Pyranose Dehydrogenase from Agaricus campestris and Agaricus xanthoderma: Characterization and Applications in Carbohydrate Conversions



Abstract
:
1. Introduction
2. Results and Discussion
2.1. Expression of Acpdh and Axpdh in P. pastoris
2.2. Multiple Sequence Alignment
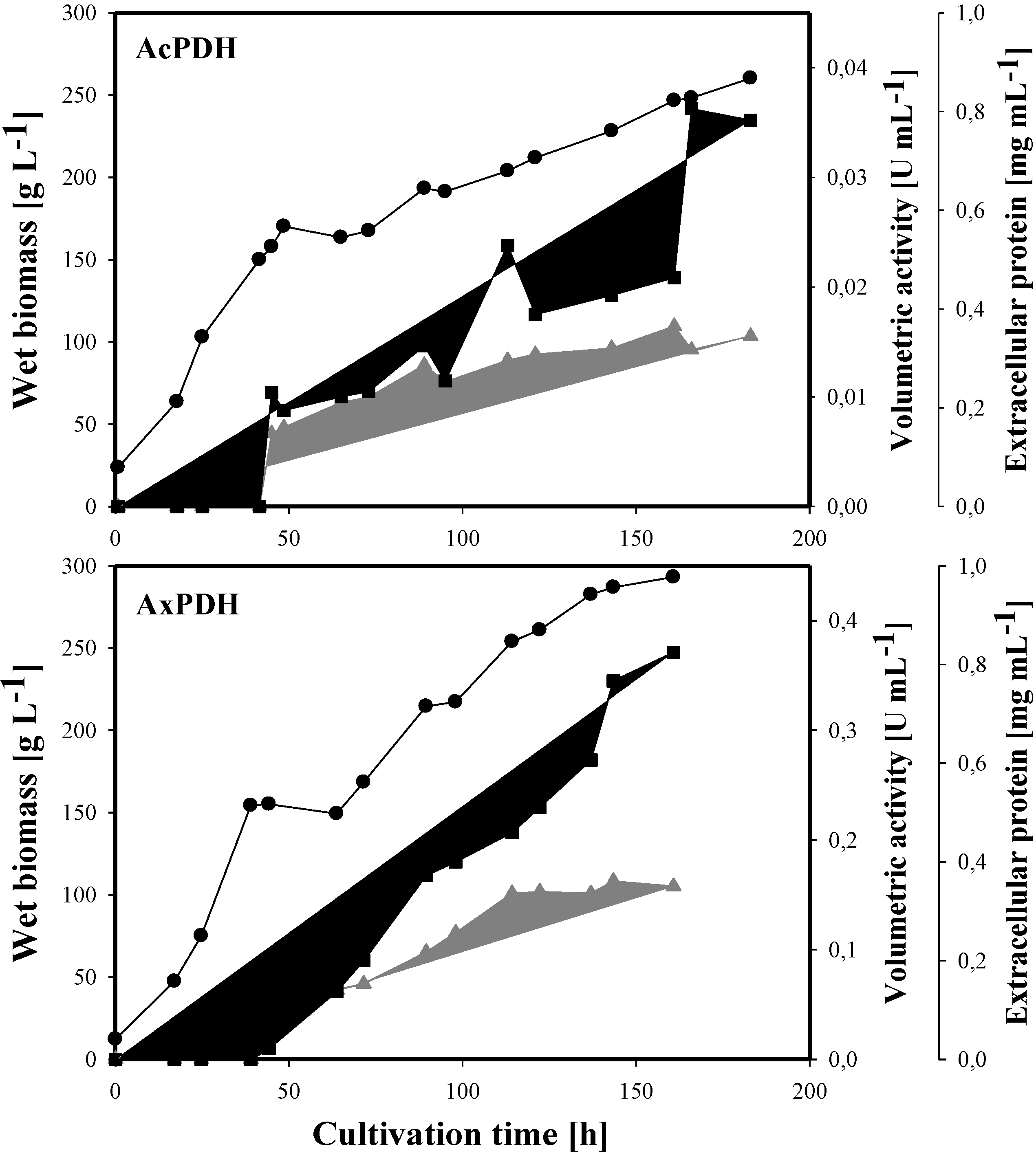
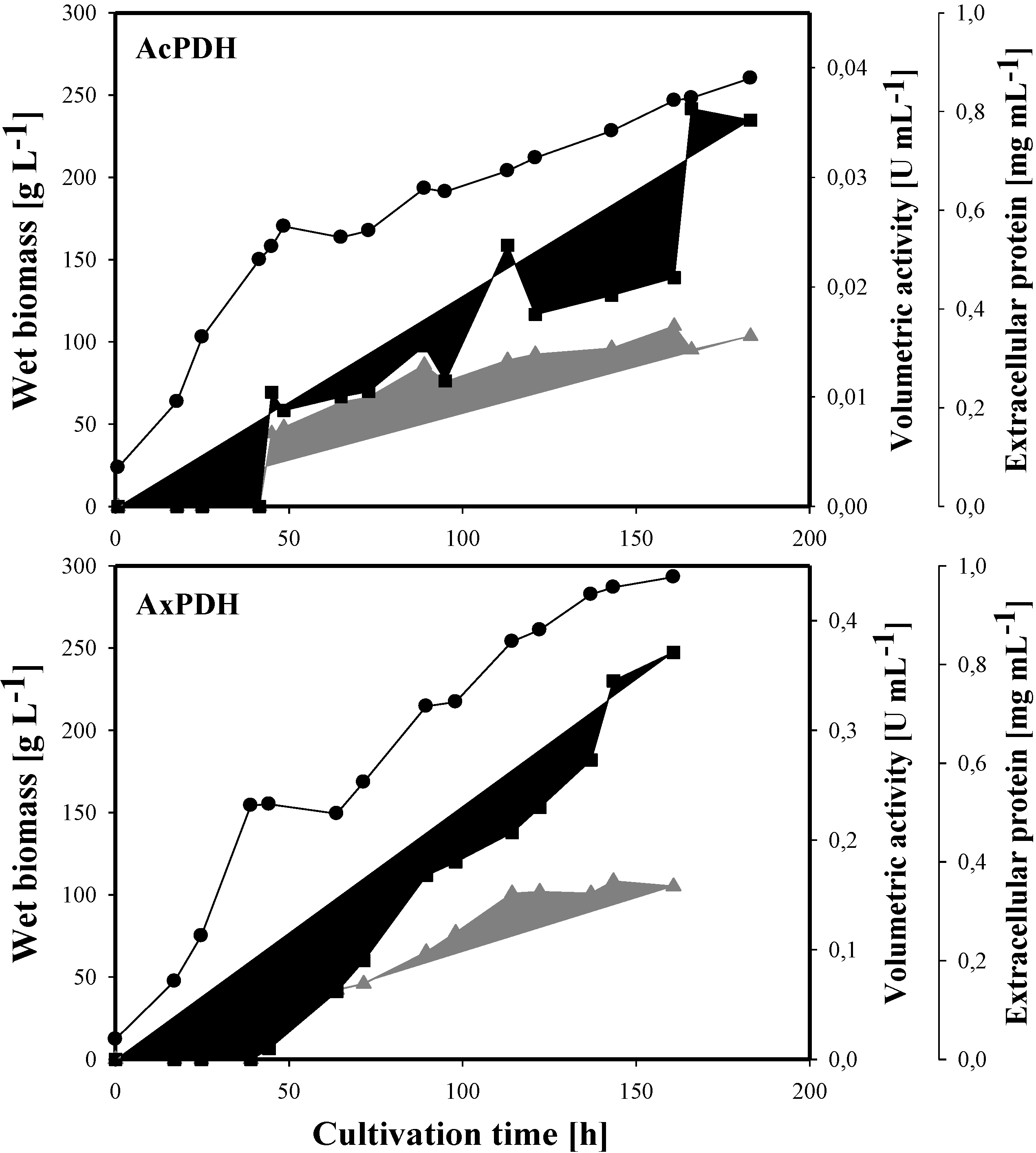
2.3. Heterologous Protein Production
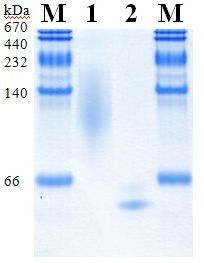
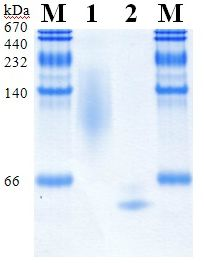
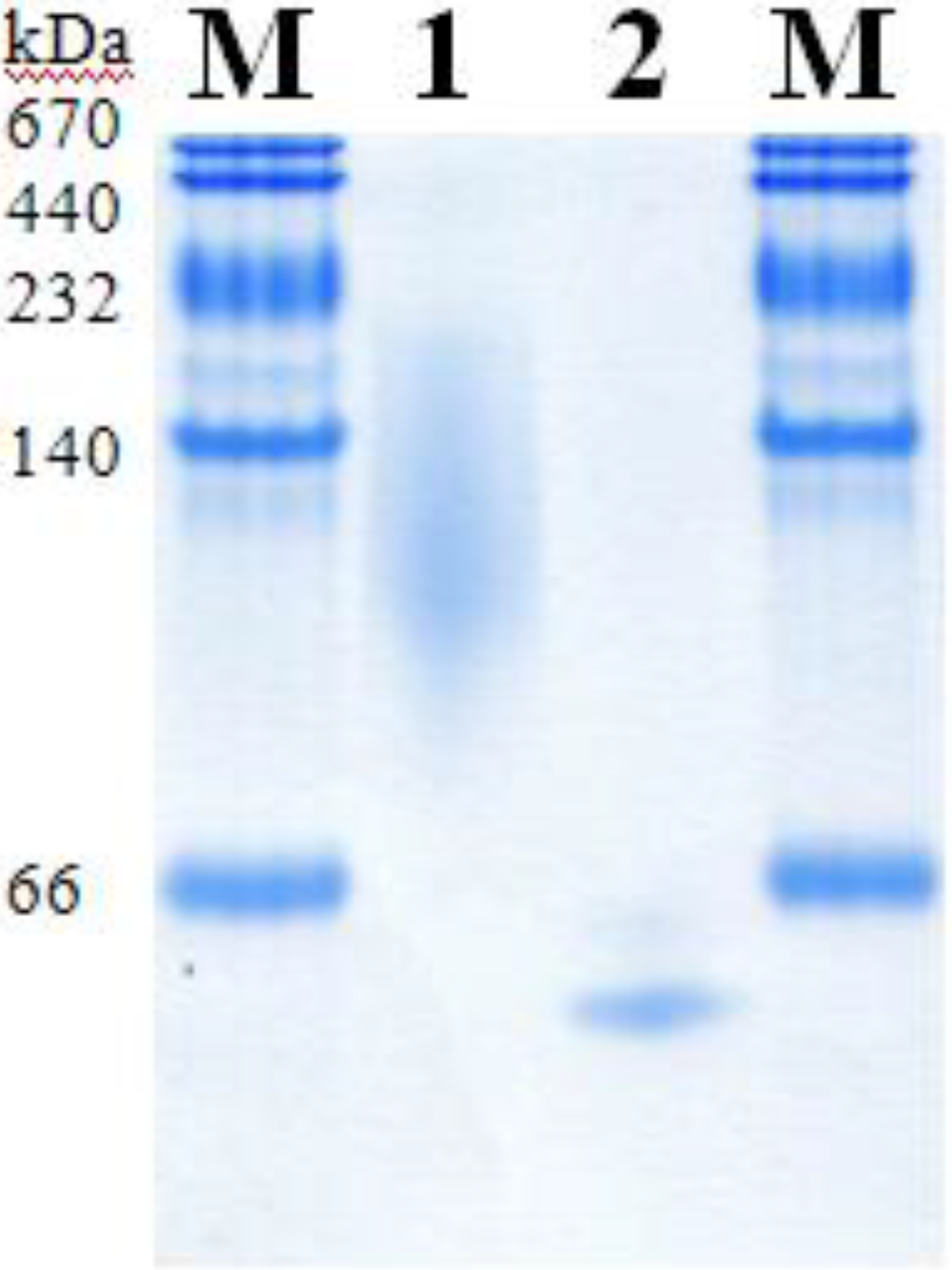
2.4. Purification of Recombinant PDHs

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Purification step | Total protein [mg] | Total activity [U] | Specific activity [U mg−1] | Purification [-fold] | Yield [%] |
|---|---|---|---|---|---|
| AcPDH | |||||
| Crude extract | 1730 | 154.4 | 0.1 | 1 | 100 |
| Phenyl sepharose | 124.4 | 27.4 | 0.2 | 2.5 | 18 |
| DEAE sepharose | 28.2 | 21.5 | 0.4 | 4.1 | 14 |
| Phenyl source | 5.5 | 15.2 | 2.8 | 31.0 | 10 |
| Gel filtration pool 1 | 1.2 | 5.6 | 4.9 | 54.5 | 4 |
| Gel filtration pool 2 | 0.4 | 1.5 | 3.7 | 41.9 | 1 |
| AxPDH | |||||
| Crude extract | 1226.4 | 1298.9 | 1.1 | 1 | 100 |
| Phenyl sepharose | 109.9 | 538.6 | 4.9 | 4.6 | 41 |
| DEAE sepharose | 40.2 | 524.9 | 13.1 | 12.3 | 40 |
| Gel filtration | 25.8 | 428.8 | 16.6 | 15.7 | 33 |
| PDH | Mass SDS-PAGE [kDa] | Mass SDS-PAGE deglyc. [kDa] | Theor. mass [kDa] | Glycan mass [%] | N-Glyc sites predicted | O-Glyc sites predicted |
|---|---|---|---|---|---|---|
| Ac | 98 | 68 | 61.8 | 31 | 6 | 3 |
| Ax | 73 | 68 | 62.3 | 7 | 5 | 0 |
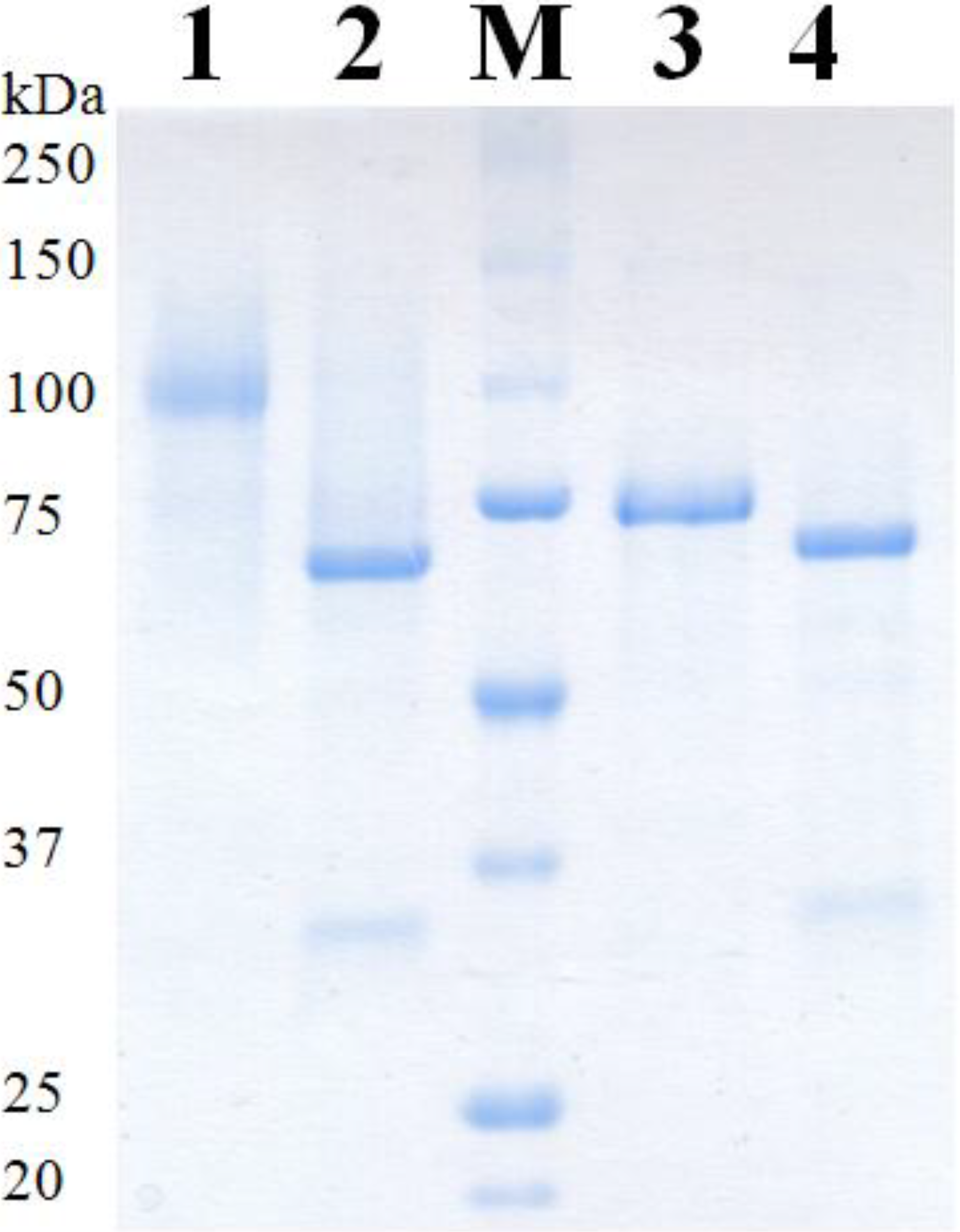
2.5. Molecular Properties
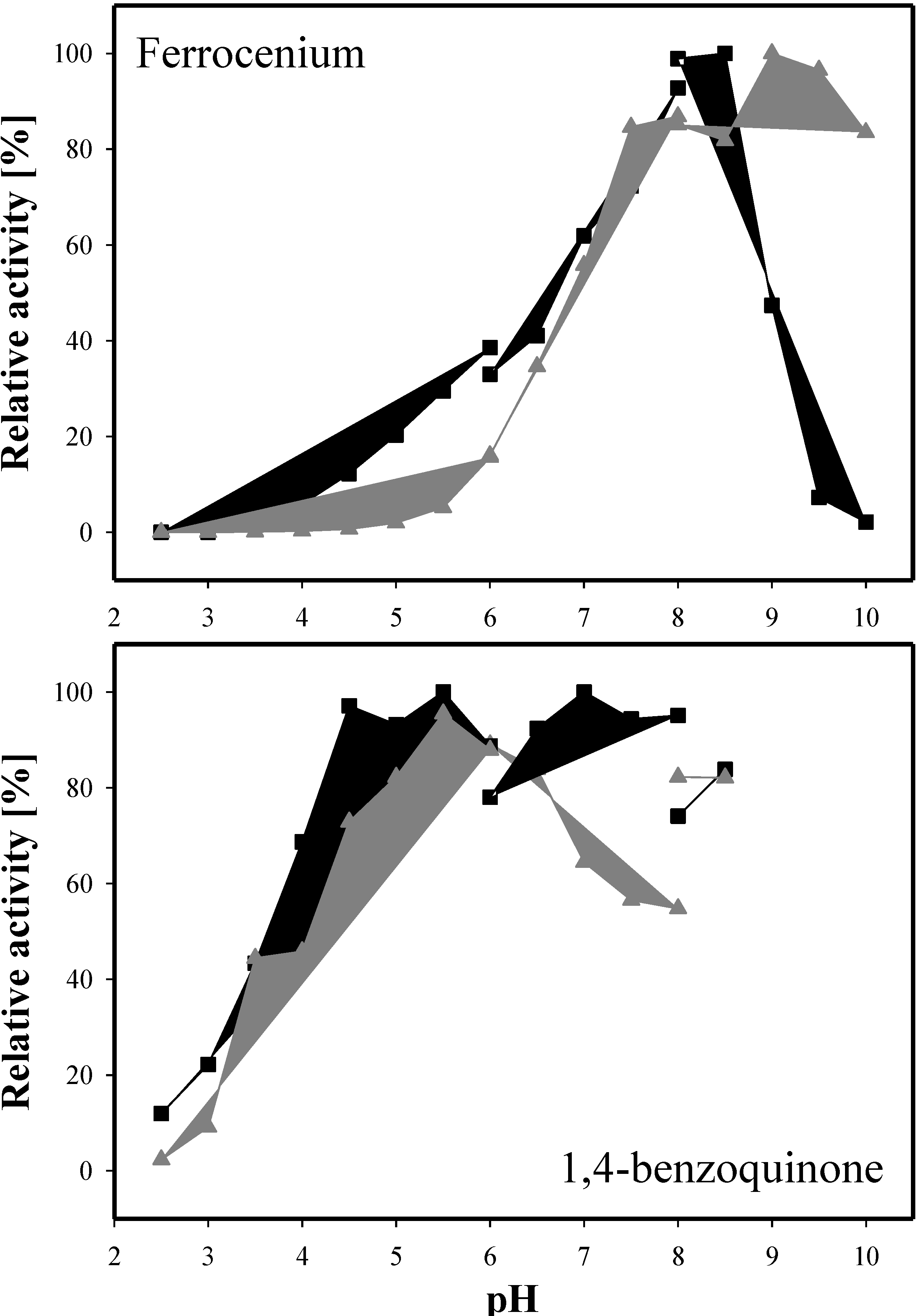
2.6. Kinetic Properties
| AcPDH | AxPDH | |||||
|---|---|---|---|---|---|---|
| Km [mM] | kcat [s−1] | kcat/Km [mM−1 s−1] | Km [mM] | kcat [s−1] | kcat/Km [mM−1 s−1] | |
| D-glucose | 0.35 ± 0.06 | 4.10 ± 0.19 | 11.7 | 0.49 ± 0.03 | 13.02 ± 0.38 | 26.6 |
| D-galactose | 7.13 ± 0.19 | 5.23 ± 0.53 | 0.7 | 4.99 ± 0.16 | 24.77 ± 2.23 | 5.0 |
| D-xylose | 4.19 ± 0.26 | 6.37 ± 0.05 | 1.5 | 1.44 ± 0.07 | 29.16 ± 0.25 | 20.3 |
| L-arabinose | 4.23 ± 0.02 | 3.05 ± 0.04 | 0.7 | 4.16 ± 0.58 | 22.90 ± 2.23 | 5.5 |
| Lactose | 53.16 ± 0.10 | 3.12 ± 0.17 | 0.1 | 293.84 ± 8.00 | 24.65 ± 0.37 | 0.1 |
| AcPDH | AxPDH | |||||
|---|---|---|---|---|---|---|
| Km [mM] | kcat [s−1] | kcat/Km [mM−1 s−1] | Km [mM] | kcat [s−1] | kcat/Km [mM−1 s−1] | |
| Fc+PF6 (pH 8.5) | 1.19 ± 0.17 | 19.92 ± 2.87 | 16.7 | 0.03 ± 0.00 | 22.07 ± 0.05 | 735.7 |
| 1,4-BQ (pH 4) | 0.12 ± 0.01 | 34.82 ± 1.02 | 302.8 | 3.25 ± 0.51 | 12.89 ± 1.56 | 4.0 |
| DCIP (pH 4) | 0.11 ± 0.00 | 10.56 ± 0.57 | 96.0 | 0.09 ± 0.01 | 7.65 ± 0.66 | 85.0 |
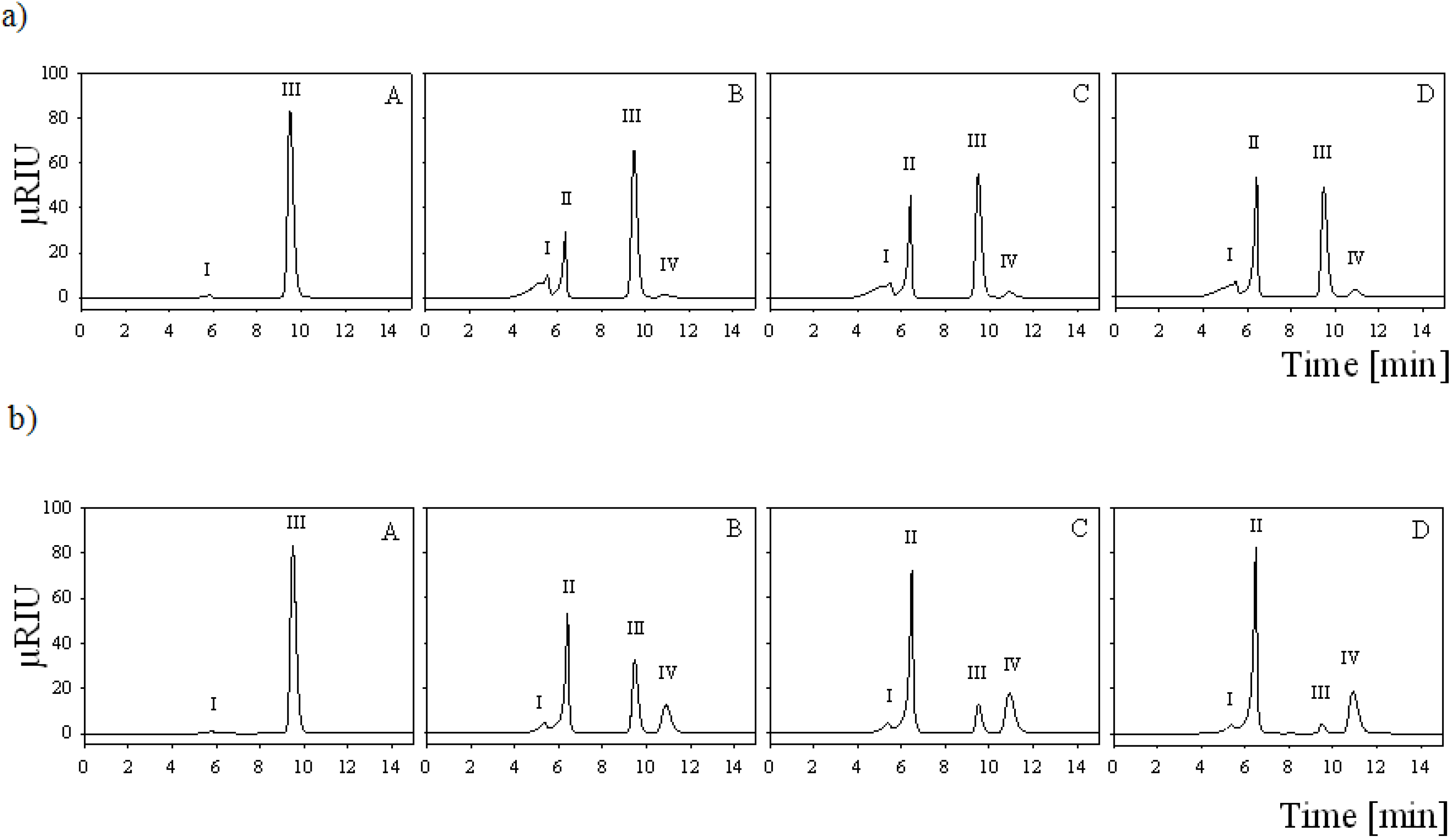
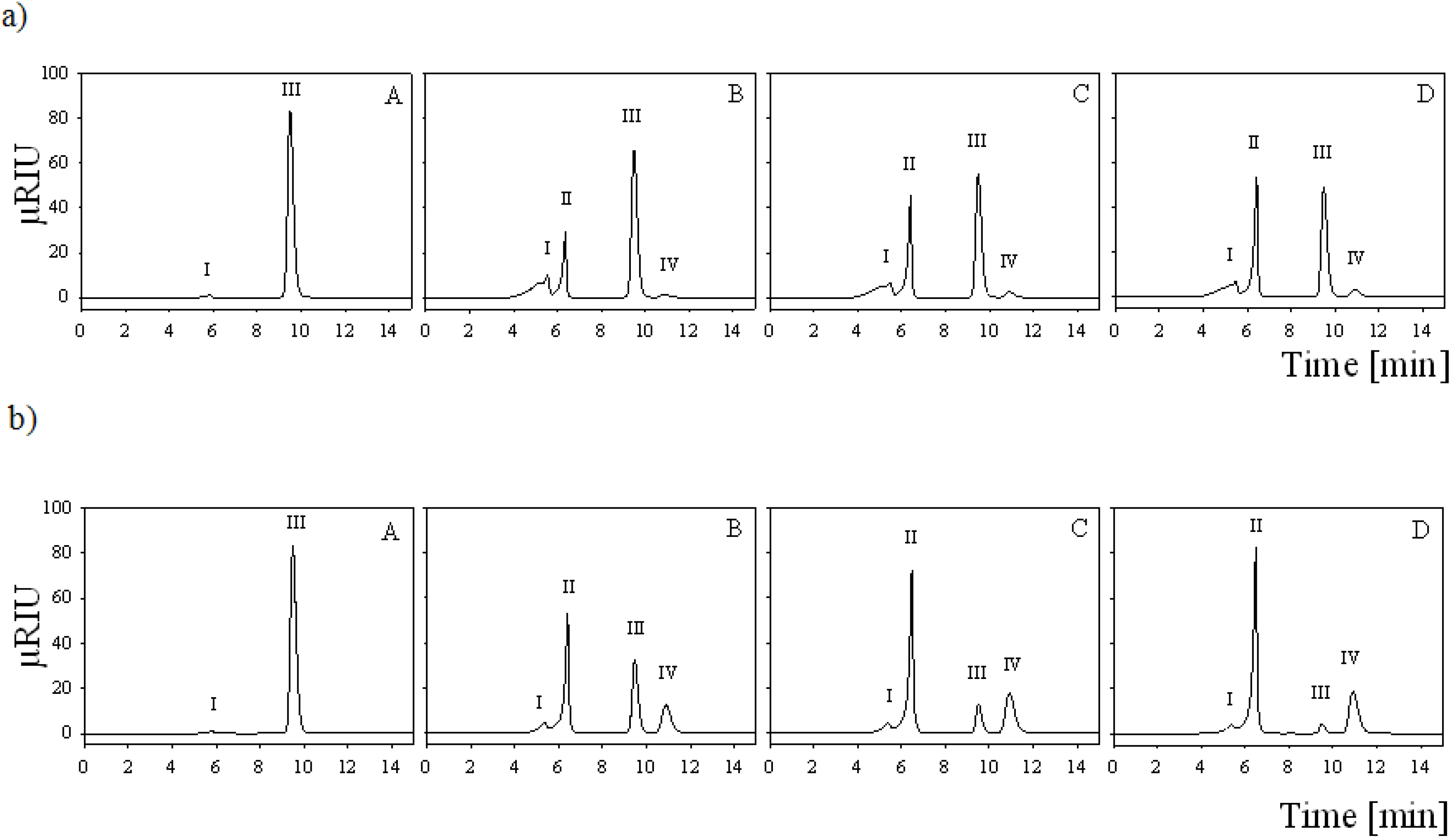
2.7. Batch Carbohydrate Conversion Experiments
3. Experimental Section
3.1. Chemicals and Microorganisms
3.2. Isolation of Genomic DNA and RNA
3.3. Cloning and Sequencing of AcPDH Encoding Gene
3.4. Cloning and Sequencing of AxPDH Encoding Gene
3.5. Construction of Expression Vectors for P. pastoris
3.6. Microscale Screening for High-Producing PDH Transformants
3.7. Sequence Analysis
3.8. Nucleotide and Protein Sequence Accession Numbers
3.9. Recombinant Protein Production in P. pastoris
3.10. Protein Purification
3.11. Enzyme Assay, Molecular Properties
3.12. Kinetic Properties
3.13. Batch Conversion Experiments
3.14. HPLC Analysis of Batch Conversion Products
4. Conclusions
Acknowledgments
Conflicts of Interest
References
- Tan, T.C.; Spadiut, O.; Wongnate, T.; Sucharitakul, J.; Krondorfer, I.; Sygmund, C.; Haltrich, D.; Chaiyen, P.; Peterbauer, C.K.; Divne, C. The 1.6 å crystal structure of pyranose dehydrogenase from Agaricus meleagris rationalizes substrate specificity and reveals a flavin intermediate. PLoS One 2013, 8, e53567. [Google Scholar] [CrossRef]
- Volc, J.; Kubátová, E.; Wood, D.A.; Daniel, G. Pyranose 2-dehydrogenase, a novel sugar oxidoreductase from the basidiomycete fungus Agaricus bisporus. Arch. Microbiol. 1997, 167, 119–125. [Google Scholar] [CrossRef]
- Volc, J.; Kubátová, E.; Daniel, G.; Sedmera, P.; Haltrich, D. Screening of basidiomycete fungi for the quinone-dependent sugar C-2/C-3 oxidoreductase, pyranose dehydrogenase, and properties of the enzyme from Macrolepiota rhacodes. Arch. Microbiol. 2001, 176, 178–186. [Google Scholar] [CrossRef]
- Kujawa, M.; Volc, J.; Halada, P.; Sedmera, P.; Divne, C.; Sygmund, C.; Leitner, C.; Peterbauer, C.; Haltrich, D. Properties of pyranose dehydrogenase purified from the litter-degrading fungus Agaricus xanthoderma. FEBS J. 2007, 274, 879–894. [Google Scholar] [CrossRef]
- Sygmund, C.; Kittl, R.; Volc, J.; Halada, P.; Kubátová, E.; Haltrich, D.; Peterbauer, C.K. Characterization of pyranose dehydrogenase from Agaricus meleagris and its application in the C-2 specific conversion of D-galactose. J. Biotechnol. 2008, 133, 334–342. [Google Scholar] [CrossRef]
- Kittl, R.; Sygmund, C.; Halada, P.; Volc, J.; Divne, C.; Haltrich, D.; Peterbauer, C.K. Molecular cloning of three pyranose dehydrogenase-encoding genes from Agaricus meleagris and analysis of their expression by real-time RT-PCR. Curr. Genet. 2008, 53, 117–127. [Google Scholar] [CrossRef]
- Peterbauer, C.K.; Volc, J. Pyranose dehydrogenases: Biochemical features and perspectives of technological applications. Appl. Microbiol. Biotechnol. 2010, 85, 837–848. [Google Scholar] [CrossRef]
- Pisanelli, I.; Kujawa, M.; Gschnitzer, D.; Spadiut, O.; Seiboth, B.; Peterbauer, C. Heterologous expression of an Agaricus meleagris pyranose dehydrogenase-encoding gene in Aspergillus spp. and characterization of the recombinant enzyme. Appl. Microbiol. Biotechnol. 2010, 86, 599–606. [Google Scholar] [CrossRef]
- Sygmund, C.; Gutmann, A.; Krondorfer, I.; Kujawa, M.; Glieder, A.; Pscheidt, B.; Haltrich, D.; Peterbauer, C.; Kittl, R. Simple and efficient expression of Agaricus meleagris pyranose dehydrogenase in Pichia pastoris. Appl. Microbiol. Biotechnol. 2012, 94, 695–704. [Google Scholar] [CrossRef]
- Volc, J.; Sedmera, P.; Halada, P.; Přikrylová, V.; Daniel, G. C-2 and C-3 oxidation of D-glc, and C-2 oxidation of D-gal by pyranose dehydrogenase from Agaricus bisporus. Carbohydr. Res. 1998, 310, 151–156. [Google Scholar] [CrossRef]
- Volc, J.; Sedmera, P.; Halada, P.; Prikrylová, V.; Haltrich, D. Double oxidation of D-xylose to D-glycero-pentos-2,3-diulose (2,3-diketo-D-xylose) by pyranose dehydrogenase from the mushroom Agaricus bisporus. Carbohydr. Res. 2000, 329, 219–225. [Google Scholar] [CrossRef]
- Volc, J.; Sedmera, P.; Kujawa, M.; Halada, P.; Kubátová, E.; Haltrich, D. Conversion of lactose to β-D-galactopyranosyl-(1→4)-D-arabino-hexos-2-ulose-(2-dehydrolactose) and lactobiono-1,5-lactone by fungal pyranose dehydrogenase. J. Mol. Catal. B Enzym. 2004, 30, 177–184. [Google Scholar] [CrossRef]
- Sedmera, P.; Halada, P.; Peterbauer, C.; Volc, J. A new enzyme catalysis: 3,4-dioxidation of some aryl β-D-glycopyranosides by fungal pyranose dehydrogenase. Tetrahedron. Lett. 2004, 45, 8677–8680. [Google Scholar]
- Sedmera, P.; Halada, P.; Kubátová, E.; Haltrich, D.; Přikrylová, V.; Volc, J. New biotransformations of some reducing sugars to the corresponding (di)dehydro(glycosyl) aldoses or aldonic acids using fungal pyranose dehydrogenase. J. Mol. Catal. B Enzym. 2006, 41, 32–42. [Google Scholar] [CrossRef]
- Haltrich, D.; Leitner, C.; Neuhauser, W.; Nidetzky, B.; Kulbe, K.D.; Volc, J. A convenient enzymatic procedure for the production of aldose-free D-tagatose. Ann. N. Y. Acad. Sci. 1998, 864, 295–299. [Google Scholar] [CrossRef]
- Leitner, C.; Neuhauser, W.; Volc, J.; Kulbe, K.D.; Nidetzky, B.; Haltrich, D. The cetus process revisited: A novel enzymatic alternative for the production of aldose-free D-fructose. Biocatal. Biotransform. 1998, 16, 365–382. [Google Scholar] [CrossRef]
- Gutiérrez, L.F.; Hamoudi, S.; Belkacemi, K. Lactobionic acid: A high value-added lactose derivative for food and pharmaceutical applications. Int. Dairy J. 2012, 26, 103–111. [Google Scholar] [CrossRef]
- Schuster-Wolff-Bühring, R.; Fischer, L.; Hinrichs, J. Production and physiological action of the disaccharide lactulose. Int. Dairy J. 2010, 20, 731–741. [Google Scholar] [CrossRef]
- Julenius, K.; Mølgaard, A.; Gupta, R.; Brunak, S. Prediction, conservation analysis, and structural characterization of mammalian mucin-type o-glycosylation sites. Glycobiology 2005, 15, 153–164. [Google Scholar]
- Berends, E.; Ohm, R.A.; de Jong, J.F.; Rouwendal, G.; Wösten, H.A.B.; Lugones, L.G.; Bosch, D. Genomic and biochemical analysis of N glycosylation in the mushroom-forming basidiomycete Schizophyllum commune. Appl. Environ. Microbiol. 2009, 75, 4648–4652. [Google Scholar] [CrossRef]
- Gemmill, T.R.; Trimble, R.B. Overview of N- and O-linked oligosaccharide structures found in various yeast species. Biochim. Biophys. Acta 1999, 1426, 227–237. [Google Scholar]
- Wilson, I.B.; Zeleny, R.; Kolarich, D.; Staudacher, E.; Stroop, C.J.; Kamerling, J.P.; Altmann, F. Analysis of asn-linked glycans from vegetable foodstuffs: Widespread occurrence of lewis a, core alpha1,3-linked fucose and xylose substitutions. Glycobiology 2001, 11, 261–274. [Google Scholar] [CrossRef]
- Duman, J.G.; Miele, R.G.; Liang, H.; Grella, D.K.; Sim, K.L.; Castellino, F.J.; Bretthauer, R.K. O-mannosylation of Pichia pastoris cellular and recombinant proteins. Biotechnol. Appl. Biochem. 1998, 28, 39–45. [Google Scholar]
- Heuts, D.P.H.M.; Scrutton, N.S.; McIntire, W.S.; Fraaije, M.W. What’s in a covalent bond? On the role and formation of covalently bound flavin cofactors. FEBS J. 2009, 276, 3405–3427. [Google Scholar] [CrossRef]
- Scrutton, S. Identification of Covalent Flavoproteins and Analysis of the Covalent Link. In Methods in Molecular Biology: Flavoprotein Protocols; Chapman, K., Reid, A., Eds.; Humana Press Inc.: Totowa, NJ, USA, 1999; Volume 131, pp. 181–193. [Google Scholar]
- Hecht, H.J.; Kalisz, H.M.; Hendle, J.; Schmid, R.D.; Schomburg, D. Crystal structure of glucose oxidase from Aspergillus niger refined at 2.3 å resolution. J. Mol. Biol. 1993, 229, 153–172. [Google Scholar] [CrossRef]
- Graf, M.M.; Bren, U.; Haltrich, D.; Oostenbrink, C. Molecular dynamics simulations give insight into D-glucose dioxidation at C2 and C3 by Agaricus meleagris pyranose dehydrogenase. J. Comput. Aided Mol. Des. 2013, 27, 295–304. [Google Scholar] [CrossRef]
- Liu, D.; Coloe, S.; Baird, R.; Pedersen, J. Rapid mini-preparation of fungal DNA for PCR [5]. J. Clin. Microbiol. 2000, 38, 471. [Google Scholar]
- Lin-Cereghino, J.; Wong, W.W.; Xiong, S.; Giang, W.; Luong, L.T.; Vu, J.; Johnson, S.D.; Lin-Cereghino, G.P. Condensed protocol for competent cell preparation and transformation of the methylotrophic yeast Pichia pastoris. BioTechniques 2005, 38, 44–48. [Google Scholar]
- Weis, R.; Luiten, R.; Skranc, W.; Schwab, H.; Wubbolts, M.; Glieder, A. Reliable high-throughput screening with Pichia pastoris by limiting yeast cell death phenomena. FEMS Yeast Res. 2004, 5, 179–189. [Google Scholar] [CrossRef]
- Artimo, P.; Jonnalagedda, M.; Arnold, K.; Baratin, D.; Csardi, G.; de Castro, E.; Duvaud, S.; Flegel, V.; Fortier, A.; Gasteiger, E.; et al. Expasy: Sib bioinformatics resource portal. Nucleic Acids Res. 2012, 40, W597–W603. [Google Scholar] [CrossRef]
- Altschul, S.F.; Madden, T.L.; Schäffer, A.A.; Zhang, J.; Zhang, Z.; Miller, W.; Lipman, D.J. Gapped blast and psi-blast: A new generation of protein database search programs. Nucleic Acids Res. 1997, 25, 3389–3402. [Google Scholar] [CrossRef]
- Laemmli, U.K. Cleavage of structural proteins during the assembly of the head of bacteriophage t4. Nature 1970, 227, 680–685. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Staudigl, P.; Krondorfer, I.; Haltrich, D.; Peterbauer, C.K. Pyranose Dehydrogenase from Agaricus campestris and Agaricus xanthoderma: Characterization and Applications in Carbohydrate Conversions. Biomolecules 2013, 3, 535-552. https://doi.org/10.3390/biom3030535
Staudigl P, Krondorfer I, Haltrich D, Peterbauer CK. Pyranose Dehydrogenase from Agaricus campestris and Agaricus xanthoderma: Characterization and Applications in Carbohydrate Conversions. Biomolecules. 2013; 3(3):535-552. https://doi.org/10.3390/biom3030535
Chicago/Turabian StyleStaudigl, Petra, Iris Krondorfer, Dietmar Haltrich, and Clemens K. Peterbauer. 2013. "Pyranose Dehydrogenase from Agaricus campestris and Agaricus xanthoderma: Characterization and Applications in Carbohydrate Conversions" Biomolecules 3, no. 3: 535-552. https://doi.org/10.3390/biom3030535
APA StyleStaudigl, P., Krondorfer, I., Haltrich, D., & Peterbauer, C. K. (2013). Pyranose Dehydrogenase from Agaricus campestris and Agaricus xanthoderma: Characterization and Applications in Carbohydrate Conversions. Biomolecules, 3(3), 535-552. https://doi.org/10.3390/biom3030535