Biophysical Characterization of α-Synuclein and Rotenone Interaction



Abstract
:1. Introduction
2. Results and Discussion
2.1. Kinetics of the α-Synuclein Fibril Formation in the Presence of Rotenone
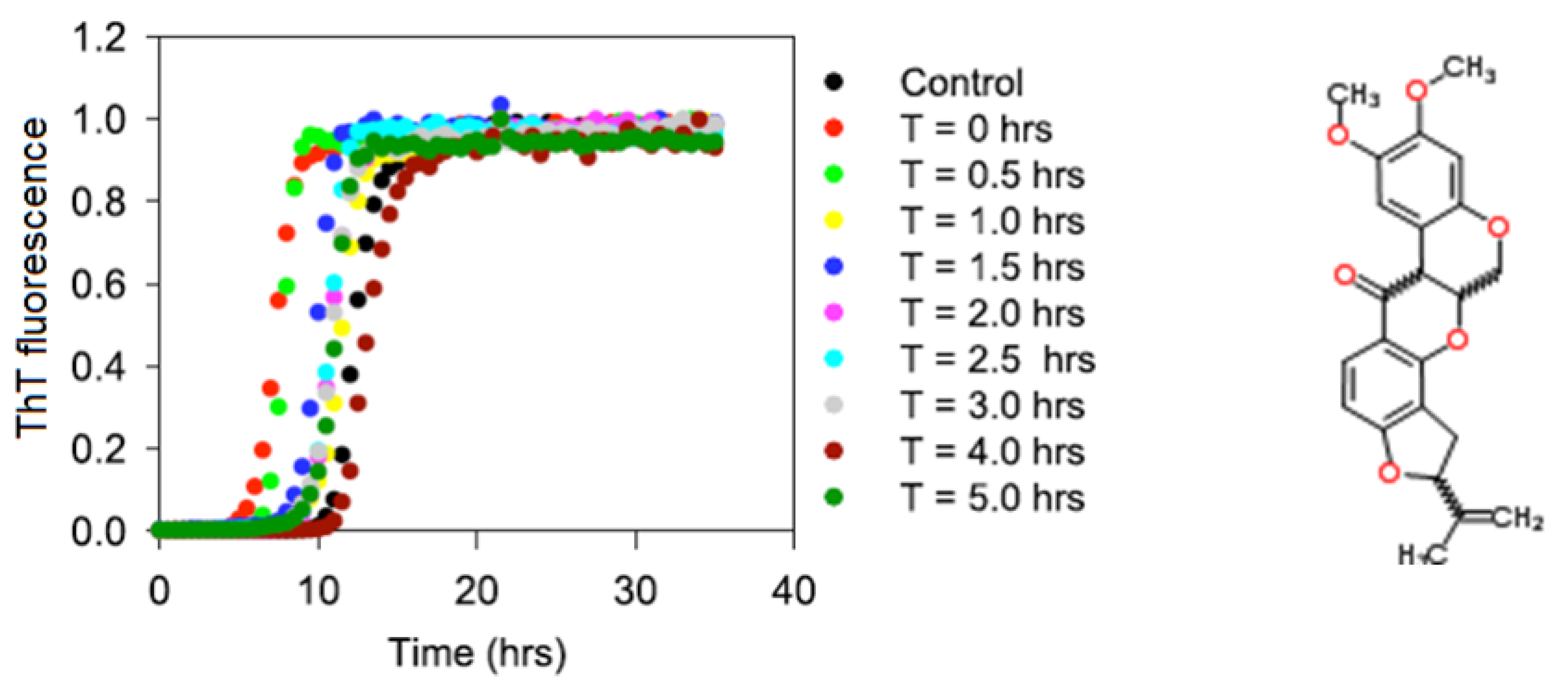

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Rotenone introduction time (h) | Lag time (h) | kapp (h−1) |
|---|---|---|
| N/A | 9.8 ± 1.8 | 1.2 ± 0.4 |
| 0.0 | 6.9 ± 1.2 | 1.7 ± 0.3 |
| 0.5 | 7.7 ± 1.0 | 2.3 ± 0.4 |
| 1.0 | 9.9 ± 2.0 | 1.7 ± 0.4 |
| 1.5 | 8.8 ± 1.1 | 1.9 ± 0.2 |
| 2.0 | 9.6 ± 0.4 | 1.8 ± 0.3 |
| 2.5 | 9.3 ± 0.7 | 1.9 ± 0.4 |
| 3.0 | 9.2 ± 1.1 | 1.7 ± 0.3 |
| 4.0 | 10.8 ± 1.0 | 1.2 ± 0.3 |
| 5.0 | 10.1 ± 0.6 | 2.1 ± 0.4 |
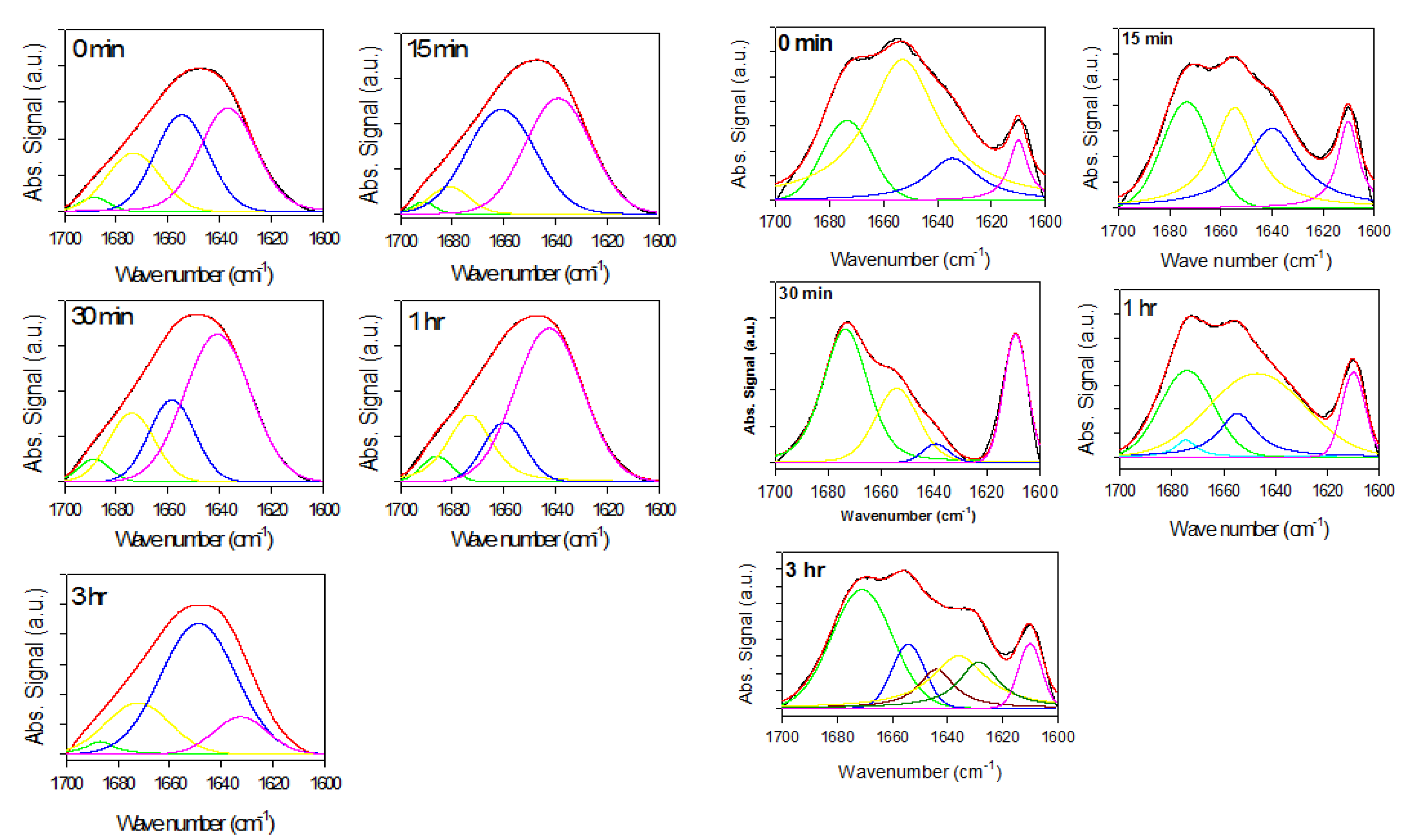
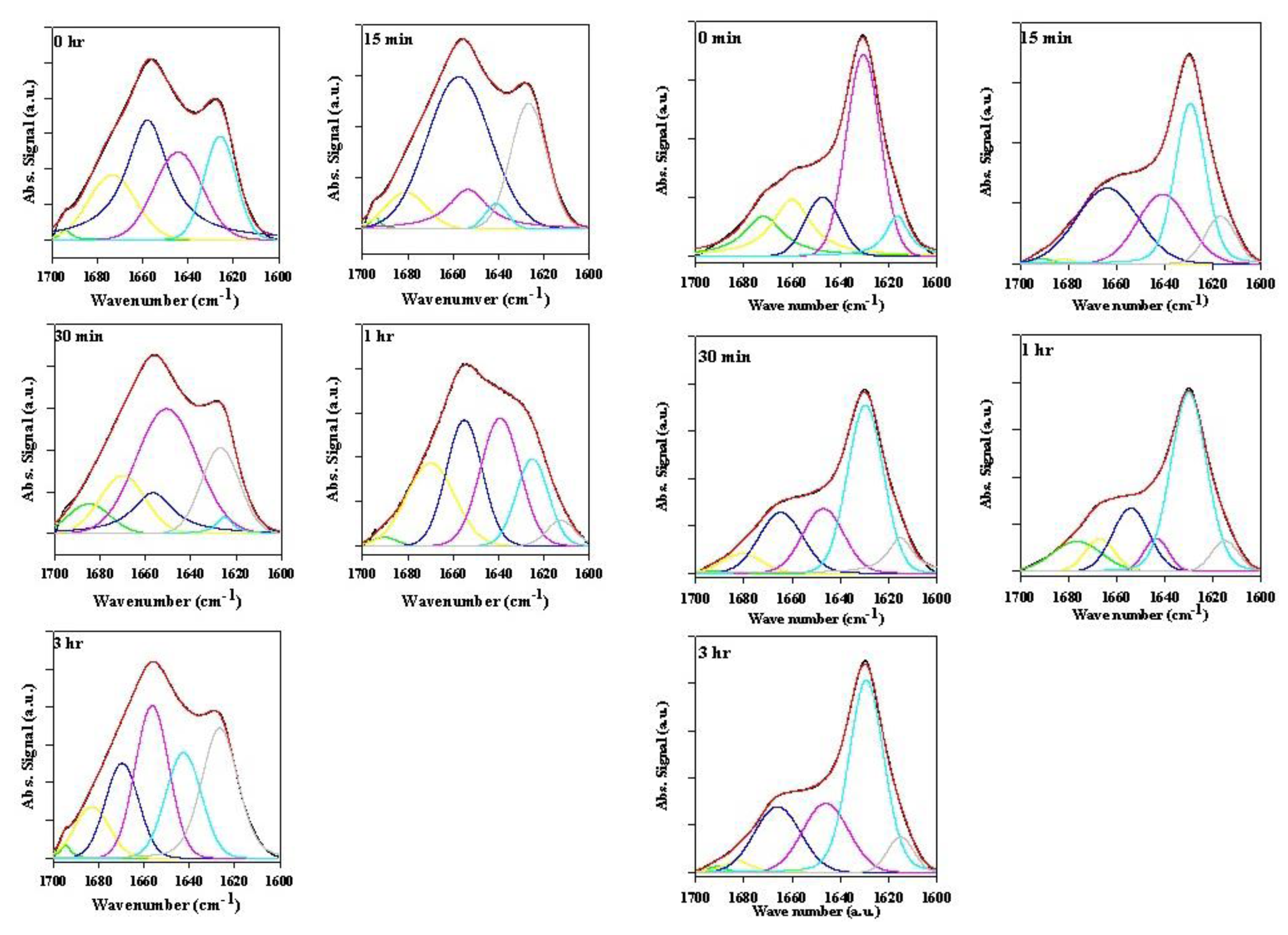
2.2. Analysis of α-Synuclein-Rotenone Fibrillation by ATR-FTIR
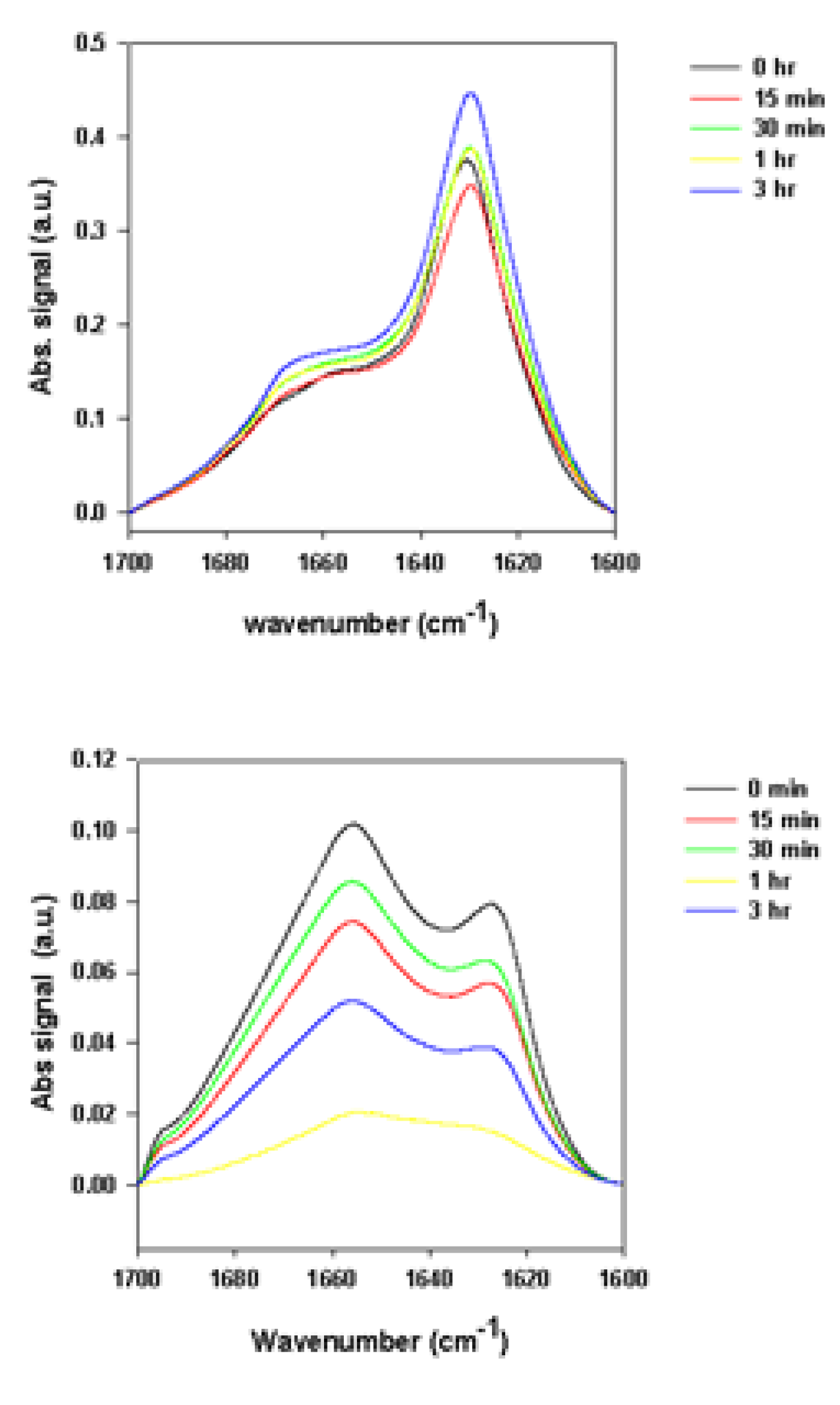
2.2.1. Effects of Rotenone on α-Synuclein ATR-FTIR Spectra during the Fibrillation Process
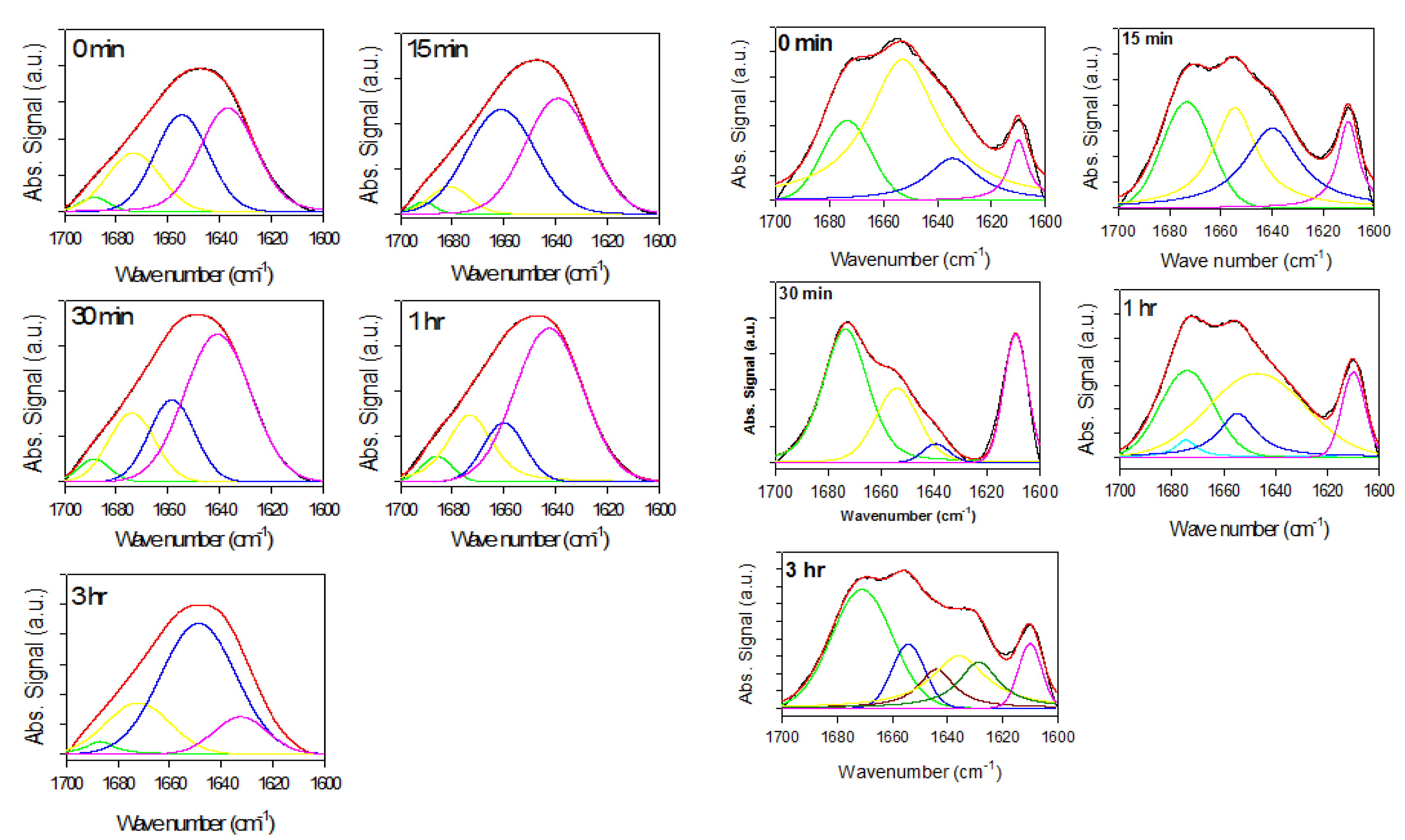
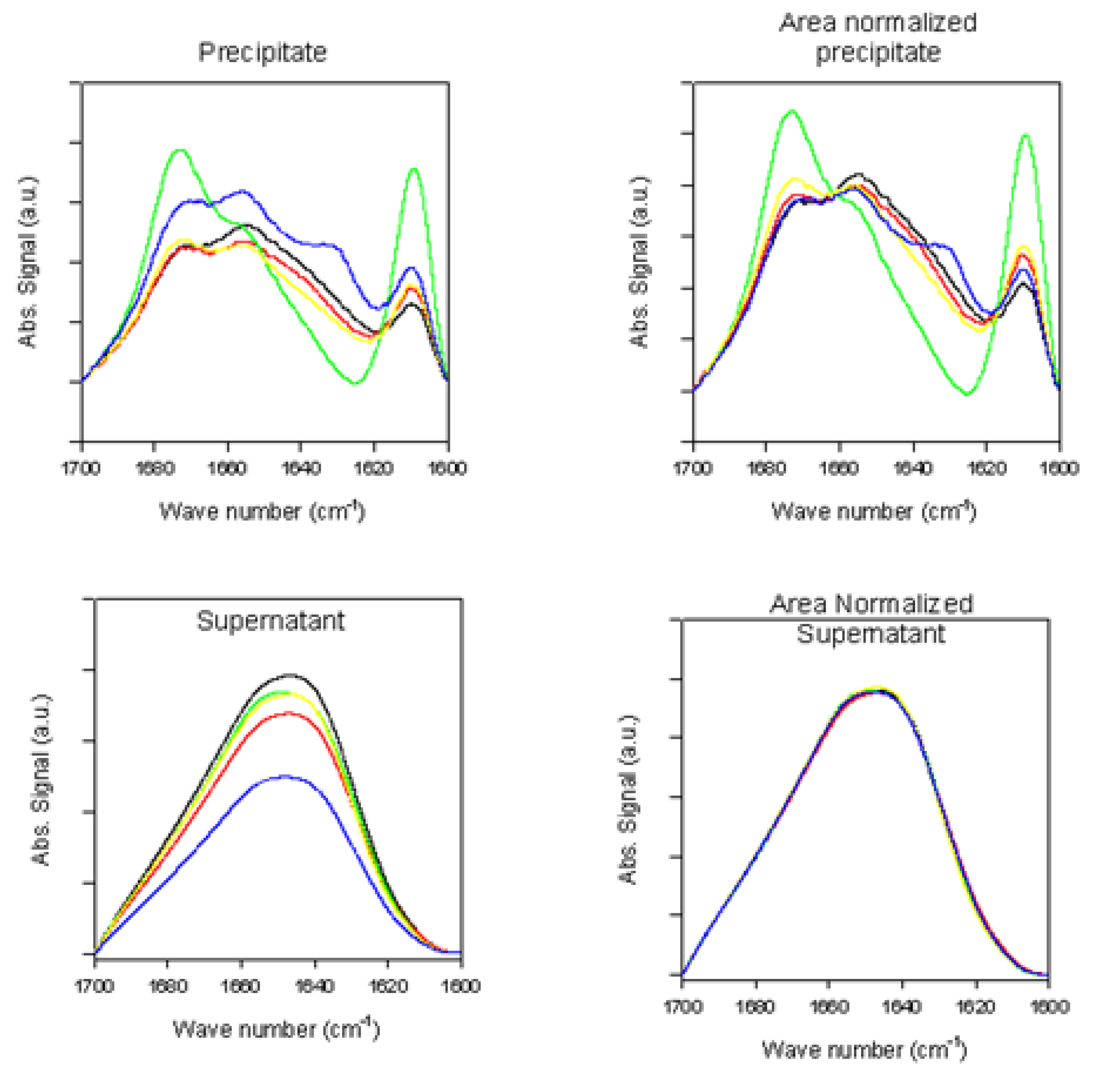
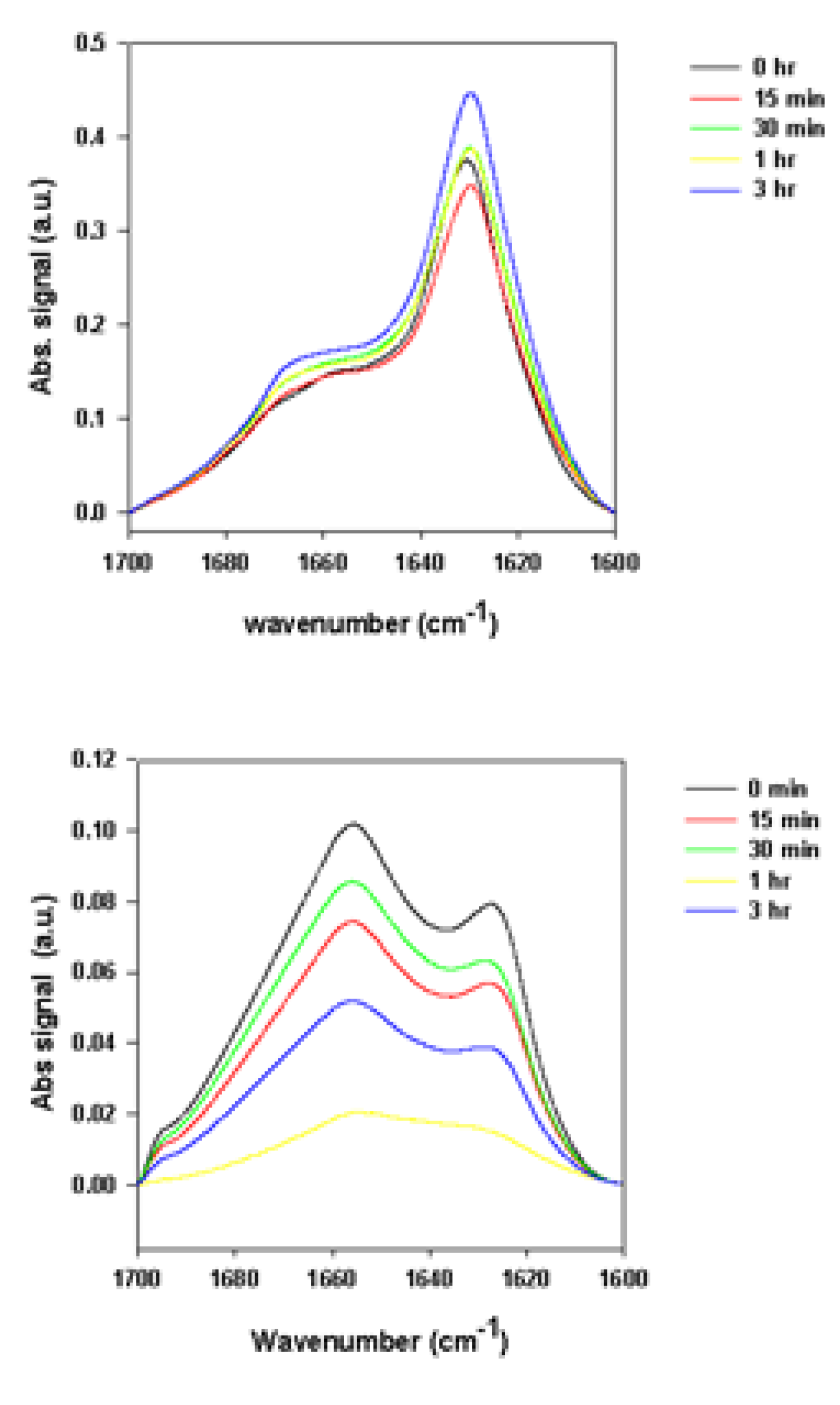
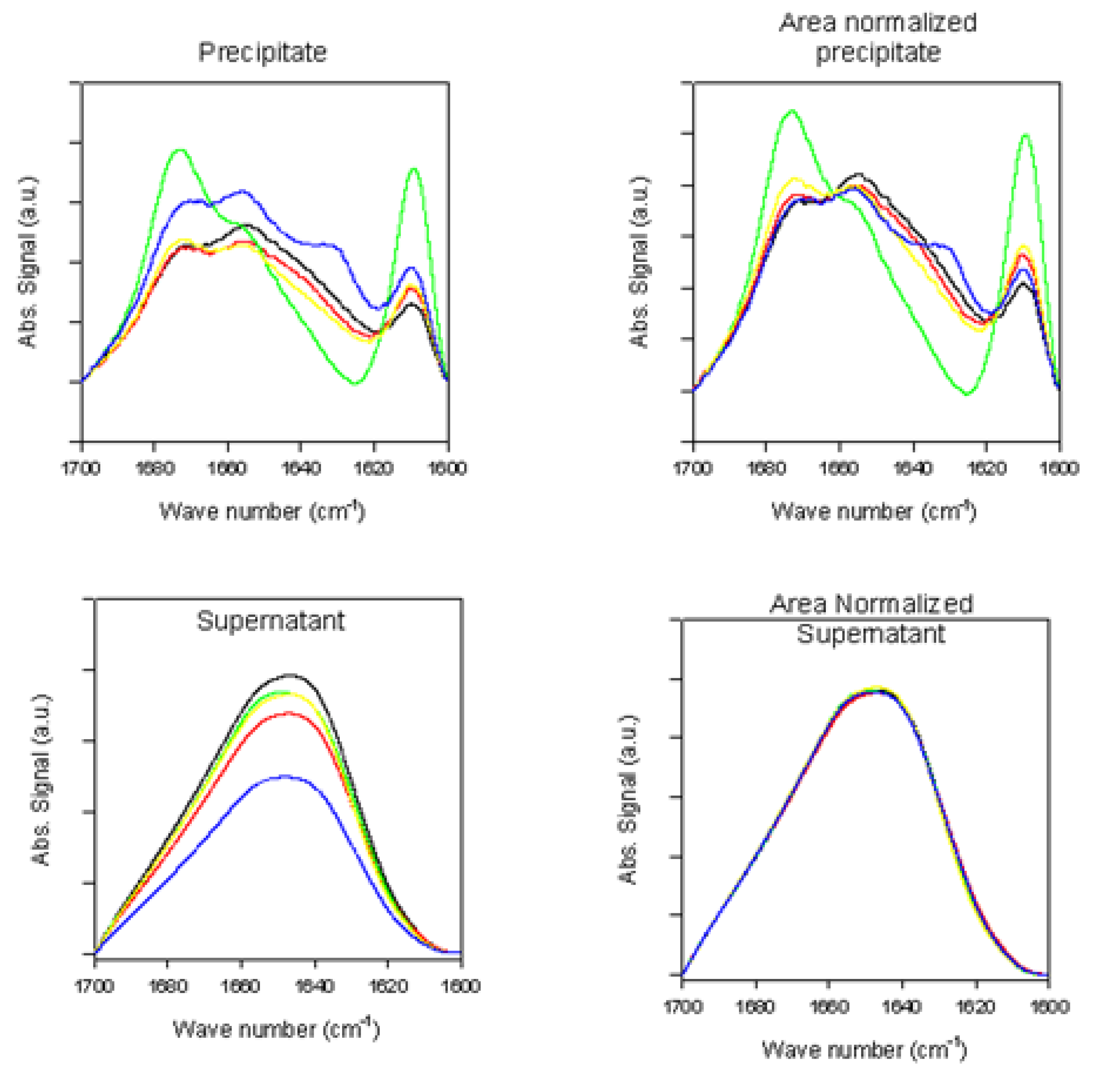
2.2.2. Effects of Rotenone on α-Synuclein ATR-FTIR Spectra at the End of the Fibrillation Process
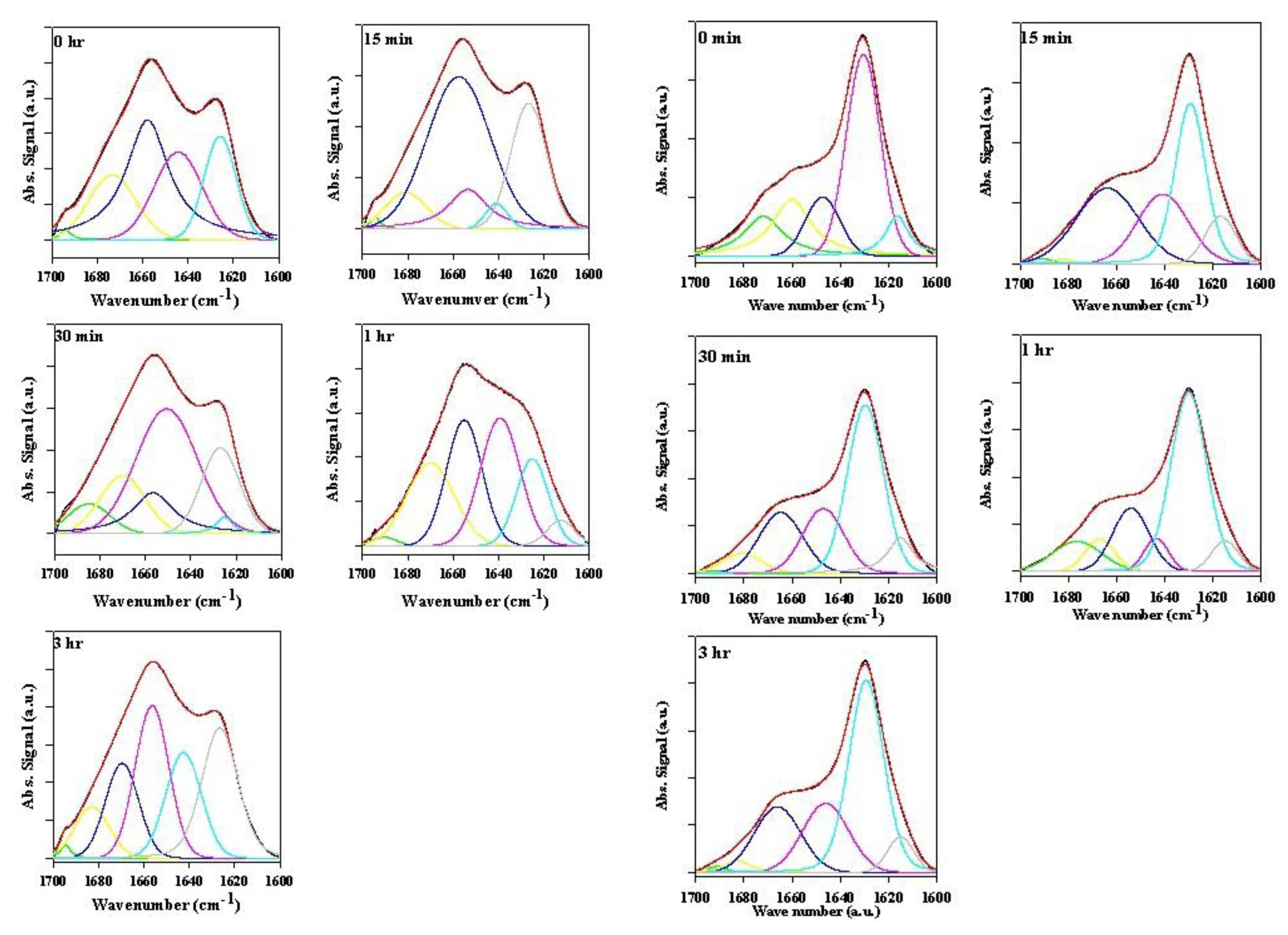
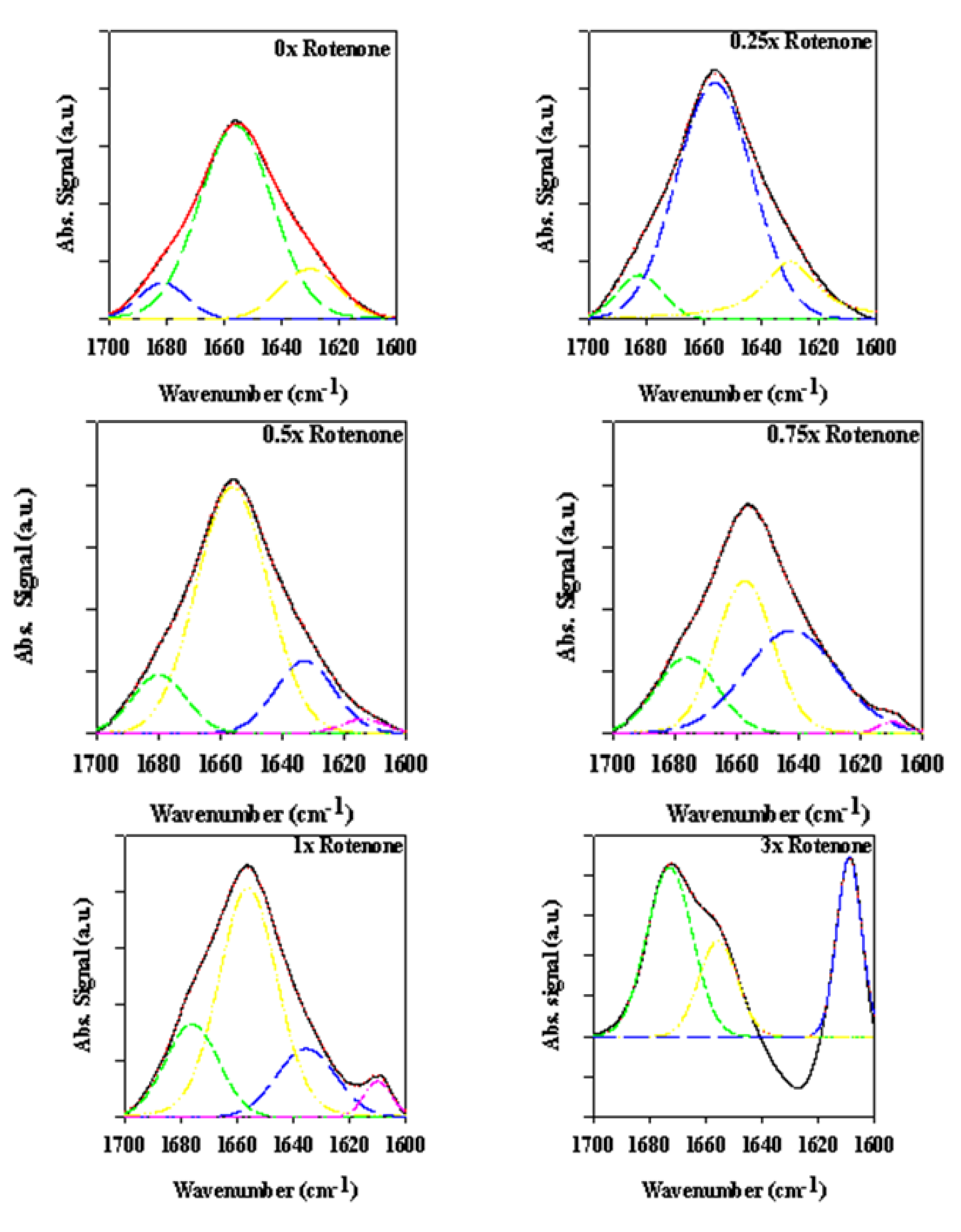
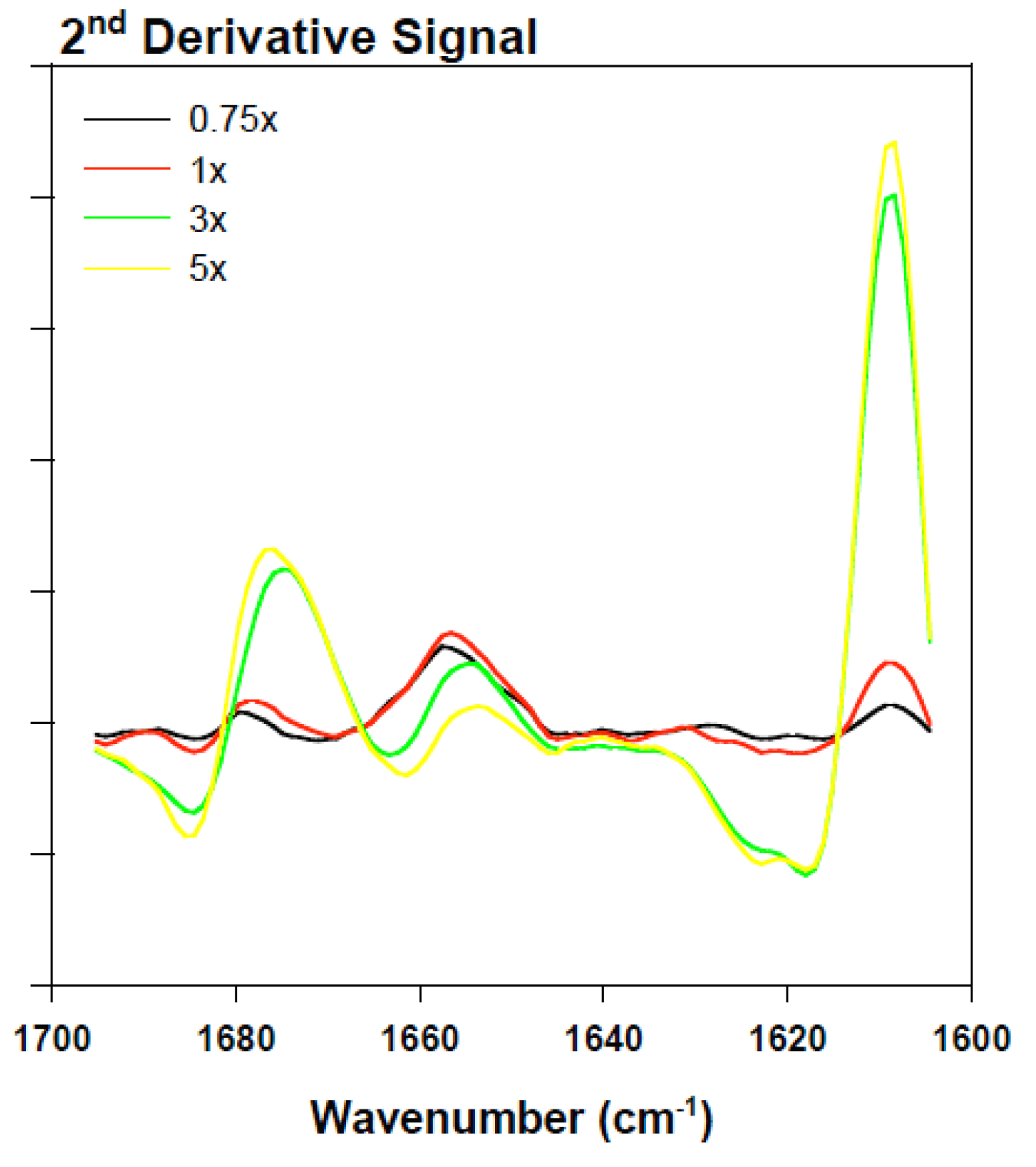
2.2.3. Secondary Structure Assessment
| Supernatant α-synuclein samples | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Time (hours) a | 0 | 15-min | 30-min | 1.0 h | 3.0 h | |||||||||
| Wave-number | % | Wave-number | % | Wave-number | % | Wave-number | % | Wave-number | % | |||||
| Structural assignment | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | |||||||||
| Turn or β-sheet | 1692 | 3 | 1692 | 2 | 1695 | 4 | 1688 | 5 | 1687 | 3 | ||||
| Turn | 1674 | 21 | 1682 | 7 | 1674 | 18 | 1673 | 20 | 1672 | 21 | ||||
| Loops/Disorder | 1655 | 35 | 1664 | 44 | 1658 | 22 | 1661 | 15 | 1650 | 63 | ||||
| Disorder/Extended | 1637 | 41 | 1639 | 47 | 1639 | 56 | 1643 | 61 | 1634 | 13 | ||||
| β-sheet | ||||||||||||||
| Side chains | ||||||||||||||
| Precipitate α-synuclein samples | ||||||||||||||
| Time (hours) a | 0 | 15-min | 30-min | 1.0 h | 3.0 h | |||||||||
| Wave-number | % | Wave-number | % | Wave-number | % | Wave-number | % | Wave-number | % | |||||
| Structural assignment | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | |||||||||
| Turn or β-sheet | 1674 | 19 | 1676 | 3 | ||||||||||
| Turn | 1674 | 28 | 1675 | 49 | 1674 | 27 | 1672 | 39 | ||||||
| Loops/Disorder | 1650 | 60 | 1655 | 30 | 1654 | 25 | 1656 | 13 | 1655 | 10 | ||||
| Disorder/Extended | 1637 | 14 | 1639 | 31 | 1639 | 4 | 1642 | 47 | 1643 | 20 | ||||
| β-sheet | 1628 | 13 | ||||||||||||
| Side chains | 1610 | 7 | 1610 | 12 | 1610 | 23 | 1610 | 11 | 1610 | 8 | ||||
| Supernatant α-synuclein samples | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Time (hours) a | 0 | 15-min | 30-min | 1.0 h | |||||||||
| Wave-number | % | Wave-number | % | Wave-number | % | Wave-number | % | Wave-number | % | ||||
| Structural assignment | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | ||||||||
| Turn or β-sheet | 1695 | 1 | 1695 | 1 | 1685 | 7 | 1692 | 1 | 1692&1683 | 1&9 | |||
| Turn | 1974 | 18 | 1681 | 8 | 1671 | 15 | 1672 | 23 | 1671 | 17 | |||
| Loops/Disorder | 1660 | 38 | 1660 | 52 | 1657 | 13 | 1656 | 26 | 1657 | 27 | |||
| Disorder/Extended | 1643 | 24 | 1652 &1642 | 11&13 | 1652 | 44 | 1639 | 30 | 1642 | 20 | |||
| β-sheet | 1626 | 19 | 1625 | 24 | 1628 | 19 | 1626 | 16 | 1626 | 26 | |||
| Side chains | 1624 | 3 | 1613 | 4 | |||||||||
| Supernatant α-synuclein samples | |||||||||||||
| Time (hours) a | 0 | 15-min | 30-min | 1.0 h | |||||||||
| Wave-number | % | Wave-number | % | Wave-number | % | Wave-number | % | Wave-number | % | ||||
| Structural assignment | cm−1 | cm−1 | cm−1 | cm−1 | cm−1 | ||||||||
| Turn or β-sheet | 1695 | 1 | 1693 | 0.5 | 1678 | 11 | 1690 | 1 | |||||
| Turn | 1674 | 14 | 1682 | 1 | 1680 | 6 | 1668 | 8 | 1684 | 3 | |||
| Loops/Disorder | 1660 | 19 | 1664 | 31 | 1665 | 20 | 1655 | 18 | 1658 | 20 | |||
| Disorder/Extended | 1649 | 14 | 1642 | 23 | 1647 | 20 | 1644 | 6 | 1644 | 22 | |||
| β-sheet | 1631 | 46 | 1629 | 34 | 1629 | 46 | 1629 | 51 | 1626 | 48 | |||
| Side chains | 1616 | 8 | 1616 | 10 | 1616 | 9 | 1613 | 6 | 1616 | 6 | |||
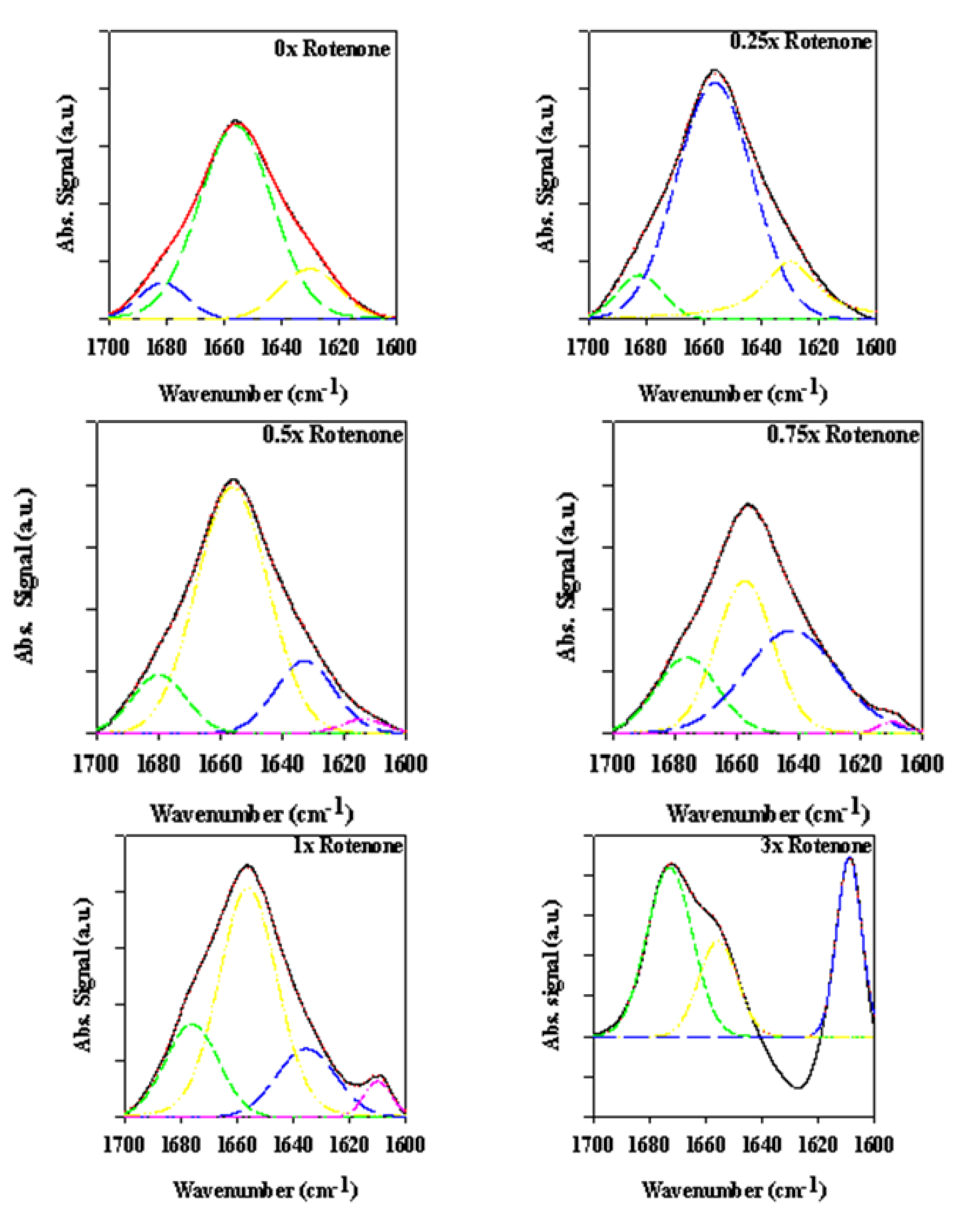
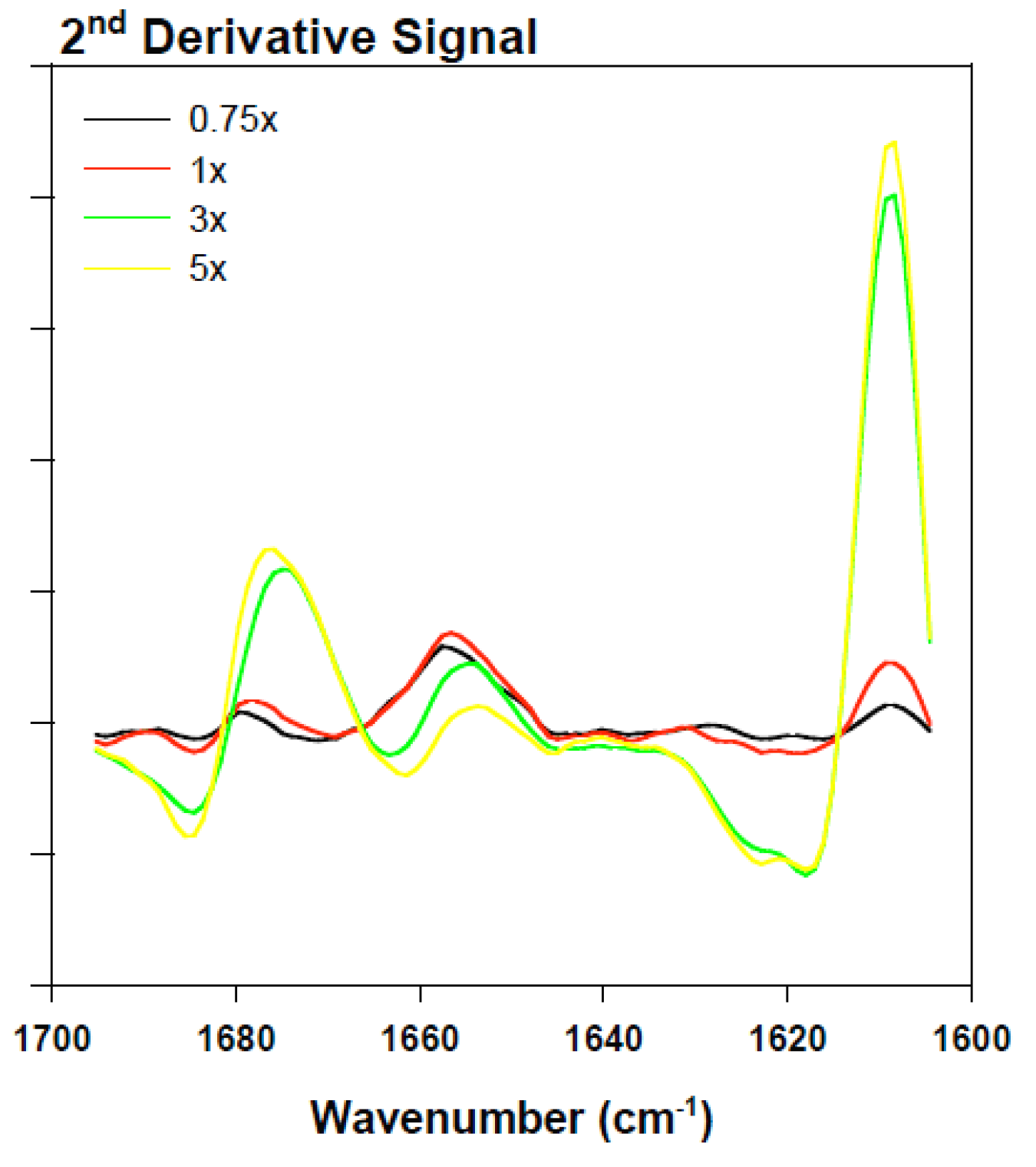
2.3. Analyzing the Effect Rotenone on Bovine Serum Albumin by ATR-FTIR

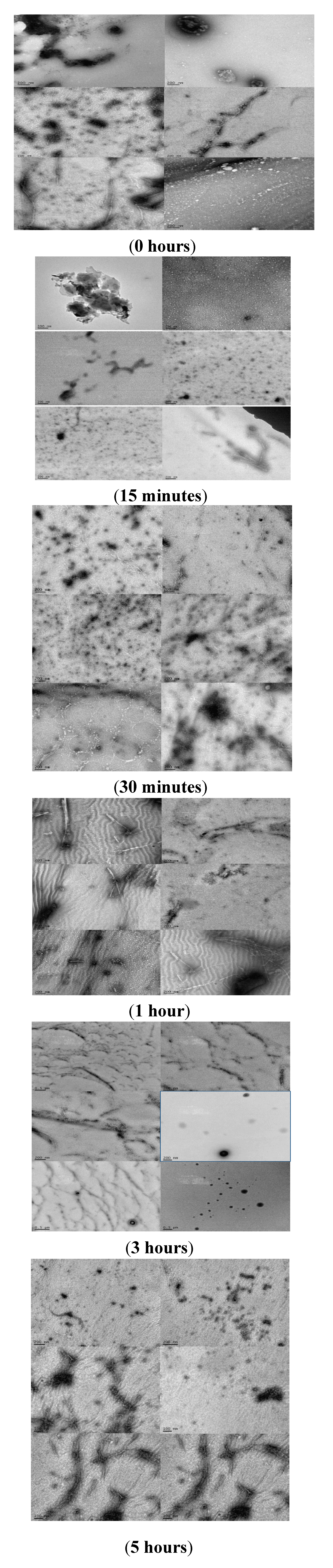
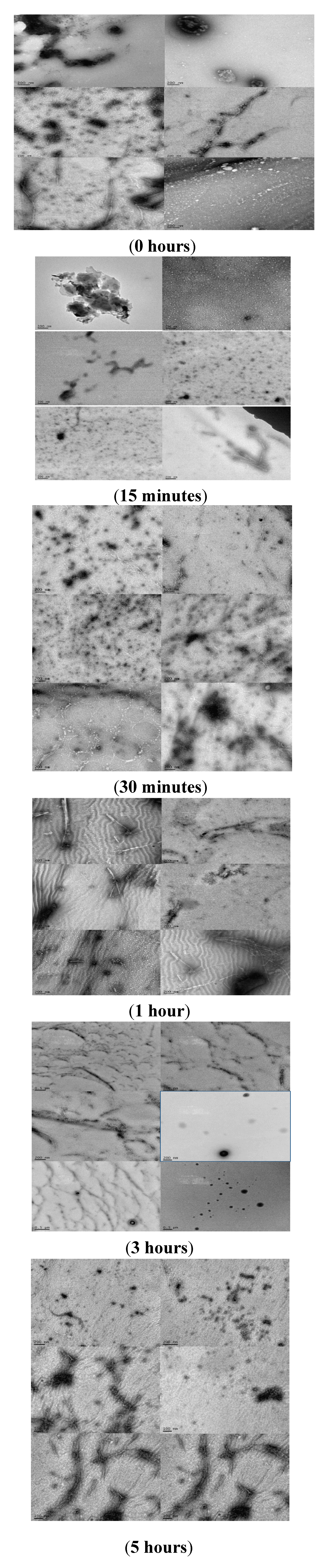
2.4. Analyzing the Morphologies of the α-Synuclein Aggregated Species by TEM and AFM
3. Experimental Section
3.1. Materials
3.1.1. Commercial Materials
3.1.2. α-Synuclein Expression and Purification
3.2. Methods
3.2.1. Sample Preparation for Aggregation Studies
3.2.2. Plate-Based Assay of α-Synuclein Aggregation in the Presence of Rotenone
3.2.3. Manual Assay for Detecting α-Synuclein Fibrils and Aggregates
3.2.4. Control Studies: Preparation of BSA-Rotenone Samples
3.2.5. Sample Preparation for ATR-FTIR Measurements
3.2.6. Protein-Rotenone ATR-FTIR Spectroscopy
3.2.7. Transmission Electron Microscopy
3.2.8. Atomic Force Microscopy
4. Conclusions
Conflicts of Interest
References
- Brown, R.C.; Lockwood, A.H.; Sonawane, B.R. Neurodegenerative diseases: An overview of environmental risk factors. Environ. Health Perspect. 2005, 113, 1250–1256. [Google Scholar] [CrossRef]
- Findley, L.J. The economic impact of Parkinson’s disease. Parkinsonism Relat. Disord. 2007, 13, S8–S12. [Google Scholar] [CrossRef]
- Uversky, V.N. Neuropathology and neurochemistry of Parkinson’s disease: The never-ending story or the story with no beginning? Minerva Psichiatr. 2009, 50, 1–26. [Google Scholar]
- Uversky, V.N.; Fink, A.L. Biophysical Properties of Human Alpha-Synuclein and its Role in Parkinson’s Disease. In Recent Research Developments in Proteins; Pandalai, S.G., Ed.; Transworld Research Network: Kerala, India, 2002; pp. 153–186. [Google Scholar]
- Tanner, C.M.; Goldman, S.M. Epidemiology of Parkinson’s disease. Neurol. Clin. 1996, 14, 317–335. [Google Scholar] [CrossRef]
- Langston, J.W.; Ballard, P.A., Jr. Parkinson’s disease in a chemist working with 1-methyl-4-phenyl-1,2,5,6-tetrahydropyridine. N. Engl. J. Med. 1983, 309, 310. [Google Scholar]
- Langston, J.W.; Ballard, P.; Tetrud, J.W.; Irwin, I. Chronic Parkinsonism in humans due to a product of meperidine-analog synthesis. Science 1983, 219, 979–980. [Google Scholar]
- Poskanzer, D.C.; Schwab, R.S. Cohort analysis of Parkinson’s syndrome: Evidence for a single etiology related to subclinical infection about 1920. J. Chron. Dis. 1963, 16, 961–973. [Google Scholar] [CrossRef]
- Jang, H.; Boltz, D.A.; Webster, R.G.; Smeyne, R.J. Viral parkinsonism. Biochim. Biophys. Acta 2009, 1792, 714–721. [Google Scholar] [CrossRef]
- Moore, D.J.; West, A.B.; Dawson, V.L.; Dawson, T.M. Molecular pathophysiology of Parkinson’s disease. Annu. Rev. Neurosci. 2005, 28, 57–87. [Google Scholar] [CrossRef]
- Cory-Slechta, D.A.; Thiruchelvam, M.; Barlow, B.K.; Richfield, E.K. Developmental pesticide models of the Parkinson disease phenotype. Environ. Health Perspect. 2005, 113, 1263–1270. [Google Scholar] [CrossRef]
- Rao, J.N.; Dua, V.; Ulmer, T.S. Characterization of alpha-synuclein interactions with selected aggregation-inhibiting small molecules. Biochemistry 2008, 47, 4651–4656. [Google Scholar] [CrossRef]
- Dauer, W.; Kholodilov, N.; Vila, M.; Trillat, A.C.; Goodchild, R.; Larsen, K.E.; Staal, R.; Tieu, K.; Schmitz, Y.; Yuan, C.A.; et al. Resistance of alpha -synuclein null mice to the parkinsonian neurotoxin MPTP. Proc. Natl. Acad. Sci. USA 2002, 99, 14524–14529. [Google Scholar] [CrossRef]
- Munishkina, L.A.; Fink, A.L. Fluorescence as a method to reveal structures and membrane-interactions of amyloidogenic proteins. Biochim. Biophys. Acta 2007, 1768, 1862–1885. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Lee, V.M. Parkinson’s disease and related alpha-synucleinopathies are brain amyloidoses. Ann. N. Y. Acad. Sci. 2003, 991, 107–110. [Google Scholar] [CrossRef]
- Stefani, M.; Dobson, C.M. Protein aggregation and aggregate toxicity: New insights into protein folding, misfolding diseases and biological evolution. J. Mol. Med. (Berl.) 2003, 81, 678–699. [Google Scholar] [CrossRef]
- Giasson, B.I.; Lee, V.M.; Trojanowski, J.Q. Interactions of amyloidogenic proteins. Neuromol. Med. 2003, 4, 49–58. [Google Scholar] [CrossRef]
- Uversky, V.N. Intrinsic disorder in proteins associated with neurodegenerative diseases. Front. Biosci. 2009, 14, 5188–5238. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Baba, M.; Nakajo, S.; Tu, P.H.; Tomita, T.; Nakaya, K.; Lee, V.M.; Trojanowski, J.Q.; Iwatsubo, T. Aggregation of alpha-synuclein in Lewy bodies of sporadic Parkinson’s disease and dementia with Lewy bodies. Am. J. Pathol. 1998, 152, 879–884. [Google Scholar]
- Polymeropoulos, M.H.; Lavedan, C.; Leroy, E.; Ide, S.E.; Dehejia, A.; Dutra, A.; Pike, B.; Root, H.; Rubenstein, J.; Boyer, R.; et al. Mutation in the alpha-synuclein gene identified in families with Parkinson’s disease. Science 1997, 276, 2045–2047. [Google Scholar] [CrossRef]
- Zarranz, J.J.; Alegre, J.; Gomez-Esteban, J.C.; Lezcano, E.; Ros, R.; Ampuero, I.; Vidal, L.; Hoenicka, J.; Rodriguez, O.; Atares, B.; et al. The new mutation, E46K, of alpha-synuclein causes Parkinson and Lewy body dementia. Ann. Neurol. 2004, 55, 164–173. [Google Scholar] [CrossRef]
- Kruger, R.; Kuhn, W.; Muller, T.; Woitalla, D.; Graeber, M.; Kosel, S.; Przuntek, H.; Epplen, J.T.; Schols, L.; Riess, O. Ala30Pro mutation in the gene encoding alpha-synuclein in Parkinson’s disease. Nat. Genet. 1998, 18, 106–108. [Google Scholar] [CrossRef]
- Singleton, A.; Gwinn-Hardy, K.; Sharabi, Y.; Li, S.T.; Holmes, C.; Dendi, R.; Hardy, J.; Crawley, A.; Goldstein, D.S. Association between cardiac denervation and parkinsonism caused by alpha-synuclein gene triplication. Brain 2004, 127, 768–772. [Google Scholar] [CrossRef]
- Singleton, A.B.; Farrer, M.; Johnson, J.; Singleton, A.; Hague, S.; Kachergus, J.; Hulihan, M.; Peuralinna, T.; Dutra, A.; Nussbaum, R.; et al. alpha-Synuclein locus triplication causes Parkinson’s disease. Science 2003, 302, 841. [Google Scholar] [CrossRef]
- Farrer, M.; Kachergus, J.; Forno, L.; Lincoln, S.; Wang, D.S.; Hulihan, M.; Maraganore, D.; Gwinn-Hardy, K.; Wszolek, Z.; Dickson, D.; et al. Comparison of kindreds with parkinsonism and alpha-synuclein genomic multiplications. Ann. Neurol. 2004, 55, 174–179. [Google Scholar] [CrossRef]
- Dickson, D.W. Alpha-synuclein and the Lewy body disorders. Curr. Opin. Neurol. 2001, 14, 423–432. [Google Scholar] [CrossRef]
- Goedert, M. Parkinson’s disease and other alpha-synucleinopathies. Clin. Chem. Lab. Med. 2001, 39, 308–312. [Google Scholar] [CrossRef]
- Goedert, M. Alpha-synuclein and neurodegenerative diseases. Nat. Rev. Neurosci. 2001, 2, 492–501. [Google Scholar] [CrossRef]
- Dev, K.K.; Hofele, K.; Barbieri, S.; Buchman, V.L.; van der Putten, H. Part II: Alpha-synuclein and its molecular pathophysiological role in neurodegenerative disease. Neuropharmacology 2003, 45, 14–44. [Google Scholar] [CrossRef]
- Trojanowski, J.Q.; Lee, V.M. Aggregation of neurofilament and alpha-synuclein proteins in Lewy bodies: Implications for the pathogenesis of Parkinson disease and Lewy body dementia. Arch. Neurol. 1998, 55, 151–152. [Google Scholar] [CrossRef]
- Uversky, V.N. Neuropathology, biochemistry, and biophysics of alpha-synuclein aggregation. J. Neurochem. 2007, 103, 17–37. [Google Scholar]
- Uversky, V.N.; Eliezer, D. Biophysics of Parkinson’s disease: Structure and aggregation of alpha-synuclein. Curr. Protein Pept. Sci. 2009, 10, 483–499. [Google Scholar] [CrossRef]
- Santner, A.; Uversky, V.N. Metalloproteomics and metal toxicology of alpha-synuclein. Metallomics 2010, 2, 378–392. [Google Scholar] [CrossRef]
- Breydo, L.; Wu, J.W.; Uversky, V.N. Alpha-synuclein misfolding and Parkinson’s disease. Biochim. Biophys. Acta 2012, 1822, 261–285. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. Alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef]
- Masliah, E.; Rockenstein, E.; Veinbergs, I.; Mallory, M.; Hashimoto, M.; Takeda, A.; Sagara, Y.; Sisk, A.; Mucke, L. Dopaminergic loss and inclusion body formation in alpha-synuclein mice: Implications for neurodegenerative disorders. Science 2000, 287, 1265–1269. [Google Scholar] [CrossRef]
- Feany, M.B.; Bender, W.W. A Drosophila model of Parkinson’s disease. Nature 2000, 404, 394–398. [Google Scholar] [CrossRef]
- Giasson, B.I.; Uryu, K.; Trojanowski, J.Q.; Lee, V.M. Mutant and wild type human alpha-synucleins assemble into elongated filaments with distinct morphologies in vitro. J. Biol. Chem. 1999, 274, 7619–7622. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Evidence for a partially folded intermediate in alpha-synuclein fibril formation. J. Biol. Chem. 2001, 276, 10737–10744. [Google Scholar] [CrossRef]
- Uversky, V.N. Neurotoxicant-induced animal models of Parkinson’s disease: Understanding the role of rotenone, maneb and paraquat in neurodegeneration. Cell Tissue Res. 2004, 318, 225–241. [Google Scholar] [CrossRef]
- Tanner, C.M.; Chen, B.; Wang, W.; Peng, M.; Liu, Z.; Liang, X.; Kao, L.C.; Gilley, D.W.; Goetz, C.G.; Schoenberg, B.S. Environmental factors and Parkinson’s disease: A case-control study in China. Neurology 1989, 39, 660–664. [Google Scholar] [CrossRef]
- Tanner, C.M. The role of environmental toxins in the etiology of Parkinson’s disease. Trends Neurosci. 1989, 12, 49–54. [Google Scholar] [CrossRef]
- Di Monte, D.A. The environment and Parkinson’s disease: Is the nigrostriatal system preferentially targeted by neurotoxins? Lancet Neurol. 2003, 2, 531–538. [Google Scholar] [CrossRef]
- McCormack, A.L.; Thiruchelvam, M.; Manning-Bog, A.B.; Thiffault, C.; Langston, J.W.; Cory-Slechta, D.A.; di Monte, D.A. Environmental risk factors and Parkinson’s disease: Selective degeneration of nigral dopaminergic neurons caused by the herbicide paraquat. Neurobiol. Dis. 2002, 10, 119–127. [Google Scholar] [CrossRef]
- Di Monte, D.A.; Lavasani, M.; Manning-Bog, A.B. Environmental factors in Parkinson’s disease. Neurotoxicology 2002, 23, 487–502. [Google Scholar] [CrossRef]
- Manning-Bog, A.B.; McCormack, A.L.; Li, J.; Uversky, V.N.; Fink, A.L.; Di Monte, D.A. The herbicide paraquat causes up-regulation and aggregation of alpha-synuclein in mice: Paraquat and alpha-synuclein. J. Biol. Chem. 2002, 277, 1641–1644. [Google Scholar]
- Ragonese, P.; Salemi, G.; Morgante, L.; Aridon, P.; Epifanio, A.; Buffa, D.; Scoppa, F.; Savettieri, G. A case-control study on cigarette, alcohol, and coffee consumption preceding Parkinson’s disease. Neuroepidemiology 2003, 22, 297–304. [Google Scholar] [CrossRef]
- Ross, G.W.; Petrovitch, H. Current evidence for neuroprotective effects of nicotine and caffeine against Parkinson’s disease. Drugs Aging 2001, 18, 797–806. [Google Scholar] [CrossRef]
- Ross, G.W.; Abbott, R.D.; Petrovitch, H.; Morens, D.M.; Grandinetti, A.; Tung, K.H.; Tanner, C.M.; Masaki, K.H.; Blanchette, P.L.; Curb, J.D.; et al. Association of coffee and caffeine intake with the risk of Parkinson disease. JAMA 2000, 283, 2674–2679. [Google Scholar] [CrossRef]
- Hirsch, E.C.; Brandel, J.P.; Galle, P.; Javoy-Agid, F.; Agid, Y. Iron and aluminum increase in the substantia nigra of patients with Parkinson’s disease: An X-ray microanalysis. J. Neurochem. 1991, 56, 446–451. [Google Scholar] [CrossRef]
- Yasui, M.; Kihira, T.; Ota, K. Calcium, magnesium and aluminum concentrations in Parkinson’s disease. Neurotoxicology 1992, 13, 593–600. [Google Scholar]
- Good, P.F.; Olanow, C.W.; Perl, D.P. Neuromelanin-containing neurons of the substantia nigra accumulate iron and aluminum in Parkinson’s disease: A LAMMA study. Brain Res. 1992, 593, 343–346. [Google Scholar] [CrossRef]
- Gorell, J.M.; Rybicki, B.A.; Johnson, C.C.; Peterson, E.L. Occupational metal exposures and the risk of Parkinson’s disease. Neuroepidemiology 1999, 18, 303–308. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Kortsha, G.X.; Brown, G.G.; Richardson, R.J. Occupational exposure to manganese, copper, lead, iron, mercury and zinc and the risk of Parkinson’s disease. Neurotoxicology 1999, 20, 239–247. [Google Scholar]
- Altschuler, E. Aluminum-containing antacids as a cause of idiopathic Parkinson’s disease. Med. Hypotheses 1999, 53, 22–23. [Google Scholar] [CrossRef]
- Seidler, A.; Hellenbrand, W.; Robra, B.P.; Vieregge, P.; Nischan, P.; Joerg, J.; Oertel, W.H.; Ulm, G.; Schneider, E. Possible environmental, occupational, and other etiologic factors for Parkinson’s disease: A case-control study in Germany. Neurology 1996, 46, 1275–1284. [Google Scholar] [CrossRef]
- Davis, L.E.; Adair, J.C. Parkinsonism from methanol poisoning: Benefit from treatment with anti-Parkinson drugs. Mov. Disord. 1999, 14, 520–522. [Google Scholar] [CrossRef]
- Hageman, G.; van der Hoek, J.; van Hout, M.; van der Laan, G.; Steur, E.J.; de Bruin, W.; Herholz, K. Parkinsonism, pyramidal signs, polyneuropathy, and cognitive decline after long-term occupational solvent exposure. J. Neurol. 1999, 246, 198–206. [Google Scholar] [CrossRef]
- Pezzoli, G.; Strada, O.; Silani, V.; Zecchinelli, A.; Perbellini, L.; Javoy-Agid, F.; Ghidoni, P.; Motti, E.D.; Masini, T.; Scarlato, G.; et al. Clinical and pathological features in hydrocarbon-induced parkinsonism. Ann. Neurol. 1996, 40, 922–925. [Google Scholar] [CrossRef]
- Uitti, R.J.; Snow, B.J.; Shinotoh, H.; Vingerhoets, F.J.; Hayward, M.; Hashimoto, S.; Richmond, J.; Markey, S.P.; Markey, C.J.; Calne, D.B. Parkinsonism induced by solvent abuse. Ann. Neurol. 1994, 35, 616–619. [Google Scholar] [CrossRef]
- Klawans, H.L.; Stein, R.W.; Tanner, C.M.; Goetz, C.G. A pure parkinsonian syndrome following acute carbon monoxide intoxication. Arch. Neurol. 1982, 39, 302–304. [Google Scholar] [CrossRef]
- Gorell, J.M.; Johnson, C.C.; Rybicki, B.A.; Peterson, E.L.; Richardson, R.J. The risk of Parkinson’s disease with exposure to pesticides, farming, well water, and rural living. Neurology 1998, 50, 1346–1350. [Google Scholar] [CrossRef]
- Fall, P.A.; Fredrikson, M.; Axelson, O.; Granerus, A.K. Nutritional and occupational factors influencing the risk of Parkinson’s disease: A case-control study in southeastern Sweden. Mov. Disord. 1999, 14, 28–37. [Google Scholar] [CrossRef]
- Semchuk, K.M.; Love, E.J.; Lee, R.G. Parkinson’s disease: A test of the multifactorial etiologic hypothesis. Neurology 1993, 43, 1173–1180. [Google Scholar] [CrossRef]
- Semchuk, K.M.; Love, E.J.; Lee, R.G. Parkinson’s disease and exposure to agricultural work and pesticide chemicals. Neurology 1992, 42, 1328–1335. [Google Scholar] [CrossRef]
- Liou, H.H.; Tsai, M.C.; Chen, C.J.; Jeng, J.S.; Chang, Y.C.; Chen, S.Y.; Chen, R.C. Environmental risk factors and Parkinson’s disease: A case-control study in Taiwan. Neurology 1997, 48, 1583–1588. [Google Scholar] [CrossRef]
- Vanacore, N.; Nappo, A.; Gentile, M.; Brustolin, A.; Palange, S.; Liberati, A.; Di Rezze, S.; Caldora, G.; Gasparini, M.; Benedetti, F.; et al. Evaluation of risk of Parkinson’s disease in a cohort of licensed pesticide users. Neurol. Sci. 2002, 23, S119–S120. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Bower, K.; Fink, A.L. Synergistic effects of pesticides and metals on the fibrillation of alpha-synuclein: Implications for Parkinson’s disease. Neurotoxicology 2002, 23, 527–536. [Google Scholar] [CrossRef]
- Uversky, V.N.; Li, J.; Fink, A.L. Pesticides directly accelerate the rate of alpha-synuclein fibril formation: A possible factor in Parkinson’s disease. FEBS Lett. 2001, 500, 105–108. [Google Scholar] [CrossRef]
- Uversky, V.N.; Fink, A.L. Conformational constraints for amyloid fibrillation: The importance of being unfolded. Biochim. Biophys. Acta 2004, 1698, 131–153. [Google Scholar] [CrossRef]
- Uversky, V.N. Mysterious oligomerization of the amyloidogenic proteins. FEBS J. 2010, 277, 2940–2953. [Google Scholar] [CrossRef]
- Yamin, G.; Uversky, V.N.; Fink, A.L. Nitration inhibits fibrillation of human alpha-synuclein in vitro by formation of soluble oligomers. FEBS Lett. 2003, 542, 147–152. [Google Scholar] [CrossRef]
- Hong, D.P.; Fink, A.L.; Uversky, V.N. Structural characteristics of alpha-synuclein oligomers stabilized by the flavonoid baicalein. J. Mol. Biol. 2008, 383, 214–223. [Google Scholar] [CrossRef]
- Hong, D.P.; Fink, A.L.; Uversky, V.N. Smoking and Parkinson’s disease: Does nicotine affect alpha-synuclein fibrillation? Biochim. Biophys. Acta 2009, 1794, 282–290. [Google Scholar] [CrossRef]
- Meng, X.; Munishkina, L.A.; Fink, A.L.; Uversky, V.N. Molecular mechanisms underlying the flavonoid-induced inhibition of alpha-synuclein fibrillation. Biochemistry 2009, 48, 8206–8224. [Google Scholar] [CrossRef]
- Zhou, W.; Gallagher, A.; Hong, D.P.; Long, C.; Fink, A.L.; Uversky, V.N. At low concentrations, 3,4-dihydroxyphenylacetic acid (DOPAC) binds non-covalently to alpha-synuclein and prevents its fibrillation. J. Mol. Biol. 2009, 388, 597–610. [Google Scholar] [CrossRef]
- Hong, D.P.; Han, S.; Fink, A.L.; Uversky, V.N. Characterization of the non-fibrillar alpha-synuclein oligomers. Protein Pept. Lett. 2011, 18, 230–240. [Google Scholar] [CrossRef]
- Zhou, W.; Long, C.; Reaney, S.H.; di Monte, D.A.; Fink, A.L.; Uversky, V.N. Methionine oxidation stabilizes non-toxic oligomers of alpha-synuclein through strengthening the auto-inhibitory intra-molecular long-range interactions. Biochim. Biophys. Acta 2010, 1802, 322–330. [Google Scholar] [CrossRef]
- Bucciantini, M.; Giannoni, E.; Chiti, F.; Baroni, F.; Formigli, L.; Zurdo, J.; Taddei, N.; Ramponi, G.; Dobson, C.M.; Stefani, M. Inherent toxicity of aggregates implies a common mechanism for protein misfolding diseases. Nature 2002, 416, 507–511. [Google Scholar] [CrossRef]
- Walsh, D.M.; Hartley, D.M.; Kusumoto, Y.; Fezoui, Y.; Condron, M.M.; Lomakin, A.; Benedek, G.B.; Selkoe, D.J.; Teplow, D.B. Amyloid beta-protein fibrillogenesis. Structure and biological activity of protofibrillar intermediates. J. Biol. Chem. 1999, 274, 25945–25952. [Google Scholar] [CrossRef]
- Militello, V.; Casarino, C.; Emanuele, A.; Giostra, A.; Pullara, F.; Leone, M. Aggregation kinetics of bovine serum albumin studied by FTIR spectroscopy and light scattering. Biophys. Chem. 2004, 107, 175–187. [Google Scholar] [CrossRef]
- Munishkina, L.A.; Fink, A.L.; Uversky, V.N. Accelerated fibrillation of alpha-synuclein induced by the combined action of macromolecular crowding and factors inducing partial folding. Curr. Alzheimer Res. 2009, 6, 252–260. [Google Scholar] [CrossRef]
- Silva, B.A.; Breydo, L.; Fink, A.L.; Uversky, V.N. Agrochemicals, alpha-synuclein, and Parkinson’s disease. Mol. Neurobiol. 2013, 47, 598–612. [Google Scholar] [CrossRef]
- Garcia, S.J.; Seidler, F.J.; Qiao, D.; Slotkin, T.A. Chlorpyrifos targets developing glia: Effects on glial fibrillary acidic protein. Brain Res. Dev. Brain Res. 2002, 133, 151–161. [Google Scholar] [CrossRef]
- Andre, C.; Truong, T.T.; Robert, J.F.; Guillaume, Y.C. Effect of metals on herbicides-alpha-synuclein association: A possible factor in neurodegenerative disease studied by capillary electrophoresis. Electrophoresis 2005, 26, 3256–3264. [Google Scholar] [CrossRef]
- Ritz, B.; Yu, F. Parkinson’s disease mortality and pesticide exposure in California 1984–1994. Int. J. Epidemiol. 2000, 29, 323–329. [Google Scholar]
- Maries, E.; Dass, B.; Collier, T.J.; Kordower, J.H.; Steece-Collier, K. The role of alpha-synuclein in Parkinson’s disease: Insights from animal models. Nat. Rev. Neurosci. 2003, 4, 727–738. [Google Scholar]
- Frigerio, R.; Sanft, K.R.; Grossardt, B.R.; Peterson, B.J.; Elbaz, A.; Bower, J.H.; Ahlskog, J.E.; de Andrade, M.; Maraganore, D.M.; Rocca, W.A. Chemical exposures and Parkinson’s disease: A population-based case-control study. Mov. Disord. 2006, 21, 1688–1692. [Google Scholar] [CrossRef]
- Seshadri, S.; Khurana, R.; Fink, A.L. Fourier transform infrared spectroscopy in analysis of protein deposits. Methods Enzymol. 1999, 309, 559–576. [Google Scholar] [CrossRef]
- Barth, A.; Zscherp, C. What vibrations tell us about proteins. Q Rev. Biophys. 2002, 35, 369–430. [Google Scholar] [CrossRef]
- Haris, P.I.; Severcan, F. FTIR spectroscopic characterization of protein structure in aqueous and non-aqueous media. J. Mol. Catal. B Enzym. 1999, 7, 207–221. [Google Scholar] [CrossRef]
- Barth, A. The infrared absorption of amino acid side chains. Progr. Biophys. Mol. Biol. 2000, 74, 141–173. [Google Scholar] [CrossRef]
- Shaw, R.A.; Mantsch, H.H. Solvent influence on the conformation of cyclosporin. An FT-IR study. Can. J. Chem. 1993, 7, 1334–1339. [Google Scholar] [CrossRef]
- Braiman, M.S.; Rothschild, K.J. Fourier transform infrared techniques for probing membrane protein structure. Annu. Rev. Biophys. Biophys. Chem. 1988, 17, 541–570. [Google Scholar] [CrossRef]
- Li, S.; Crooks, P.A.; Wei, X.; de Leon, J. Toxicity of dipyridyl compounds and related compounds. Crit. Rev. Toxicol. 2004, 34, 447–460. [Google Scholar] [CrossRef]
- Tanner, C.M.; Ottman, R.; Goldman, S.M.; Ellenberg, J.; Chan, P.; Mayeux, R.; Langston, J.W. Parkinson disease in twins: An etiologic study. JAMA 1999, 281, 341–346. [Google Scholar] [CrossRef]
- Ulmer, T.S.; Bax, A.; Cole, N.B.; Nussbaum, R.L. Structure and dynamics of micelle-bound human alpha-synuclein. J. Biol. Chem. 2005, 280, 9595–9603. [Google Scholar]
- Murphy, R.M.; Tsai, A.M. Misbehaving Proteins: Protein (Mis)Folding, Aggregation, and Stability; Springer: New York, NY, USA, 2006; p. 356. [Google Scholar]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar]
- Keane, P.C.; Kurzawa, M.; Blain, P.G.; Morris, C.M. Mitochondrial dysfunction in Parkinson's disease. Parkinson's Disease 2011, 2011. Article ID 716871, 18 pages. [Google Scholar]
- Sherer, T.B.; Richardson, J.R.; Testa, C.M.; Seo, B.B.; Panov, A.V.; Yagi, T.; Matsuno-Yagi, A.; Miller, G.W.; Greenamyre, J.T. Mechanism of toxicity of pesticides acting at complex I: relevance to environmental etiologies of Parkinson's disease. J. Neurochem. 2007, 100, 1469–1479. [Google Scholar]
- Uversky, V.N.; Li, J.; Fink, A.L. Metal-triggered structural transformations, aggregation, and fibrillation of human alpha-synuclein. A possible molecular NK between Parkinson’s disease and heavy metal exposure. J. Biol. Chem. 2011, 276, 44284–44296. [Google Scholar] [CrossRef]
- Bernstein, S.L.; Liu, D.; Wyttenbach, T.; Bowers, M.T.; Lee, J.C.; Gray, H.B.; Winkler, J.R. Alpha-synuclein: Stable compact and extended monomeric structures and pH dependence of dimer formation. J. Am. Soc. Mass Spectrom. 2004, 15, 1435–1443. [Google Scholar] [CrossRef]
- Byler, D.M.; Susi, H. Examination of the secondary structure of proteins by deconvolved FTIR spectra. Biopolymers 1986, 25, 469–487. [Google Scholar] [CrossRef]
- Venyaminov, S.; Kalnin, N.N. Quantitative IR spectrophotometry of peptide compounds in water (H2O) solutions. I. Spectral parameters of amino acid residue absorption bands. Biopolymers 1990, 30, 1243–1257. [Google Scholar] [CrossRef]
- Uversky, V.N.; Lee, H.J.; Li, J.; Fink, A.L.; Lee, S.J. Stabilization of partially folded conformation during alpha-synuclein oligomerization in both purified and cytosolic preparations. J. Biol. Chem. 2001, 276, 43495–43498. [Google Scholar]
- Li, J.; Zhu, M.; Rajamani, S.; Uversky, V.N.; Fink, A.L. Rifampicin inhibits alpha-synuclein fibrillation and disaggregates fibrils. Chem. Biol. 2004, 11, 1513–1521. [Google Scholar] [CrossRef]
- Oberg, K.; Chrunyk, B.A.; Wetzel, R.; Fink, A.L. Nativelike secondary structure in interleukin-1 beta inclusion bodies by attenuated total reflectance FTIR. Biochemistry 1994, 33, 2628–2634. [Google Scholar] [CrossRef]
- Fink, A.L.; Oberg, K.A.; Seshadri, S. Discrete intermediates versus molten globule models for protein folding: Characterization of partially folded intermediates of apomyoglobin. Fold. Des. 1998, 3, 19–25. [Google Scholar] [CrossRef]
- Oberg, K.A.; Fink, A.L. A new attenuated total reflectance Fourier transform infrared spectroscopy method for the study of proteins in solution. Anal. Biochem. 1998, 256, 92–106. [Google Scholar] [CrossRef]
- Seshadri, S.; Oberg, K.A.; Fink, A.L. Thermally denatured ribonuclease A retains secondary structure as shown by FTIR. Biochemistry 1994, 33, 1351–1355. [Google Scholar] [CrossRef]
- Fandrich, M.; Fletcher, M.A.; Dobson, C.M. Amyloid fibrils from muscle myoglobin. Nature 2001, 410, 165–166. [Google Scholar] [CrossRef]
- Zerovnik, E. Amyloid-fibril formation. Proposed mechanisms and relevance to conformational disease. Eur. J. Biochem. 2002, 269, 3362–3371. [Google Scholar] [CrossRef]
- Wouters, M.A.; Curmi, P.M.G. Analysis of side chain interactions and pair correlations within antiparallel b-sheets: The differences between backbone hydrogen-bonded and non-hydrrogen-bonded residue pairs. Proteins Struct. Funct. Genet. 1995, 22, 119–131. [Google Scholar] [CrossRef]
- Harper, J.D.; Lansbury, P.T., Jr. Models of amyloid seeding in Alzheimer’s disease and scrapie: Mechanistic truths and physiological consequences of the time-dependent solubility of amyloid proteins. Annu. Rev. Biochem. 1997, 66, 385–407. [Google Scholar] [CrossRef]
- Harper, J.D.; Lieber, C.M.; Lansbury, P.T., Jr. Atomic force microscopic imaging of seeded fibril formation and fibril branching by the Alzheimer’s disease amyloid-beta protein. Chem. Biol. 1997, 4, 951–959. [Google Scholar] [CrossRef]
- Harper, J.D.; Wong, S.S.; Lieber, C.M.; Lansbury, P.T. Observation of metastable Abeta amyloid protofibrils by atomic force microscopy. Chem. Biol. 1997, 4, 119–125. [Google Scholar] [CrossRef]
- Yamin, G.; Munishkina, L.A.; Karymov, M.A.; Lyubchenko, Y.L.; Uversky, V.N.; Fink, A.L. Forcing nonamyloidogenic beta-synuclein to fibrillate. Biochemistry 2005, 44, 9096–9107. [Google Scholar]
- Krebs, M.R.H.; Bromley, E.H.C.; Donald, A.M. The binding of thioflavin-T to amyloid fibrils: Localisation and implications. J. Struct. Biol. 2005, 149, 30–37. [Google Scholar] [CrossRef]
- Voropai, E.S.; Samtsov, M.P.; Kaplevskii, K.N.; Maskevich, A.A.; Stepuro, V.I.; Povarova, O.I.; Kuznetsova, J.M.; Turoverov, K.K.; Fink, A.L.; Uverskii, V.N. Spectral Properties of Thioflavine T and its Complexes with Amyloid Fibrils. J. Appl. Spectrosc. 2003, 70, 868–874. [Google Scholar] [CrossRef]
- Munishkina, L.A.; Phelan, C.; Uversky, V.N.; Fink, A.L. Conformational behavior and aggregation of alpha-synuclein in organic solvents: Modeling the effects of membranes. Biochemistry 2003, 42, 2720–2730. [Google Scholar] [CrossRef]
- Nielsen, L.; Khurana, R.; Coats, A.; Frokjaer, S.; Brange, J.; Vyas, S.; Uversky, V.N.; Fink, A.L. Effect of environmental factors on the kinetics of insulin fibril formation: Elucidation of the molecular mechanism. Biochemistry 2001, 40, 6036–6046. [Google Scholar]
- Munishkina, L.A.; Cooper, E.M.; Uversky, V.N.; Fink, A.L. The effect of macromolecular crowding on protein aggregation and amyloid fibril formation. J. Mol. Recognit. 2004, 17, 456–464. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Silva, B.A.; Einarsdóttir, Ó.; Fink, A.L.; Uversky, V.N. Biophysical Characterization of α-Synuclein and Rotenone Interaction. Biomolecules 2013, 3, 703-732. https://doi.org/10.3390/biom3030703
Silva BA, Einarsdóttir Ó, Fink AL, Uversky VN. Biophysical Characterization of α-Synuclein and Rotenone Interaction. Biomolecules. 2013; 3(3):703-732. https://doi.org/10.3390/biom3030703
Chicago/Turabian StyleSilva, Blanca A., Ólöf Einarsdóttir, Anthony L. Fink, and Vladimir N. Uversky. 2013. "Biophysical Characterization of α-Synuclein and Rotenone Interaction" Biomolecules 3, no. 3: 703-732. https://doi.org/10.3390/biom3030703