5-(5-Aryl-1,3,4-oxadiazole-2-carbonyl)furan-3-carboxylate and New Cyclic C-Glycoside Analogues from Carbohydrate Precursors with MAO-B, Antimicrobial and Antifungal Activities

Abstract
:1. Introduction
2. Results and Discussion
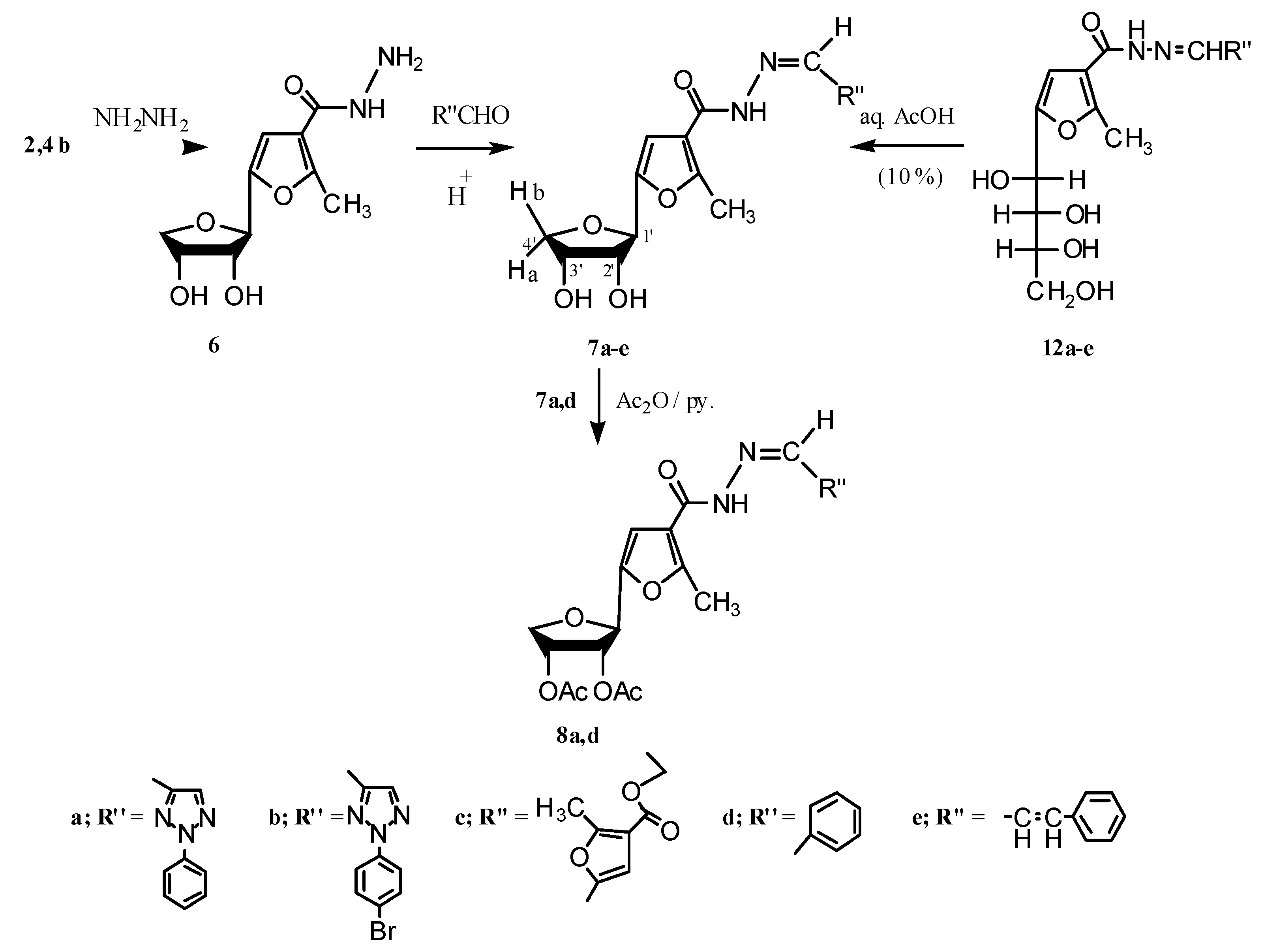
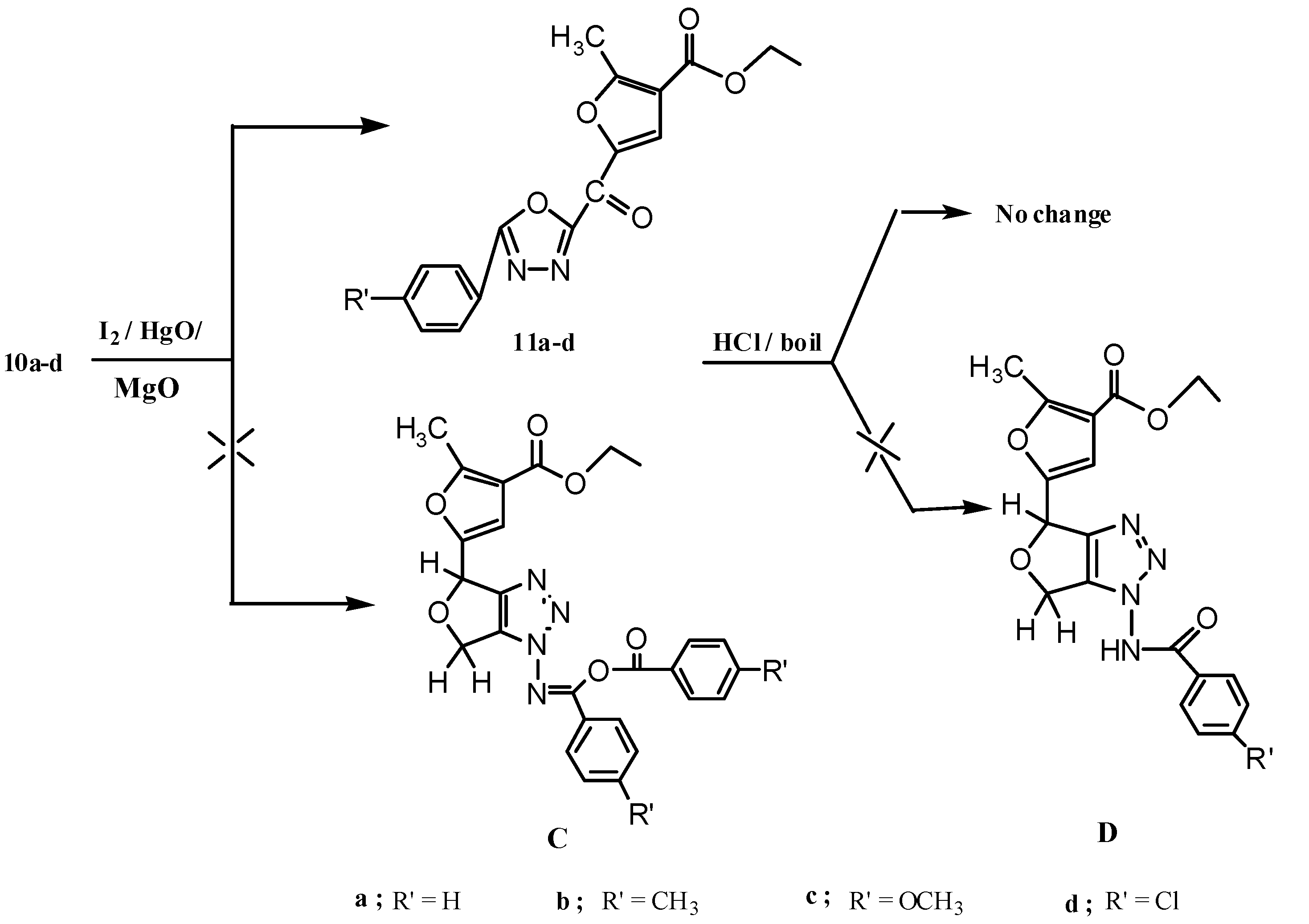
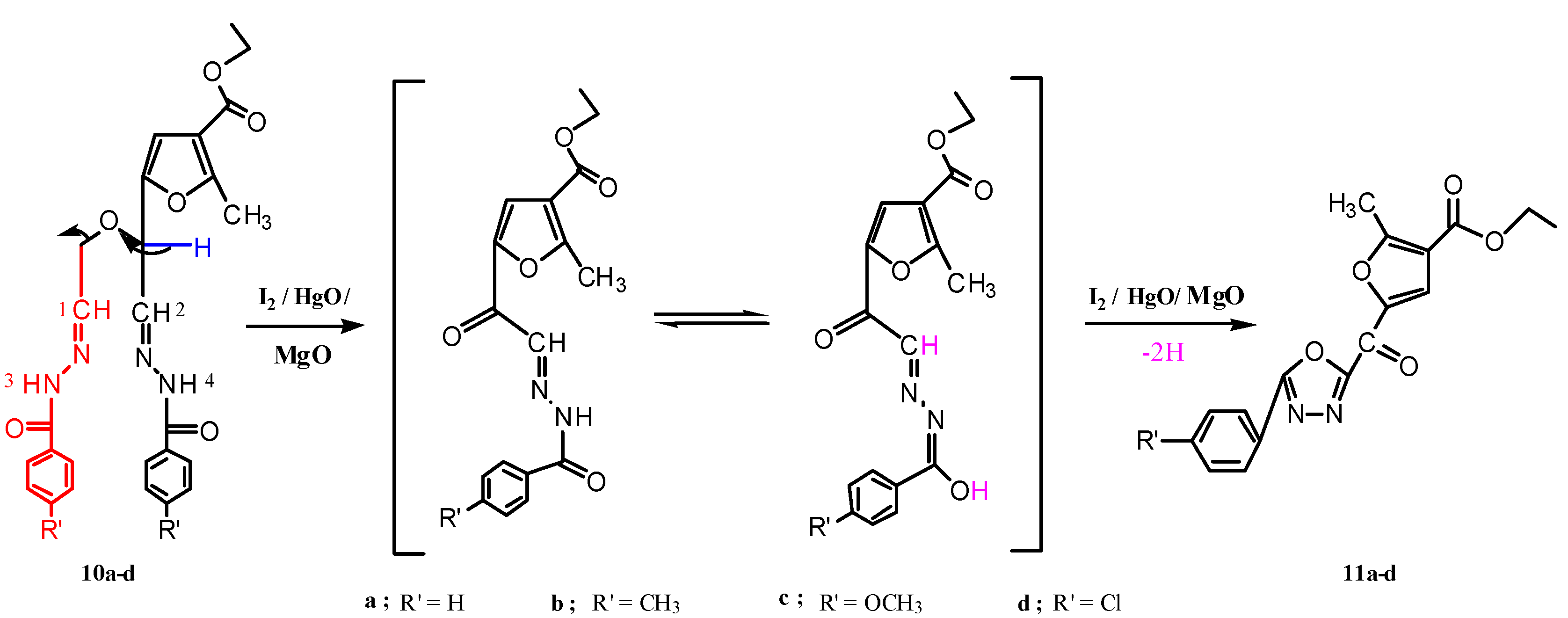
2.1. Chemistry
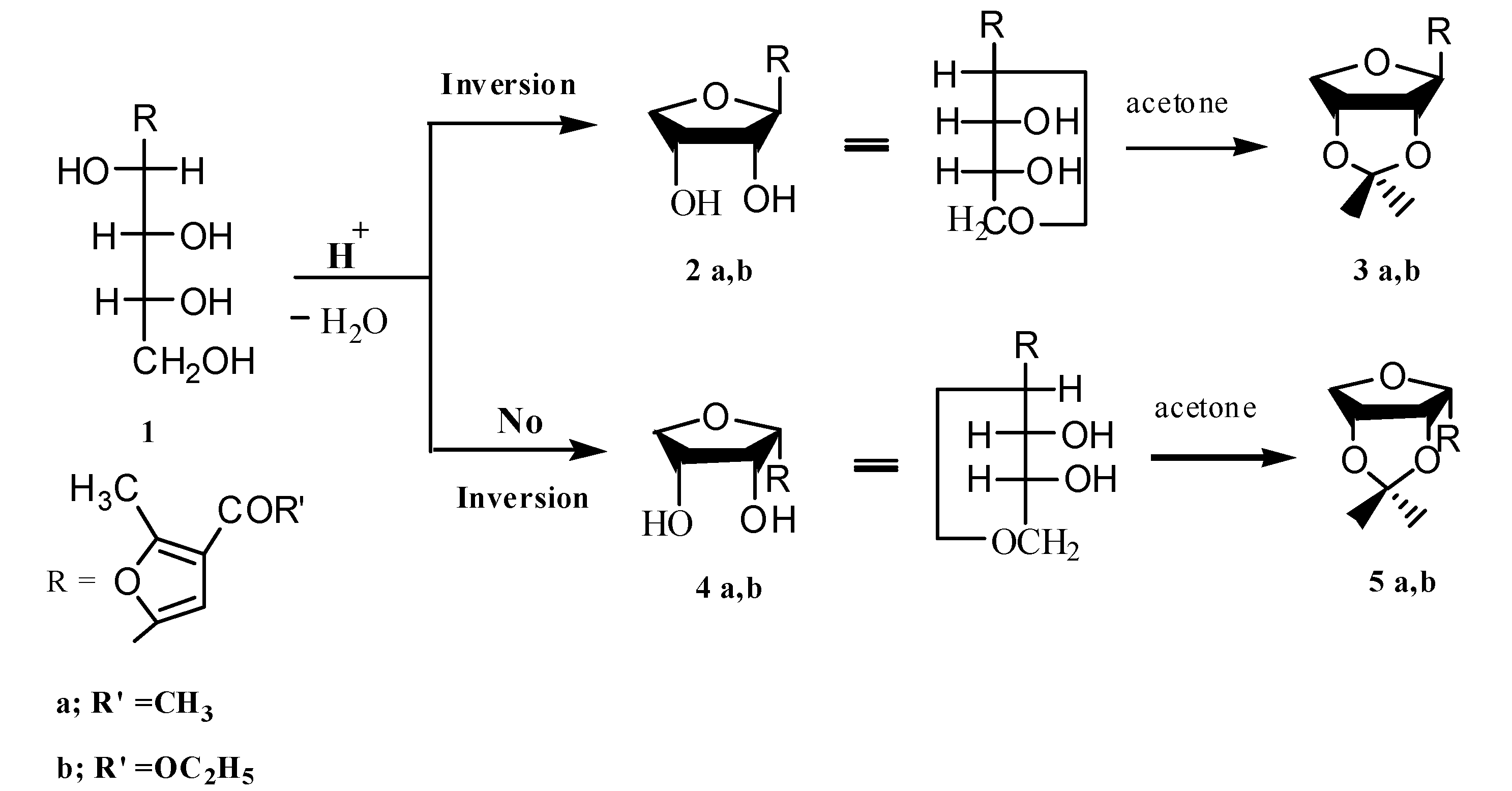
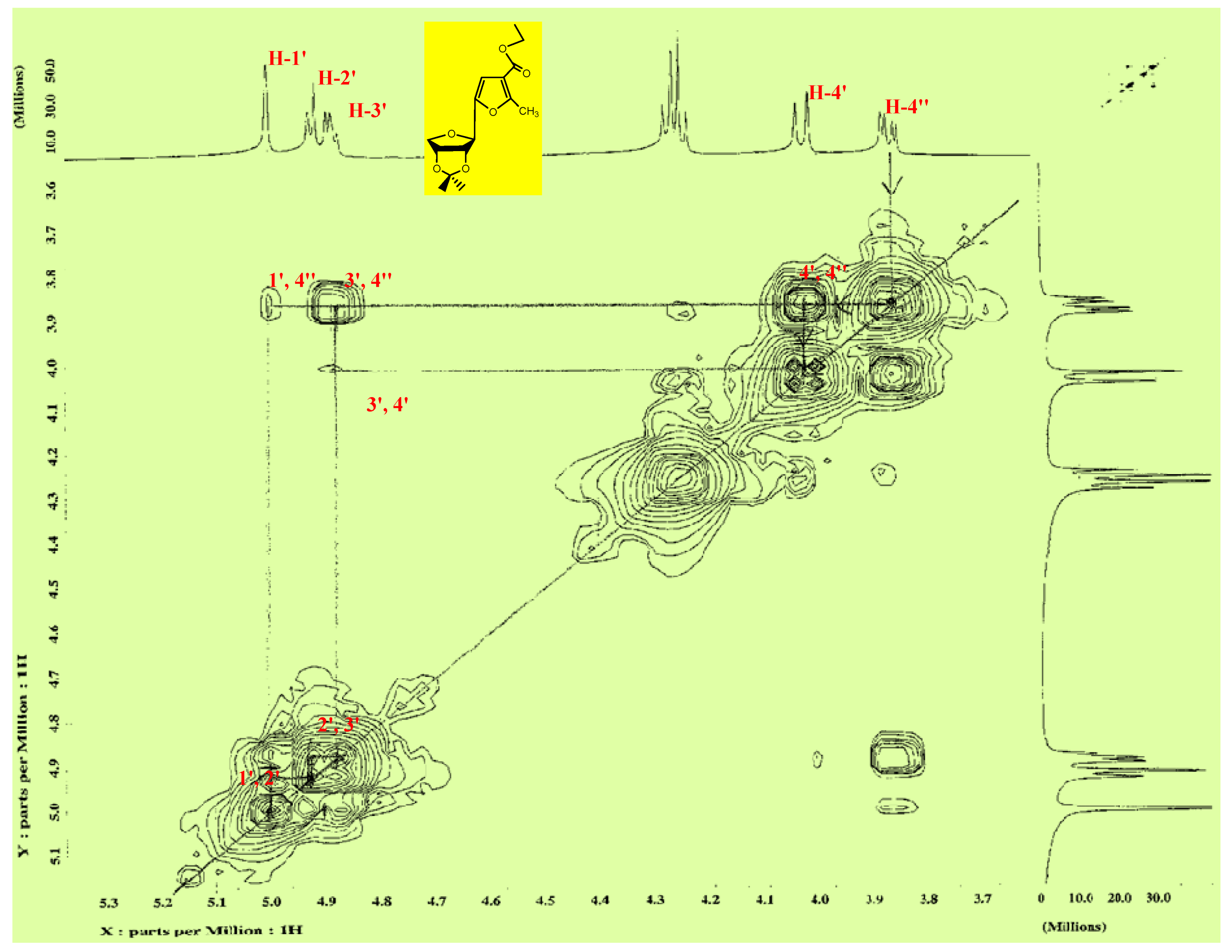
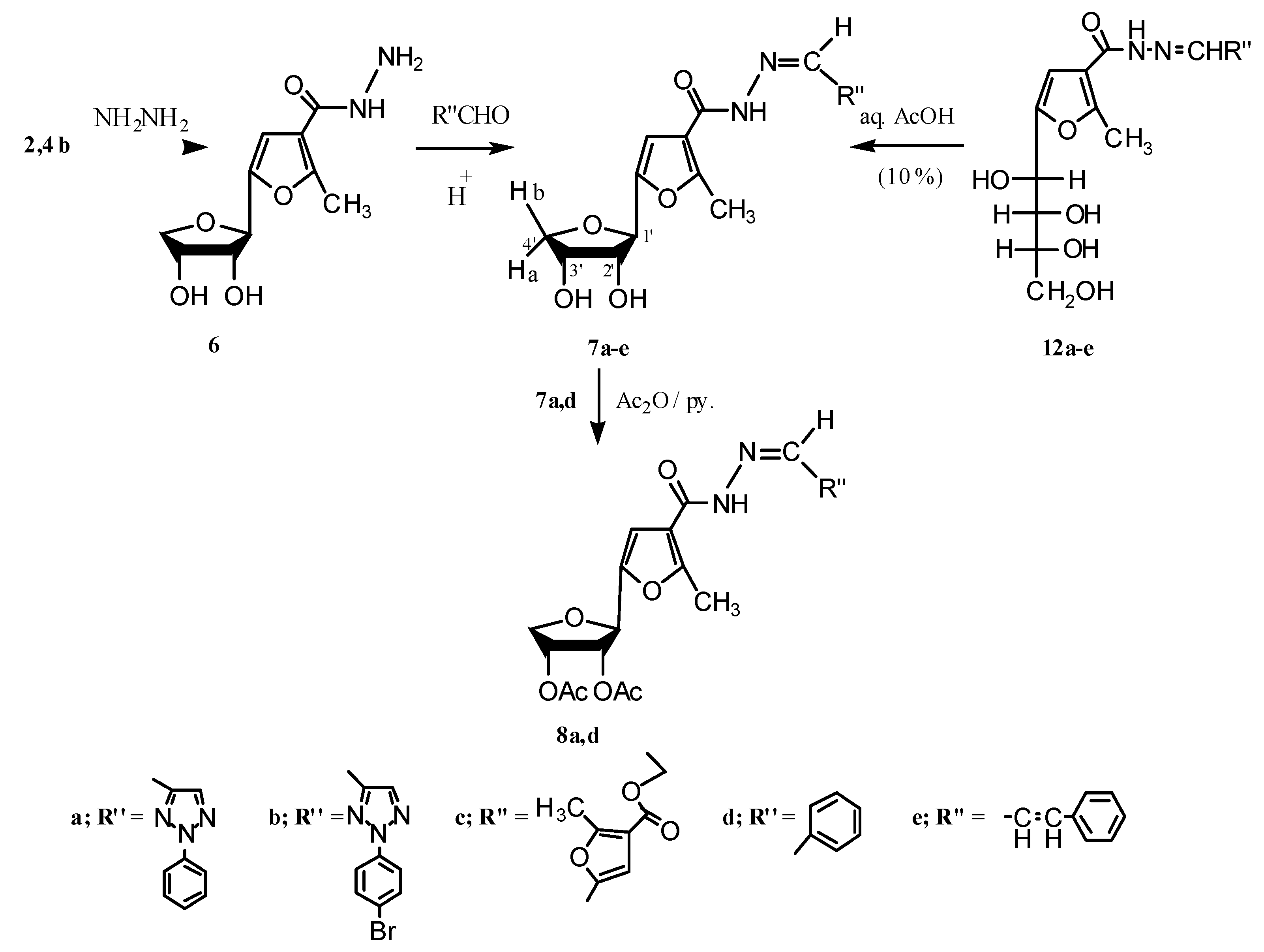

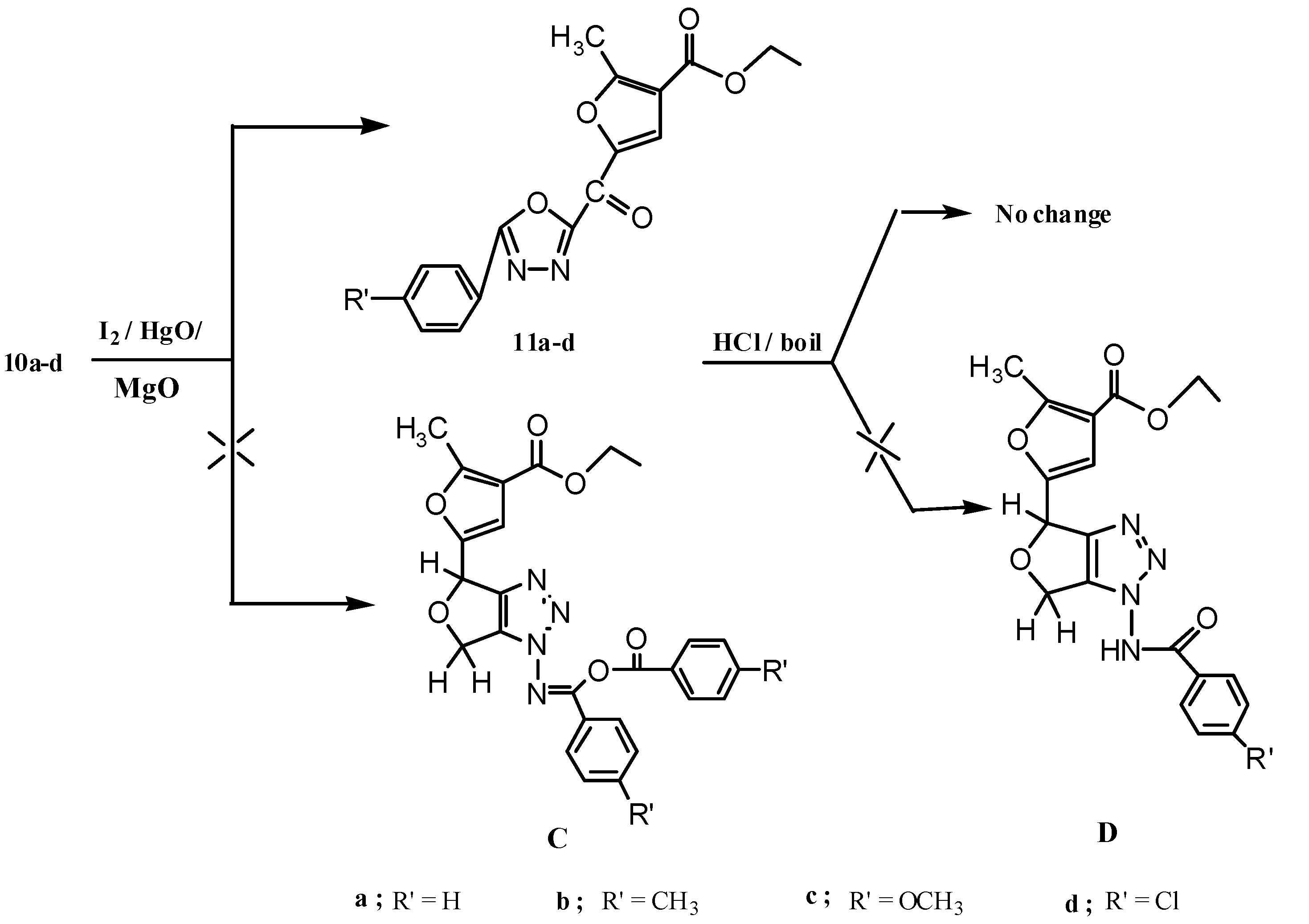
2.2. Pharmacological Screening
2.2.1. MAO-B Activity
2.2.1.1. Effect of Tested Compounds on MAO-B
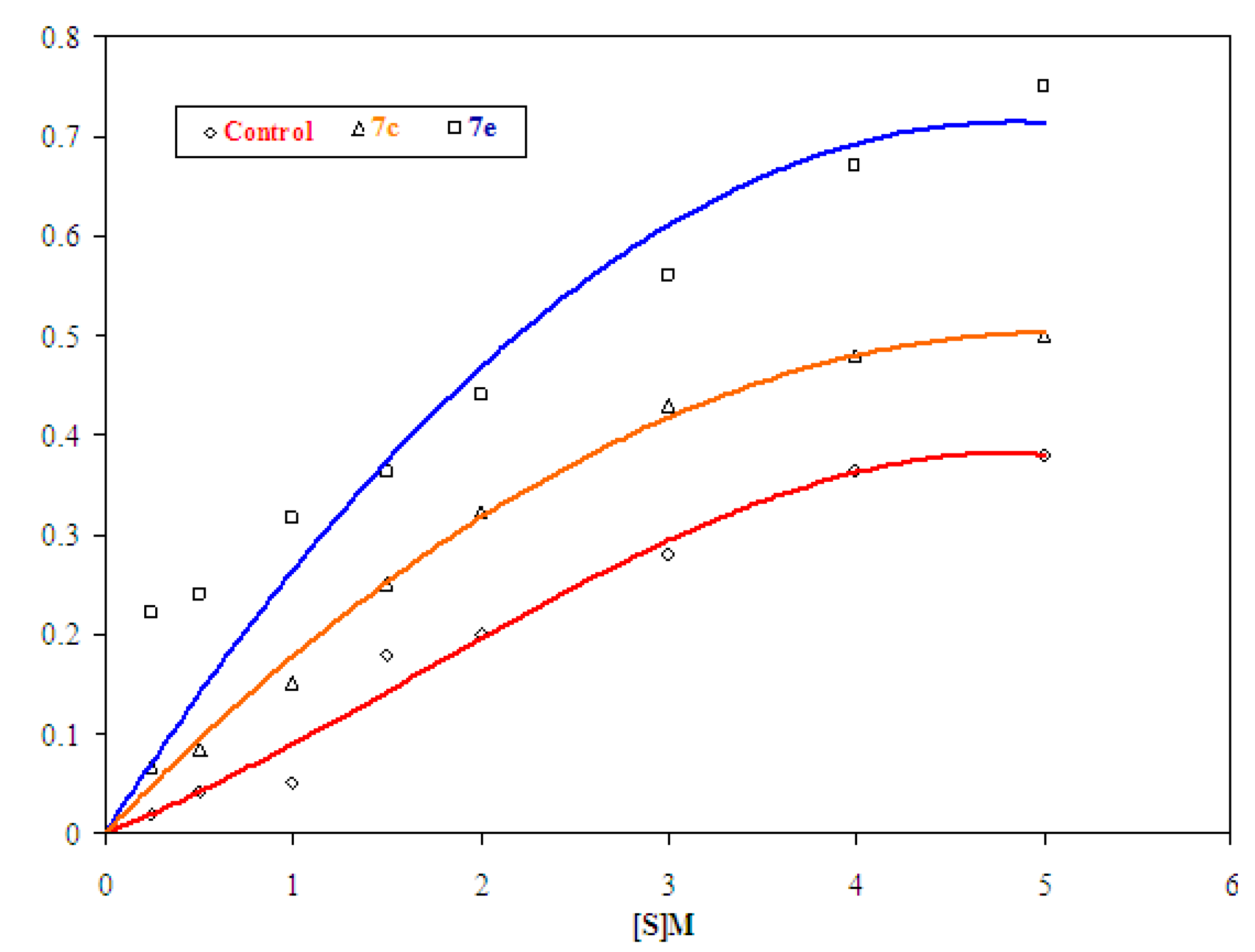
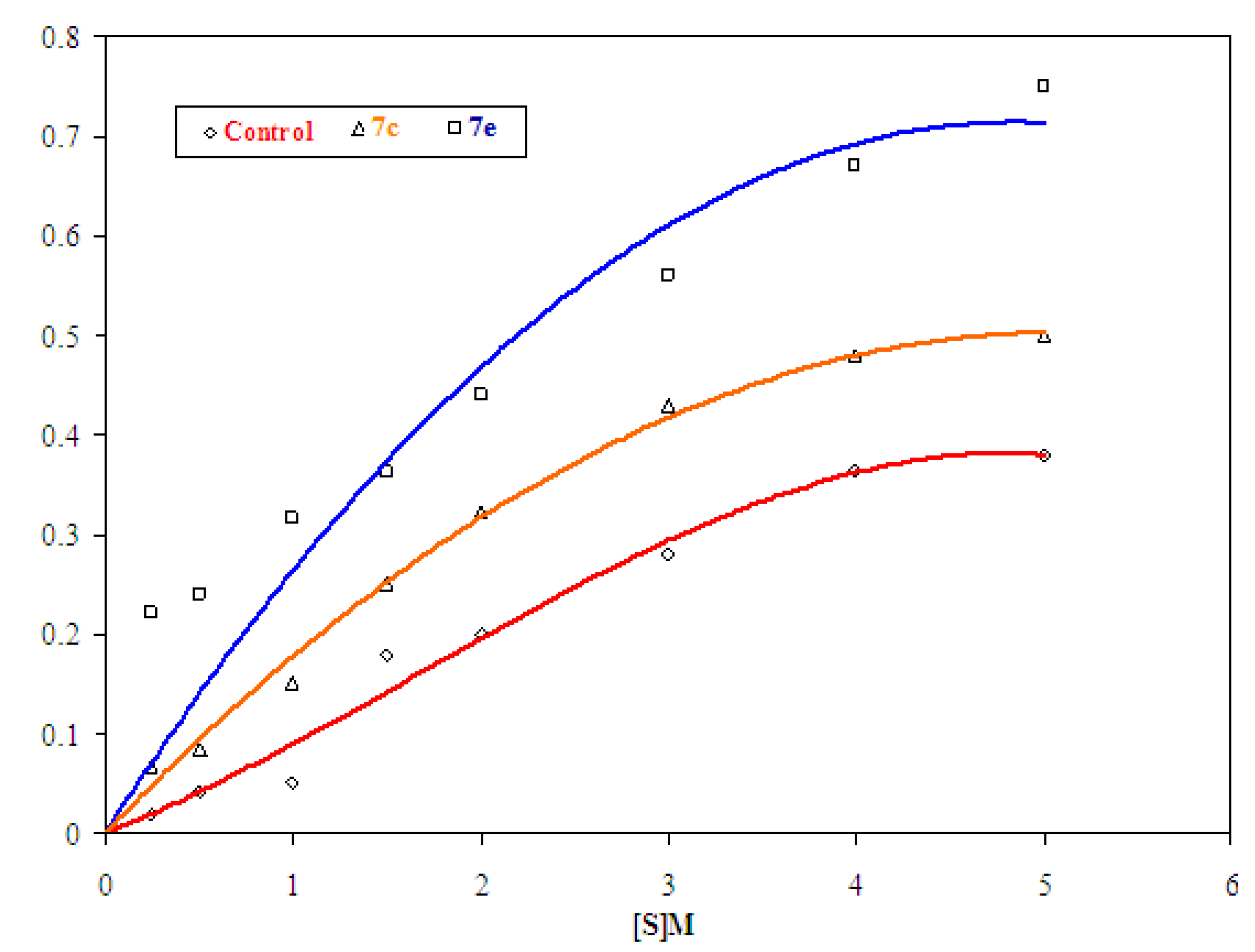
2.2.1.2. Determination of Vmax and Km

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Substrate conc. | Rate | ||
|---|---|---|---|
| Control | 7c | 7e | |
| 0.25 × 10−3 | 0.020 | 0.068 | 0.222 |
| 0.50 × 10−3 | 0.042 | 0.085 | 0.240 |
| 1.00 × 10−3 | 0.050 | 0.151 | 0.315 |
| 1.50 × 10−3 | 0.180 | 0.250 | 0.363 |
| 2.00 × 10−3 | 0.200 | 0.322 | 0.440 |
| 3.00 × 10−3 | 0.280 | 0.430 | 0.560 |
| 4.00 × 10−3 | 0.365 | 0.481 | 0.674 |
| 5.00 × 10−3 | 0.380 | 0.498 | 0.700 |
2.2.2. Antibacterial and Antifungal Activities
| Compound No. | Bacteria | |||||
| Gram positive | Gram negative | |||||
| Bacillus sp. | Sarcina sp. | Staphylococcus sp. | E. coli | |||
| 3b | …. | …. | …. | …. | ||
| 6 | …. | …. | 2 | …. | ||
| 7c | …. | …. | 0.5 | …. | ||
| 7e | …. | …. | 2 | …. | ||
| 10a | …. | …. | …. | …. | ||
| 10b | …. | …. | …. | …. | ||
| 10c | …. | …. | …. | …. | ||
| 11a | …. | …. | 1 | …. | ||
| 11b | …. | …. | …. | …. | ||
| 11d | …. | …. | …. | …. | ||
| Compound No. | Fungi | |||||
| Aspergillus niger | Aspergillus fumigatus | Alternariasp. | Fusarium sp. | Chaetomium sp. | Penicilliu sp. | |
| 3b | 1 | 1 | …. | 2 | …. | …. |
| 6 | …. | …. | 1 | 1 | 2 | 2 |
| 7c | 1 | 1 | 0.5 | 2 | …. | 0.5 |
| 7e | …. | 2.5 | 0.5 | 2 | …. | 1 |
| 10a | …. | …. | …. | …. | …. | …. |
| 10b | 1 | …. | …. | …. | …. | …. |
| 10c | 1 | …. | …. | …. | …. | …. |
| 11a | 0.5 | …. | 0.5 | …. | …. | 2 |
| 11b | …. | …. | …. | …. | …. | …. |
| 11d | …. | …. | …. | 2 | …. | …. |
3. Experimental
3.1. Chemistry
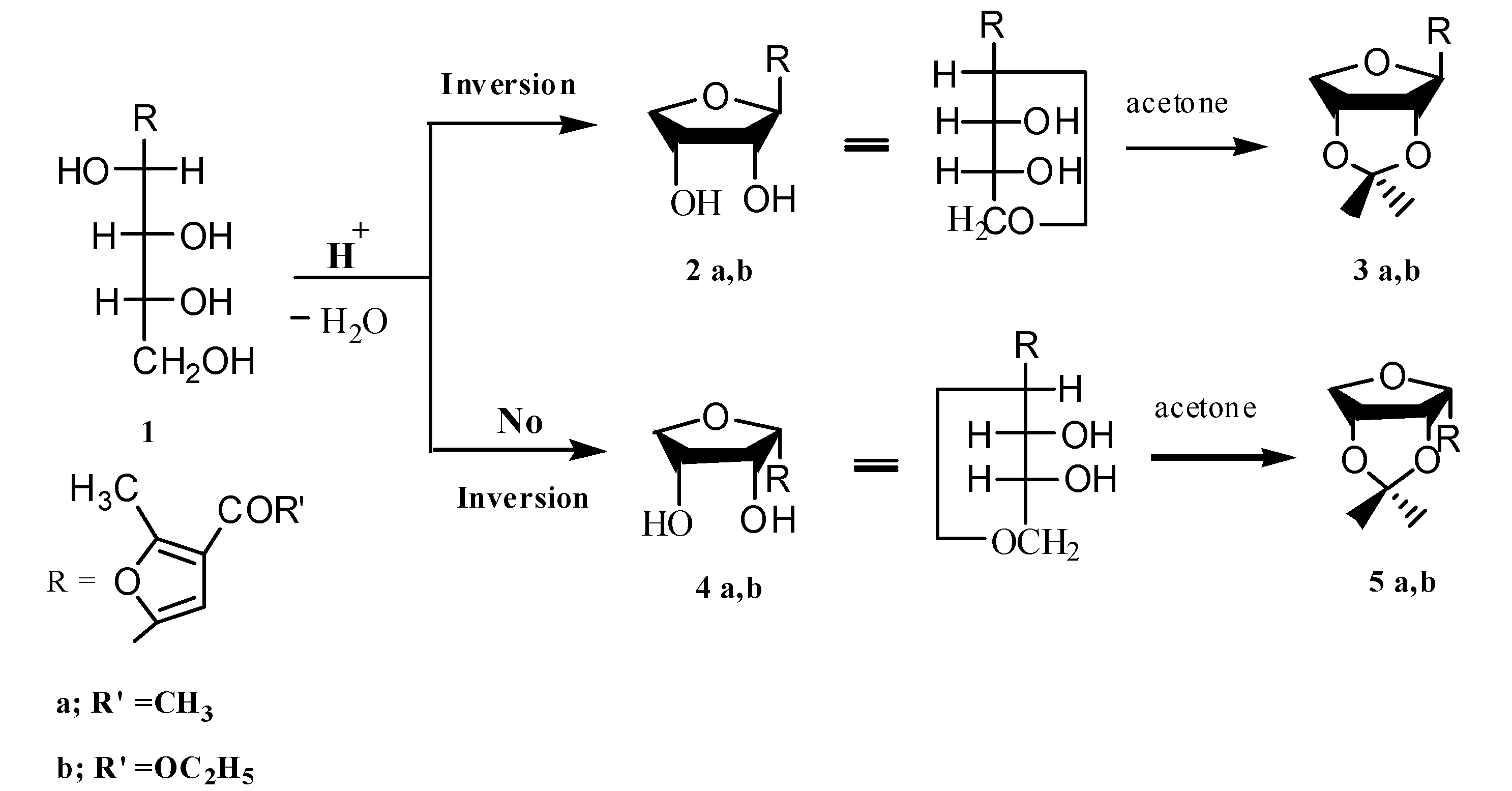
3.1.1. Ethyl 5-(2',3'-Dihydroxytetrahydrofuran-1'-yl)-2-methylfuran-3-carboxylates 2b,4b
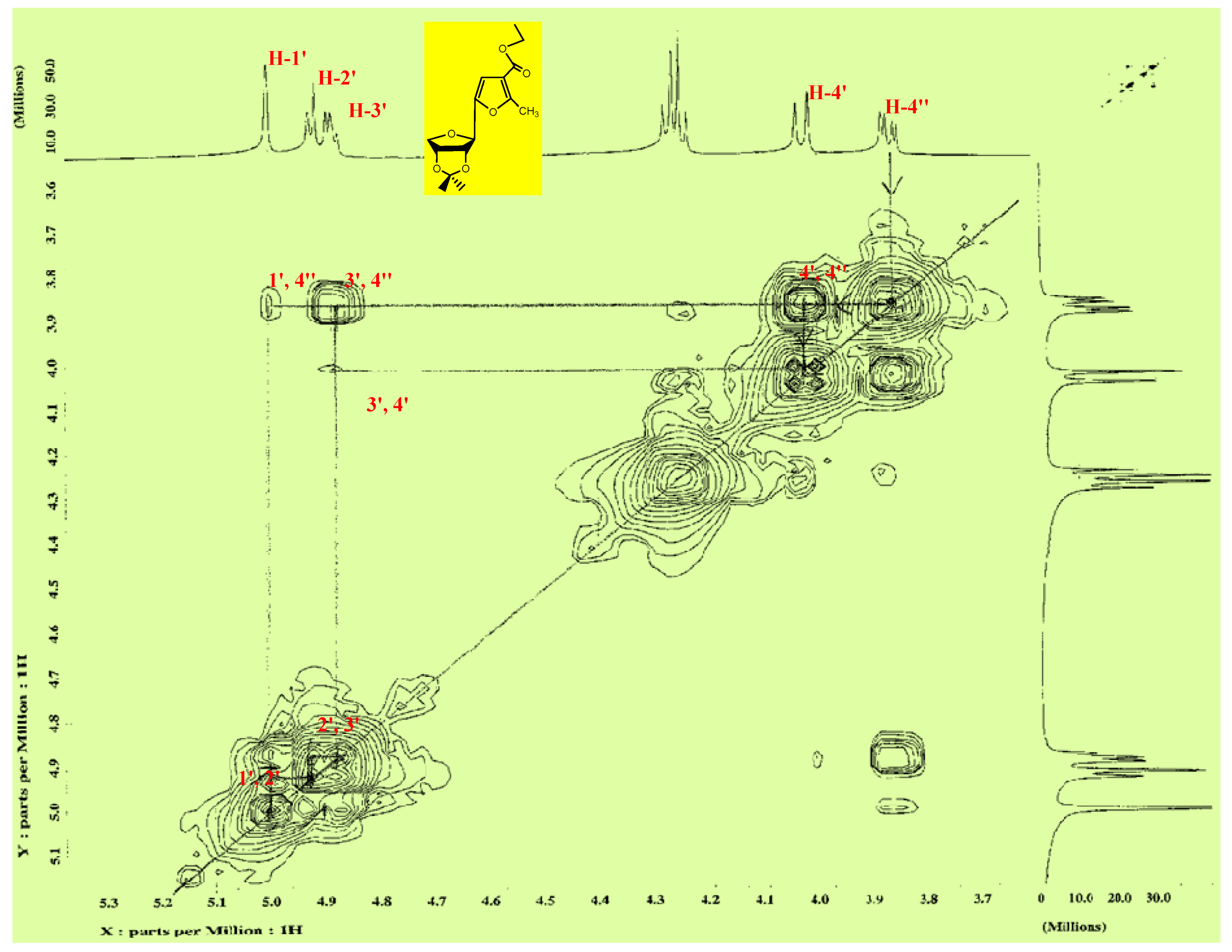
3.1.2. Synthesis of Isopropylidene Derivatives
 −69.03; IR (KBr): 1,709 (CO-ester) 1,613, 1,583 cm−1 (C=C); 1H-NMR (CDCl3); δ: 1.324 (t, 3H, CH3(ester), J = 6.90 Hz), 1.352, 1.538 (2s, 6H, CMe2, Δδ 0.187), 2.542 (s, 3H, CH3(furan)), 3.870 (dd, 1H, H-4'a, J3',4'a = 3.80 Hz, J4'b,4'a = 10.70 Hz), 4.029 (d, 1H, H-4'b, J4'b,4'a = 10.70 Hz), 4.260 (q, 2H, CH2(ester), J = 6.90 Hz), 4.876–4.928 (m, 2H, H-2', H-3'), 5.003 (s, 1H, H-1'), 6.486 (s, 1H, CHfuran); MS: m/z (%), 297 (6.3, M++1), 296 (20.6, M+), 182 (100), 181 (30.6), 145 (60), 153 (43), 137 (25), 136 (25.6), 105 (21.3), 80 (27.5), 79 (28.8), 69 (38.1), 65 (28.8), 60 (13.8), 59 (76.3), 56 (25), 55 (30.6), 53 (33.8), 52 (44.4), 51 (60), 50 (25). −58.98; IR (KBr): 3,384, 3,314 (OH and NH2), 3272 (NH), 1,636 cm−1 (CO-amide); 1H-NMR (DMSO-d6); δ: 2.463 (s, 3H, CH3(furan)), 3.570 (dd, 1H, H-4'b, J3',4'b = 2.30 Hz, J4'b,4'a = 9.15 Hz), 3.979 (dd, 1H, H-4'a, J3',4'a = 4.60 Hz, J4'b,4'a = 9.15 Hz), 4.029–4.066 (m, 2H, H-3', H-2'), 4.312 (s, 2H, NH2), 4.395 (d, 1H, H-1', J1',2' = 6.90 Hz), 4.985 (d, 1H, 2'-OH, J2',OH = 4.60 Hz), 5.076 (d, 1H, 3'-OH, J3',OH = 6.85 Hz), 6.715 (s, 1H, CHfuran), 9.243 (s, 1H, NH). After shaking with D2O, the NH, NH2, and the two hydroxyl protons disappeared; MS: m/z (%), 243 (2.19, M++1), 242 (16.89, M+), 224 (0.47, M+-H2O), 212 (11.54, M+-2NH), 211 (100, M+-NHNH2); Anal. Calcd for C10H14N2O5: C, 49.58; H, 5.84; N, 1.55%; found: C, 49.58; H, 5.83; N, 1.56%.
−69.03; IR (KBr): 1,709 (CO-ester) 1,613, 1,583 cm−1 (C=C); 1H-NMR (CDCl3); δ: 1.324 (t, 3H, CH3(ester), J = 6.90 Hz), 1.352, 1.538 (2s, 6H, CMe2, Δδ 0.187), 2.542 (s, 3H, CH3(furan)), 3.870 (dd, 1H, H-4'a, J3',4'a = 3.80 Hz, J4'b,4'a = 10.70 Hz), 4.029 (d, 1H, H-4'b, J4'b,4'a = 10.70 Hz), 4.260 (q, 2H, CH2(ester), J = 6.90 Hz), 4.876–4.928 (m, 2H, H-2', H-3'), 5.003 (s, 1H, H-1'), 6.486 (s, 1H, CHfuran); MS: m/z (%), 297 (6.3, M++1), 296 (20.6, M+), 182 (100), 181 (30.6), 145 (60), 153 (43), 137 (25), 136 (25.6), 105 (21.3), 80 (27.5), 79 (28.8), 69 (38.1), 65 (28.8), 60 (13.8), 59 (76.3), 56 (25), 55 (30.6), 53 (33.8), 52 (44.4), 51 (60), 50 (25). −58.98; IR (KBr): 3,384, 3,314 (OH and NH2), 3272 (NH), 1,636 cm−1 (CO-amide); 1H-NMR (DMSO-d6); δ: 2.463 (s, 3H, CH3(furan)), 3.570 (dd, 1H, H-4'b, J3',4'b = 2.30 Hz, J4'b,4'a = 9.15 Hz), 3.979 (dd, 1H, H-4'a, J3',4'a = 4.60 Hz, J4'b,4'a = 9.15 Hz), 4.029–4.066 (m, 2H, H-3', H-2'), 4.312 (s, 2H, NH2), 4.395 (d, 1H, H-1', J1',2' = 6.90 Hz), 4.985 (d, 1H, 2'-OH, J2',OH = 4.60 Hz), 5.076 (d, 1H, 3'-OH, J3',OH = 6.85 Hz), 6.715 (s, 1H, CHfuran), 9.243 (s, 1H, NH). After shaking with D2O, the NH, NH2, and the two hydroxyl protons disappeared; MS: m/z (%), 243 (2.19, M++1), 242 (16.89, M+), 224 (0.47, M+-H2O), 212 (11.54, M+-2NH), 211 (100, M+-NHNH2); Anal. Calcd for C10H14N2O5: C, 49.58; H, 5.84; N, 1.55%; found: C, 49.58; H, 5.83; N, 1.56%.3.1.3. Reactions of 6 with a Number of Aldehydes
General Methods
−37.59; IR (KBr): 3,395 (OH), 3,108 (NH), 1,669 (CO-amide), 1,638 cm−1 (C=N); 1H-NMR (DMSO-d6); δ: 2.524 (s, 3H, CH3(furan)), 3.616 (dd, 1H, H-4'b, J3',4'b = 2.30 Hz, J4'b,4'a = 9.20 Hz), 4.008–4.026 (m, 1H, H-4'a), 4.036–4.095 (m, 2H, H-3', H-2'), 4.493 (d, 1H, H-1', J1',2' = 6.90 Hz), 5.043 (ss, 1H, 2'-OH), 5.142 (d, 1H, 3'-OH, J3',OH = 6.15 Hz), 6.860 (s, 1H, CHfuran), phenyl protons: 7.432 (t, 1H,p-H), 7.568 (t, 2H, O-H), 8.017 (d, 2H, m-H), 8.401 (s, 1H, CH=N), 8.537 (s, 1H, CHtriazole), 11.610 (s, 1H, NH). After shaking with D2O, the NH proton and the two hydroxyl protons disappeared; MS: m/z (%), 399 (0.89, M++2), 398 (5.36, M++1), 397 (23.99, M+), 379 (3.55, M+-H2O), 324 (21.85), 253 (11.94, M+-phenyltriazole moiety), 211 (100), 137 (35.81), 77 (20.56, C6H5); Anal. Calcd for C19H19N5O5: C, 57.42; H, 4.80; N, 17.64%; found: C, 57.43; H, 4.82; N, 17.62%. −31.39; IR (KBr): 3,423 (OH), 3,249 (NH), 1,653 (CO-amide), 1,618 cm−1 (C=N); 1H-NMR (DMSO-d6); δ: 2.519 (s, 3H, CH3(furan)), 3.617 (dd, 1H, H-4'b, J3',4'b = 2.30 Hz, J4'b,4'a = 9.95 Hz), 4.019 (dd, 1H, H-4'a, J3',4'a = 3.85 Hz, J4'b,4'a = 9.95 Hz), 4.080–4.109 (m, 2H, H-3', H-2'), 4.493 (d, 1H, H-1', J1',2' = 6.90 Hz), 5.051 (ss, 1H, 2'-OH), 5.147 (d, 1H, 3'-OH, J3',OH = 6.10 Hz), 6.857 (s, 1H, CHfuran), p-bromophenyl protons: 7.752 (d, 2H, m-H), 7.954 (d, 2H, o-H), 8.422 (s, 1H, CH=N), 8.518 (s, 1H, CH(triazole)), 11.630 (s, 1H, NH). After shaking with D2O, the NH proton and the two hydroxyl protons disappeared; 13C-NMR; δ: 14.00 (C-13), 70.91 (C-4'), 73.32 (C-3'), 75.33 (C-2'), 76.50 (C-1'), 107.79 (C-12), 115.11 (C-11), 120.92 (C-10), 121.23 (C-9), 133.27 (C-8), 135.16 (C-7), 137.74 (C-6), 138.52 (C-5), 146.50 (C-4), 151.71 (C-3), 158.19 (C-2), 159.84 (C-1); MS: m/z (%), 478/476 (2.03, 2.17, M++1), 477/475 (8.85, 9.10, M+), 459/457 (1.25, 1.24, M+-H2O), 253 (10.01, M+-p-bromophenyl triazole moiety), 211 (100), 137 (34); Anal. Calcd for C19H18BrN5O5: C, 47.90; H, 3.83; N, 14.71; Br, 16.77%; found: C, 47.91; H, 3.81; N, 14.70; Br, 16.78%. −44.42; IR (KBr): 3,426 (OH), 3,208 (NH), 1,694 (CO-ester), 1,655 (CO-amide), 1,616 cm−1 (C=N); 1H-NMR (DMSO-d6); δ: 1.254 (t, 3H, CH3(ester), J = 7.65 Hz), 2.501 (s, 3H, CH3(furan-1)), 2.580 (s, 3H, CH3(furan-2)), 3.603 (dd, 1H, H-4'b, J3',4'b = 2.30 Hz, J4'b,4'a = 9.90 Hz), 4.004 (dd, 1H, H-4'a, J3',4'a = 4.60 Hz, J4'b,4'a = 9.90 Hz), 4.043–4.088 (m, 2H, H-3', H-2'), 4.209 (q, 2H, CH2(ester), J = 7.65 Hz), 4.467 (d, 1H, H-1', J1',2' = 6.85 Hz), 5.028 (d, 1H, 2'-OH, J2',OH = 3.80 Hz), 5.124 (d, 1H, 3'-OH, J3',OH = 6.10 Hz), 6.830 (s, 1H, CHfuran-1), 7.059 (s, 1H, CHfuran-2), 8.150 (s, 1H, CH=N), 11.409 (s, 1H, NH). After shaking with D2O, the NH proton and the two hydroxyl protons disappeared; MS: m/z (%), 408 (0.79, M++2), 407 (4.16, M++1), 406 (18.26, M+), 211 (100), 154 (32.75); Anal. Calcd for C19H22N2O8: C, 56.13; H, 5.47; N, 6.86%; found: C, 56.15; H, 5.46; N, 6.89%. −42.16; IR (KBr): 3,380 (OH and NH), 1,661 (CO-amide), 1,616 cm−1 (C=N); 1H-NMR (DMSO-d6); δ: 2.513 (s, 3H, CH3(furan)), 3.608 (dd, 1H, H-4'b, J3',4'b = 2.30 Hz, J4'b,4'a = 9.95 Hz), 4.012 (dd, 1H, H-4'a, J3',4'a = 4.60 Hz, J4'b,4'a = 9.95 Hz), 4.065–4.089 (m, 2H, H-3', H-2'), 4.476 (d, 1H, H-1', J1',2' = 6.90 Hz), 5.043 (d, 1H, 2'-OH, J2',OH = 3.85 Hz), 5.141 (d, 1H, 3'-OH, J3',OH = 6.10 Hz), 6.869 (s, 1H, CH(furan)), phenyl protons: 7.395–7.436 (m, 3H, p-H, O-H), 7.672 (d, 2H, m-H), 8.340 (s, 1H, CH=N), 11.389 (s, 1H, NH). After addition of D2O, the NH proton and the two hydroxyl protons disappeared; MS: m/z (%), 331 (2.25, M++1), 330 (10.80, M+), 227 (9.18, M+-C6H5CN), 211 (100), 154 (59.63), 77 (9.32, C6H5); Anal. Calcd for C17H18N2O5: C, 61.80; H, 5.50; N, 8.45%; found: C, 61.8; H, 5.49; N, 8.48%. −42.55; IR (KBr): 3,409 (OH), 3,241 (NH), 1,657 (CO-amide), 1,628 cm−1 (C=N); 1H-NMR (DMSO-d6); δ: 2.499 (s, 3H, CH3(furan)), 3.598–3.617 (m, 1H, H-4'b), 4.013 (dd, 1H, H-4'a, J3',4'a = 3.80 Hz, J4'b,4'a = 9.90 Hz), 4.026–4.104 (m, 2H, H-3', H-2'), 4.476 (d, 1H, H-1', J1',2' = 6.90 Hz), 5.025 (d, 1H, 2'-OH, J2',OH = 3.80 Hz), 5.121 (d, 1H, 3'-OH, J3',OH = 6.15 Hz); 6.849 (s, 1H,CHfuran), 7.00 (bs, 2H, CH=CH), phenyl protons: 7.273–7.302 (m, 1H, p-H), 7.355 (t, 2H, O-H), 7.585 (d, 2H, m-H); 8.124 (d, 1H, CH=N, J = 6.15 Hz), 11.264 (s, 1H, NH); MS: m/z (%), 358 (0.21, M++2), 357 (0.95, M++1), 356 (3.22, M+), 338 (0.11, M+-H2O), 320 (0.21, M+-2H2O), 279 (0.52, M+-C6H5), 211 (100), 154 (55.50), 130 (35.56), 77 (8.65, C6H5), 43 (49, COCH3); Anal. Calcd for C19H20N2O5: C, 64.03; H, 5.64; N, 7.84%; found: C, 64.04; H, 5.66; N, 7.86%.3.1.4. Reactions of Compounds 7a,d with Acetic Anhydride
−47.75; IR (KBr): 3,217 (NH), 1,754 (OAc), 1,647 (CO-amide), 1,601 cm−1 (C=N); 1H-NMR (CDCl3); δ: 2.061, 2.139 (2s, 6H, 2OAc), 2.677 (s, 3H, CH3(furan)), 3.984–3.968 (m, 1H, H-4'b), 4.371 (dd, 1H, H-4'a, J3',4'a = 4.60 Hz, J4'b,4'a = 9.95 Hz), 4.925 (d, 1H, H-1', J1',2' = 6.10 Hz), 5.492–5.532 (m, 2H, H-3', H-2'), 6.744 (bs, 1H, CHfuran), 7.462–7.478 (m, 1H, p-H), 7.531–7.625 (m, 2H, O-H), 8.019–8.141 (m, 4H, m-H, CH=N, CHtriazole), and 12.239 (s, 1H, NH); MS: m/z (%), 482 (0.61, M++1), 481 (1.97, M+), 363 (22.58), 362 (100), 295 (67.47), 137 (21.53), 115 (19.79), 77 (15.78); Anal. Calcd for C23H23N5O7: C, 57.35; H, 4.82; N, 14.53%; found: C, 57.38; H, 4.82; N, 14.55%. −48.37; IR (KBr): 3,248 (NH), 1,752 (OAc), 1,654 (CO-amide), 1,582 cm−1 (C=N); 1H-NMR (CDCl3); δ: 2.070, 2.125 (2s, 6H, 2OAc), 2.602 (s, 3H, CH3(furan)), 3.954–3.974 (m, 1H, H-4'b), 4.381–4.350 (m, 1H, H-4'a), 4.950 (bs, 1H, H-1'), 5.417–5.524 (m, 2H, H-3', H-2'), 6.620 (s, 1H, CHfuran), 7.519–7.549 (m, 5H, phenyl protons), 7.883 (d, 1H, CH=N, J = 7.65 Hz), 9.550 (s, 1H, NH); MS: m/z (%), 415 (0.39, M++1), 414 (0.81, M+), 295 (100), 77 (2.61); Anal. Calcd for C21H22N2O7: C, 60.86; H, 5.37; N, 6.77%; found: C, 60.86; H, 5.35; N, 6.76%.3.1.5. Reactions of 9 with a Number of Aroylhydrazines
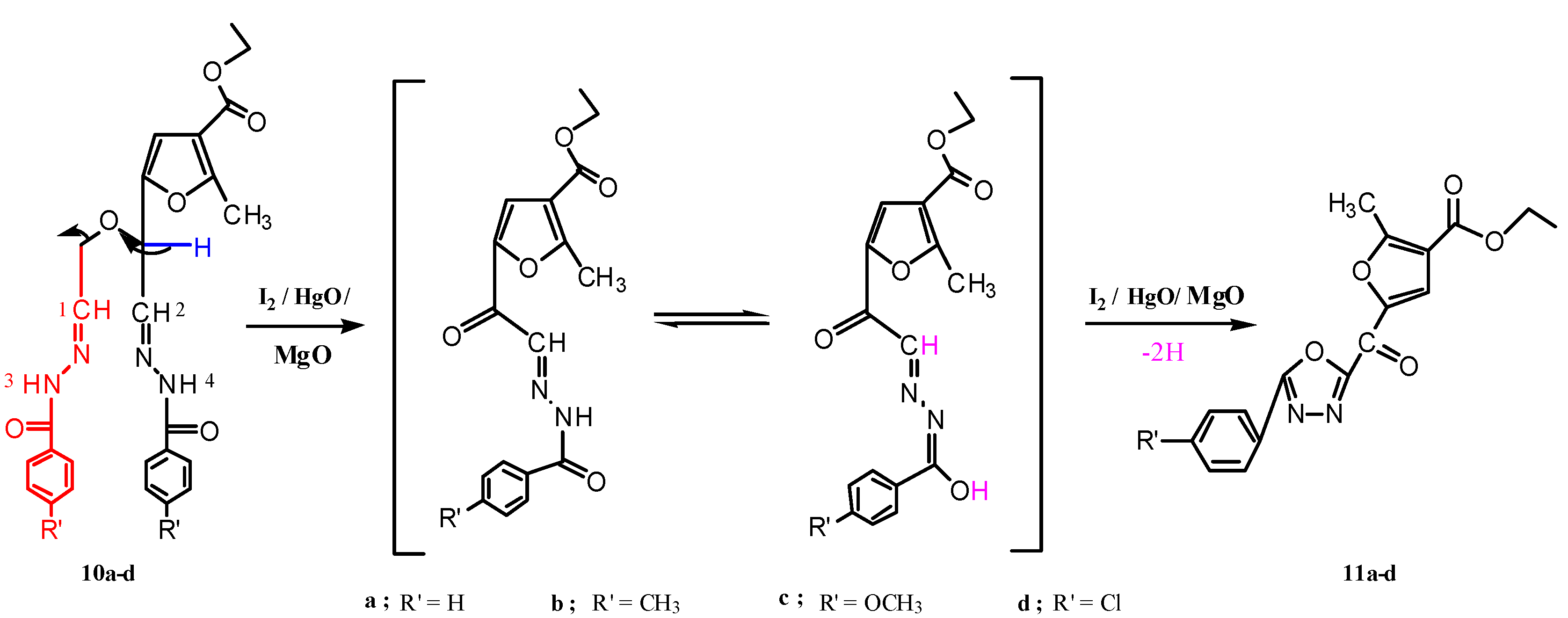
3.1.6. Reactions of Compounds 10a–d with Yellow Mercuric Oxide
3.2. Pharmacological Screening
3.2.1. MAO-B Activity
3.2.1.1. Enzyme Preparation
3.2.1.2. Enzyme Assay
3.2.1.3. Determination of Vmax and Km
3.2.2. Antibacterial and Antifungal Screening
4. Conclusions
Acknowledgements
- Sample Availability: Samples of the compounds are available from the authors.
References
- Rainier, J.D.; Cox, J.M. Aluminum- and boron-mediated C-glycoside synthesis from 1,2-anhydro-glycosides. Org. Lett. 2000, 2, 2707–2709. [Google Scholar] [CrossRef]
- Yale, H.L.; Losee, K. 2-Amino-5-substituted 1,3,4-oxadiazoles and 5-imino-2-substituted Δ2-1,3,4-oxadiazolines. A group of novel muscle relaxants. J. Med. Chem. 1966, 9, 478–483. [Google Scholar] [CrossRef]
- Omar, F.A.; Mahfouz, N.M.; Rahman, M.A. Design, synthesis and anti-inflammatory activity of some 1,3,4-oxadiazole derivatives. Eur. J. Med. Chem. 1996, 31, 819–825. [Google Scholar] [CrossRef]
- Palusa, S.K.G.; Udupi, R.H.; Himabindu, V.; Sridhara, A.M. Synthesis and evaluation of a series of pyrimidine substituted 1,3,4-oxadiazole derivatives as antimicrobial and anti-inflammatory agents. Org. Commun. 2011, 4, 82–93. [Google Scholar]
- Tan, T.M.C.; Chen, Y.; Kong, K.H.; Bai, J.; Li, Y.; Lim, S.G.; Ang, T.H.; Lam, Y. Synthesis and the biological evaluation of 2-benzenesulfonyl alkyl-5-substituted-sulfanyl-(1,3,4)-oxadiazoles as potential anti-hepatitis B virus agents. Antiviral Res. 2006, 71, 7–14. [Google Scholar] [CrossRef]
- Adelstein, G.W.; Yen, C.H.; Dajani, E.Z.; Bianchi, R.G. 3,3-Diphenyl-3-(2-alkyl-1,3,4-oxadiazol-5-yl)propylcycloalkylamines, a novel series of antidiarrheal agents. Med. Chem. 1976, 19, 1221–1225. [Google Scholar] [CrossRef]
- Jha, K.K.; Samad, A.; Kumar, Y.; Shaharyar, M.; Khosa, R.; Jain, J.; Bansal, S. 3D-QSAR studies of 1,3,4-oxadiazole derivatives as antimycobacterial agents. Iran. J. Pharm. Res. 2009, 8, 163–167. [Google Scholar]
- Macaev, F.; Rusu, G.; Pogrebnoi, S.; Gudima, A.; Stingaci, E.; Vlad, L.; Shvets, N.; Kandemirli, F.; Dimoglo, A.; Reynolds, R. Synthesis of novel 5-aryl-2-thio-1,3,4-oxadiazoles and the study of their structure-anti-mycobacterial activities. Bioorg. Med. Chem. 2005, 13, 4842–4850. [Google Scholar] [CrossRef]
- Navarrete-Vazquez, G.; Molina-Salinas, G.M.; Duarte-Fajardo, Z.V.; Vargas-Villarreal, J.; Estrada-Soto, S.; Gonzalez-Salazar, F.; Hernandez-Nunez, E.; Said-Fernandez, S. Synthesis and antimycobacterial activity of 4-(5-substituted-1,3,4-oxadiazol-2-yl)pyridines. Bioorg. Med. Chem. 2007, 15, 5502–5508. [Google Scholar]
- Zarghi, A.; Tabatabai, S.A.; Faizi, M.; Ahadian, A.; Navabi, P.; Zanganeh, V.; Shafiee, A. Synthesis and anticonvulsant activity of new 2-substituted-5-(2-benzyloxyphenyl)-1,3,4-oxadiazoles. Bioorg. Med. Chem. Lett. 2005, 15, 1863–1865. [Google Scholar] [CrossRef]
- Jin, L.; Chen, J.; Song, B.; Chen, Z.; Yang, S.; Li, Q.; Hu, D.; Xu, R. Synthesis, structure, and bioactivity of N'-substituted benzylidene-3,4,5-trimethoxybenzohydrazide and 3-acetyl-2 substituted phenyl-5-(3,4,5-trimethoxyphenyl)-2,3-dihydro-1,3,4-oxadiazole derivatives. Bioorg. Med. Chem.Lett. 2006, 16, 5036–5040. [Google Scholar]
- Sawant, R.; Lanke, P.; Jadhav, G.; Bhangale, L. QSAR analysis of structurally similar 1,3,4-oxadiazoles as enzyme tyrosinase inhibitors. Drug Invent. Today 2010, 2, 169–172. [Google Scholar]
- Sánchez, A.G.; Roldán, A.R. Dehydraion of 2-(D-arabino-tetrahyroxybutyl)furans. Carbohydr. Res. 1972, 22, 53–62. [Google Scholar]
- González, F.G. Reactions of monosaccharides with beta-ketonic esters and related substances. In Advances in Carbohydrate Chemistry; Wolfrom, L.M., Tipson, S.R., Eds.; Academic Press Inc.: New York, NY, USA, 1956; Volume 11, pp. 97–143. [Google Scholar]
- El-Sallam, M.A.; Hegazy, E.I.A. Studies on 3-epimeric 2-hexulose phenylosazones. Structure and anomeric configuration of the 3,6-anhydro-osazone derivatives obtained from D-arabino- and D-ribo-2-hexulose phenylosazone. Carbohydr. Res. 1981, 95, 177–188. [Google Scholar]
- El-Sallam, M.A.; Ibrahim, E.S.I.; El-Eter, K.A.A.; Cassady, J.M. Synthesis and anomeric configuration of 2-(erythrofuranosyl)benzimidazole C-nucleoside analogues. Carbohydr. Res. 1997, 298, 93–104. [Google Scholar] [CrossRef]
- Sable, H.Z.; Ritchey, W.M.; Nordlander, J.E. Studies on cyclic polyols part IV. Nuclear magnetic resonance spectra and configurational assignments of polyhydroxycyclopentanes. Carbohydr. Res. 1965, 1, 10–21. [Google Scholar] [CrossRef]
- MacCoss, M.; Robins, M.J.; Rayner, B.; Imbach, J.L. A new aspect of the use of 2',3'-O-isopropylidene ribonucleosides for investigation of anomeric configuration. Carbohydr. Res. 1977, 59, 575–579. [Google Scholar] [CrossRef]
- Maeba, I.; Iwata, K.; Usami, F.; Furukawa, H. C-Nucleosides 1. Synthesis of 3-(β-D-ribofuranosyl) pyridazines. J. Org. Chem. 1983, 48, 2998–3002. [Google Scholar]
- Hassan, S.Y.; Yaccout, G.A.; Abd El-Dayem, N.S.; El Sadek, M.M. Chemistry Department, Faculty of Science, Alexandria University, Alexandria, Egypt. Unpublished work, 2009.
- El Sadek, M.M.; Zagzoug, N.B.; El Soccary, N.N. Reactions of 2-methyl-5-(D-arabino-tetrahydroxybutyl)-3-furoylhydrazine. Carbohydr. Res. 1993, 250, 323–326. [Google Scholar] [CrossRef]
- Sánchez, A.G.; Roldán, A.R. Dehydration of 2-(D-arabino-tetrahydroxybutyl)furans. Carbohydr. Res. 1972, 22, 53–62. [Google Scholar] [CrossRef]
- Guthrie, R.D.; Honeyman, J. Periodate oxidation of methyl 4:6-O-benzylidene-a-D-glucoside. Chem. Ind. 1958, 29, 388–389. [Google Scholar]
- Alexandrou, N.E. Reassignment of structures of the dihydro-v-tetrazines. II: Mechanism of oxidation of diacylhydrazones. Tetrahedron 1966, 22, 1309–1316. [Google Scholar]
- El Khadem, H.; Shaban, M.A.E. Carbohydrate derivatives of 1-substituted 1,2,3-triazoles. J. Chem. Soc. C 1967, 519–520. [Google Scholar] [CrossRef]
- El-Khadem, H.; Shaban, M.A.E.; Nassr, M.A.M. Oxidation of mesoxalaldehyde bis- and tris-(benzoylhydrazones). J. Chem. Soc. C 1969, 1969, 1416–1418. [Google Scholar]
- El-Khadem, H.; El-Sadik, M.M.; Meshreki, M.H. Reactions of phenylglyoxal bisarylhydrazones. J. Chem. Soc. C 1968, 1968, 2097–2099. [Google Scholar]
- Ni, X.; Chan, D.; Chan, K.; McMain, S.; Kennedy, J.L. Serotonin genes and gene-gene interactions in borderline personality disorder in a matched case-control study. Prog. Neuropsychopharmacol. Biol. Psychiatry 2009, 33, 128–133. [Google Scholar] [CrossRef]
- Upadhyay, A.K.; Edmondson, D.E. Characterization of detergent purified recombinant rat liver monoamine oxidase B expressed in Pichia pastoris. Protein Expr. Purif. 2008, 59, 349–356. [Google Scholar] [CrossRef]
- Setini, A.; Pierucci, F.; Senatori, O.; Nicotra, A. Molecular characterization of monoamine oxidase in zebrafish (Danio rerio). Comp. Biochem. Physiol. 2005, 140, 153–161. [Google Scholar] [CrossRef]
- Kaneda, Y.; Fujii, A.; Nagamine, I. Platelet serotonin concentrations in medicated schizophrenic patients. Prog. Neuropsychopharmacol. Biol. Psychiatry 2001, 25, 983–992. [Google Scholar] [CrossRef]
- Garpenstrand, H.; Longato-Stadler, E.; Klinteberg, B.A.; Grigorenko, E.; Damberg, M.; Oreland, L.; Hallman, J. Low platelet monoamine oxidase activity in Swedish imprisoned criminal offenders. Eur. Neuropsychopharmacol. 2002, 12, 135–140. [Google Scholar]
- Parsian, A.; Racette, B.; Zhang, Z.H.; Rundle, M.; Perlmutter, J.S. Association of variations in monoamine oxidases A and B with Parkinson’s disease subgroups. Genomics 2004, 83, 454–460. [Google Scholar] [CrossRef]
- Paris, J.; Zweig-Frank, H.; Ying Kin, N.M.K.N.; Schwartz, G.; Steiger, H.; Nair, N.P.V. Neurobiological correlates of diagnosis and underlying traits in patients with borderline personality disorder compared with normal controls. Psychiatry Res. 2004, 121, 239–252. [Google Scholar] [CrossRef]
- Vindis, C.; Séguélas, M.H.; Bianchi, P.; Parini, A.; Cambon, C. Monoamine oxidase B induces ERK-dependent cell mitogenesis by hydrogen peroxide generation. Biochem. Biophys. Res. Commun. 2000, 271, 181–185. [Google Scholar] [CrossRef]
- Carpéné, C.; Daviaud, D.; Boucher, J.; Bour, S.; Visentin, V.; Grès, S.; Duffaut, C.; Fontana, E.; Testar, X.; Saulnier-Blache, J.S.; et al. Short- and long-term insulin-like effects of monoamine oxidases and semicarbazide-sensitive amine oxidase substrates in cultured adipocytes. Metabolism 2006, 55, 1397–1405. [Google Scholar] [CrossRef]
- Rommelspacher, H.; Meier-Henco, M.; Smolka, M.; Kloft, C. The levels of norharman are high enough after smoking to affect monoamineoxidase B in platelets. Eur. J. Pharmachol. 2002, 441, 115–125. [Google Scholar] [CrossRef]
- Petzer, J.P.; Castagnoli, N., Jr; Schwarzschild, M.A.; Chen, J.F.; Schyf, C.J.V.D. Dual-target-directed drugs that block monoamine oxidase B and adenosine A2A receptors for Parkinson’s disease. Neurotherapeutics 2009, 6, 141–151. [Google Scholar]
- Rahman, M.A. Chalcone: A valuable insight into the recent advances and potential pharmacological activities. Chem. Sci. J. 2011. CSJ-29. [Google Scholar]
- Green, A.L.; El Hait, M.A.S. p-Methoxyamphetamine, a potent reversible inhibitor of type-A monoamine oxidase in vitro and in vivo. J. Pharm. Pharmacol. 1980, 32, 262–266. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
El-Sadek, M.M.; Hassan, S.Y.; El-Dayem, N.S.A.; Yacout, G.A. 5-(5-Aryl-1,3,4-oxadiazole-2-carbonyl)furan-3-carboxylate and New Cyclic C-Glycoside Analogues from Carbohydrate Precursors with MAO-B, Antimicrobial and Antifungal Activities. Molecules 2012, 17, 7010-7027. https://doi.org/10.3390/molecules17067010
El-Sadek MM, Hassan SY, El-Dayem NSA, Yacout GA. 5-(5-Aryl-1,3,4-oxadiazole-2-carbonyl)furan-3-carboxylate and New Cyclic C-Glycoside Analogues from Carbohydrate Precursors with MAO-B, Antimicrobial and Antifungal Activities. Molecules. 2012; 17(6):7010-7027. https://doi.org/10.3390/molecules17067010
Chicago/Turabian StyleEl-Sadek, Mohamed Mohamed, Seham Yassen Hassan, Nagwa Said Abd El-Dayem, and Galila Ahmed Yacout. 2012. "5-(5-Aryl-1,3,4-oxadiazole-2-carbonyl)furan-3-carboxylate and New Cyclic C-Glycoside Analogues from Carbohydrate Precursors with MAO-B, Antimicrobial and Antifungal Activities" Molecules 17, no. 6: 7010-7027. https://doi.org/10.3390/molecules17067010
APA StyleEl-Sadek, M. M., Hassan, S. Y., El-Dayem, N. S. A., & Yacout, G. A. (2012). 5-(5-Aryl-1,3,4-oxadiazole-2-carbonyl)furan-3-carboxylate and New Cyclic C-Glycoside Analogues from Carbohydrate Precursors with MAO-B, Antimicrobial and Antifungal Activities. Molecules, 17(6), 7010-7027. https://doi.org/10.3390/molecules17067010