric transfer hydrogenation of imines and ketones using chiral RuIICl(η6-p-cymene)[(S,S)-N-TsDPEN] as a catalyst: A computational study. Organometallics 2011, 30, 4822–4829. [Google Scholar] [CrossRef]
- Šot, P.; Kuzma, M.; Václavík, J.; Pecháček, J.; Přech, J.; Januščák, J.; Kačer, P. Asymmetric transfer hydrogenation of acetophenone N-benzylimine using [RuIICl(S,S)-N-(TsDPEN)η6-(p-cymene)]: A DFT study. Organometallics 2012, 31, 6496–6499. [Google Scholar] [CrossRef]
- Xue, D.; Chen, Y.-C.; Cui, X.; Wang, Q.-W.; Zhu, J.; Deng, J.-G. Transfer hydrogenation of activated C=C bonds catalyzed by ruthenium amido complexes: Reaction scope, limitation, and enantioselectivity. J. Org. Chem. 2005, 70, 3584–3591. [Google Scholar] [CrossRef]
- Tang, W.; Zhang, X. New chiral phosphorus ligands for enantioselective hydrogenation. Chem. Rev. 2003, 103, 3029–3069. [Google Scholar] [CrossRef]
- Václavík, J.; Kačer, P.; Červený, L. Rational design of chiral ruthenium complexes for asymmetric hydrogenations. In Homogeneous Catalysts: Types, Reactions and Applications; Poehler, A.C., Ed.; Nova Science Publishers, Inc.: Hauppauge, NY, USA, 2011; pp. 127–153. [Google Scholar]
- Lühr, S.; Holz, J.; Börner, A. The synthesis of chiral phosphorus ligands for use in homogeneous metal catalysis. ChemCatChem 2011, 3, 1708–1730. [Google Scholar] [CrossRef]
- Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Murata, K.; Sandoval, C.; Noyori, R. The hydrogenation/transfer hydrogenation network: Asymmetric hydrogenation of ketones with chiral η6-arene/N-tosylethylenediamine–ruthenium(II) catalysts. J. Am. Chem. Soc. 2006, 128, 8724–8725. [Google Scholar]
- Sandoval, C.A.; Ohkuma, T.; Utsumi, N.; Tsutsumi, K.; Murata, K.; Noyori, R. Mechanism of asymmetric hydrogenation of acetophenone catalyzed by chiral η6-arene-N-tosylethylenediamine-ruthenium(II) complexes. Chem. Asian J. 2006, 1, 102–110. [Google Scholar] [CrossRef]
- Chen, Y.; Tang, Y.; Liu, S.; Lei, M.; Fang, W. Mechanism and influence of acid in hydrogenation of ketones by η6-arene/N-tosylethylenediamine ruthenium (II). Organometallics 2009, 28, 2078–2084. [Google Scholar]
- Ohkuma, T.; Tsutsumi, K.; Utsumi, K.; Arai, N.; Noyori, R.; Murata, K. Asymmetric hydrogenation of α-chloro aromatic ketones catalyzed by η6-arene/TsDPEN-ruthenium(II) complexes. Org. Lett. 2007, 9, 255–257. [Google Scholar]
- Ohkuma, T.; Utsumi, N.; Watanabe, M.; Tsutsumi, K.; Arai, N.; Murata, K. Asymmetric hydrogenation of α-hydroxy aromatic ketones catalyzed by MsDPEN–Cp*Ir(III) Complex. Org. Lett. 2007, 13, 2565–2567. [Google Scholar]
- Li, C.; Xiao, J. Asymmetric hydrogenation of cyclic imines with an ionic Cp*Rh(III) catalyst. J. Am. Chem. Soc. 2008, 130, 13208–13209. [Google Scholar] [CrossRef]
- Chen, F.; Ding, Z.; He, Y.; Qin, J.; Wang, T.; Fan, Q.-H. Asymmetric hydrogenation of N-alkyl and N-aryl ketimines using chiral cationic Ru(diamine) complexes as catalysts: The counteranion and solvent effects, And substrate scope. Tetrahedron 2012, 68, 5248–5257. [Google Scholar] [CrossRef]
- Chen, F.; Ding, Z.; Qin, J.; Wang, T.; He, Y.; Fan, Q. Highly effective asymmetric hydrogenation of cyclic N-alkyl imines with chiral cationic Ru-MsDPEN catalysts. Org. Lett. 2011, 13, 4348–4351. [Google Scholar] [CrossRef]
- Kaldor, I.; Feldman, P.L.; Mook, R.A.; Ray, J.A.; Samano, V.; Sefler, A.M.; Thompson, J.B.; Travis, B.R.; Boros, E.E. Stereocontrolled synthesis of cis-dibenzoquinolizine chlorofumarates: curare-like agents of ultrashort duration. J. Org. Chem. 2001, 66, 3495–3501. [Google Scholar] [CrossRef]
- Boros, E.E.; Samano, V.; Ray, J.A.; Thompson, J.B.; Jung, D.K.; Kaldor, I.; Koble, C.S.; Martin, M.T.; Styles, V.L.; Mook, R.A.; et al. Neuromuscular blocking activity and therapeutic potential of mixed-tetrahydroisoquinolinium halofumarates and halosuccinates in rhesus monkeys. J. Med. Chem. 2003, 46, 2502–2515. [Google Scholar] [CrossRef]
- Li, Y.; Li, Z.; Li, F.; Wang, Q.; Tao, F. Preparation of polymer-supported Ru-TsDPEN catalysts and use for enantioselective synthesis of (S)-fluoxetine. Org. Biomol. Chem. 2005, 3, 2513–2518. [Google Scholar] [CrossRef]
- Liu, J.; Zhou, D.; Jia, X.; Huang, L.; Li, X.; Chan, A.S.C. A convenient synthesis of (R)-salmeterol via Rh-catalyzed asymmetric transfer hydrogenation. Tetrahedron: Asymmetry 2008, 19, 1824–1828. [Google Scholar]
- Huang, L.; Liu, J.; Shan, W.; Liu, B. A. O.; Shi, A.; Li, X. The asymmetric synthesis of (R,R)-formoterol via transfer hydrogenation with polyethylene glycol bound Rh catalyst in PEG2000 and water. Chirality 2010, 211, 206–211. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Václavík, J.; Šot, P.; Vilhanová, B.; Pecháček, J.; Kuzma, M.; Kačer, P. Practical Aspects and Mechanism of Asymmetric Hydrogenation with Chiral Half-Sandwich Complexes. Molecules 2013, 18, 6804-6828. https://doi.org/10.3390/molecules18066804
Václavík J, Šot P, Vilhanová B, Pecháček J, Kuzma M, Kačer P. Practical Aspects and Mechanism of Asymmetric Hydrogenation with Chiral Half-Sandwich Complexes. Molecules. 2013; 18(6):6804-6828. https://doi.org/10.3390/molecules18066804
Chicago/Turabian StyleVáclavík, Jiří, Petr Šot, Beáta Vilhanová, Jan Pecháček, Marek Kuzma, and Petr Kačer. 2013. "Practical Aspects and Mechanism of Asymmetric Hydrogenation with Chiral Half-Sandwich Complexes" Molecules 18, no. 6: 6804-6828. https://doi.org/10.3390/molecules18066804
APA StyleVáclavík, J., Šot, P., Vilhanová, B., Pecháček, J., Kuzma, M., & Kačer, P. (2013). Practical Aspects and Mechanism of Asymmetric Hydrogenation with Chiral Half-Sandwich Complexes. Molecules, 18(6), 6804-6828. https://doi.org/10.3390/molecules18066804