The Impact of Sphingosine Kinase-1 in Head and Neck Cancer


{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Sphingolipid Overview
3. Head and Neck Cancer Background
4. SphK1 Is a Major Player in HNSCC
5. SphK1 Influence in Head and Neck Cancer
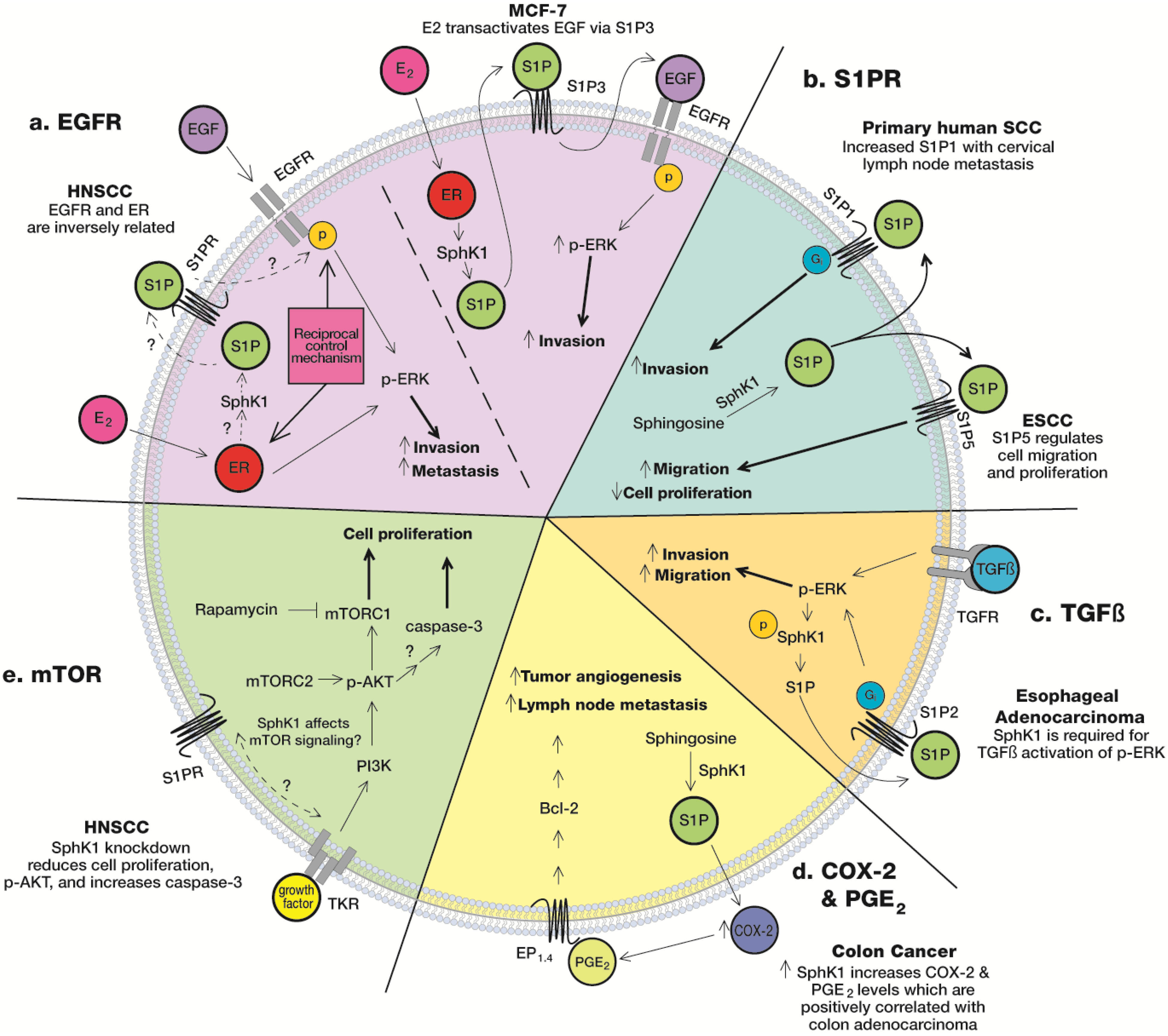
5.1. S1PR
5.2. Epidermal Growth Factor Receptor (EGFR)
5.3. mTOR
5.4. COX-2 & PGE2
5.5. TGFβ
6. SphK1/S1P Pathway and Thyroid Cancer: Mechanism of Action
7. Other Sphingolipid Mediators and HNSCC
7.1. Sphingosine Kinase 2
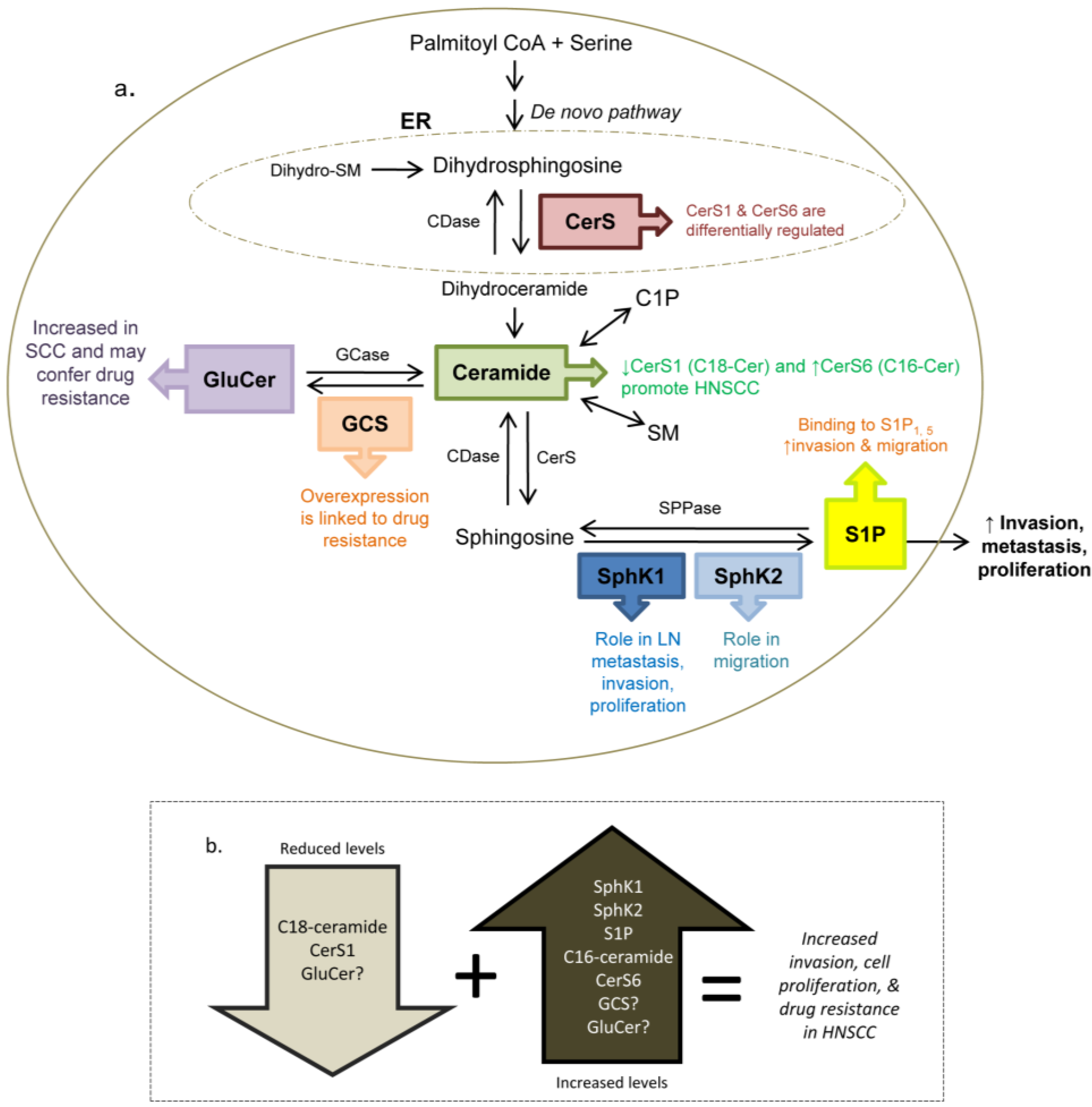
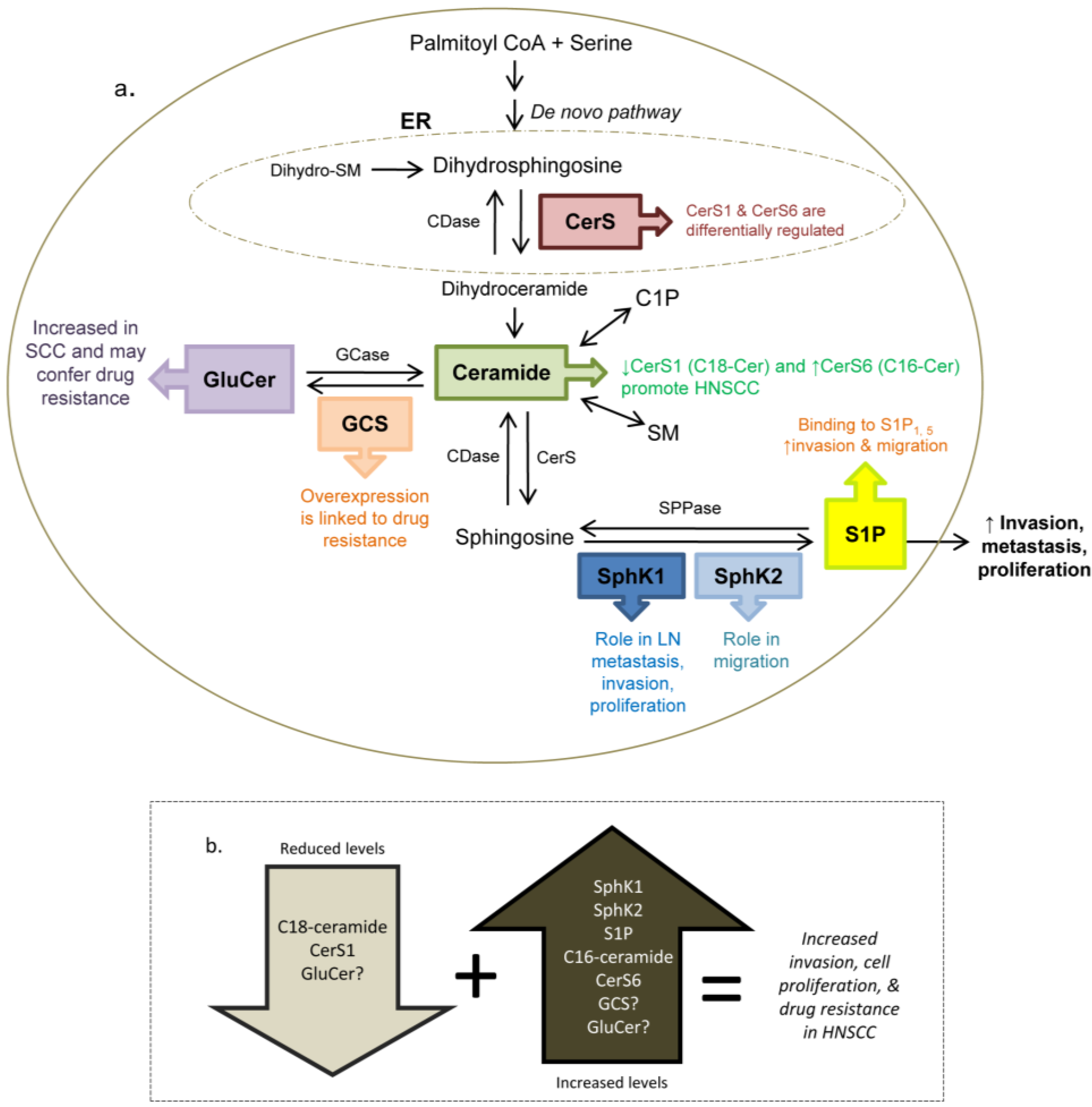
7.2. Ceramide
7.3. Glucosylceramide
8. Therapies
9. Summary and Conclusions
Acknowledgments
Conflict of Interest
References
- Argiris, A.; Karamouzis, M.V.; Raben, D.; Ferris, R.L. Head and neck cancer. Lancet 2008, 371, 1695–1709. [Google Scholar] [CrossRef]
- Patel, V.; Marsh, C.A.; Dorsam, R.T.; Mikelis, C.M.; Masedunskas, A.; Amornphimoltham, P.; Nathan, C.A.; Singh, B.; Weigert, R.; Molinolo, A.A.; et al. Decreased lymphangiogenesis and lymph node metastasis by mtor inhibition in head and neck cancer. Cancer Res. 2011, 71, 7103–7112. [Google Scholar] [CrossRef]
- Ogretmen, B.; Hannun, Y.A. Biologically active sphingolipids in cancer pathogenesis and treatment. Nat. Rev. Cancer 2004, 4, 604–616. [Google Scholar] [CrossRef]
- Van Meer, G.; Lisman, Q. Sphingolipid transport: Rafts and translocators. J. Biol. Chem. 2002, 277, 25855–25858. [Google Scholar] [CrossRef]
- Sillence, D.J. New insights into glycosphingolipid functions--storage, lipid rafts, and translocatorS. Int. Rev. Cytol. 2007, 262, 151–189. [Google Scholar] [CrossRef]
- Hannun, Y.A.; Obeid, L.M. Principles of bioactive lipid signalling: Lessons from sphingolipids. Nat. Rev. Mol. Cell Biol. 2008, 9, 139–150. [Google Scholar] [CrossRef]
- Hla, T. Physiological and pathological actions of sphingosine 1-phosphate. Semin. Cell Dev. Biol. 2004, 15, 513–520. [Google Scholar] [CrossRef]
- Smith, E.R.; Merrill, A.H.; Obeid, L.M.; Hannun, Y.A. Effects of sphingosine and other sphingolipids on protein kinase C. Methods Enzymol. 2000, 312, 361–373. [Google Scholar] [CrossRef]
- Liu, H.; Sugiura, M.; Nava, V.E.; Edsall, L.C.; Kono, K.; Poulton, S.; Milstien, S.; Kohama, T.; Spiegel, S. Molecular cloning and functional characterization of a novel mammalian sphingosine kinase type 2 isoform. J. Biol. Chem. 2000, 275, 19513–19520. [Google Scholar] [CrossRef]
- Pitson, S.M.; D’Andrea, R.J.; Vandeleur, L.; Moretti, P.A.; Xia, P.; Gamble, J.R.; Vadas, M.A.; Wattenberg, B.W. Human sphingosine kinase: Purification, molecular cloning and characterization of the native and recombinant enzymes. Biochem. J. 2000, 350, 429–441. [Google Scholar] [CrossRef]
- Shirai, K.; Kaneshiro, T.; Wada, M.; Furuya, H.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M.; Ogretmen, B.; Kawamori, T. A role of sphingosine kinase 1 in head and neck carcinogenesis. Cancer Prev. Res. 2011, 4, 454–462. [Google Scholar] [CrossRef]
- Koybasi, S.; Senkal, C.E.; Sundararaj, K.; Spassieva, S.; Bielawski, J.; Osta, W.; Day, T.A.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; et al. Defects in cell growth regulation by c18:0-ceramide and longevity assurance gene 1 in human head and neck squamous cell carcinomas. J. Biol. Chem. 2004, 279, 44311–44319. [Google Scholar] [CrossRef]
- Spiegel, S.; Milstien, S. The outs and the ins of sphingosine-1-phosphate in immunity. Nat. Rev. Immunol. 2011, 11, 403–415. [Google Scholar] [CrossRef]
- Hu, W.; Bielawski, J.; Samad, F.; Merrill, A.H., Jr.; Cowart, L.A. Palmitate increases sphingosine-1-phosphate in c2c12 myotubes via upregulation of sphingosine kinase message and activity. J. Lipid Res. 2009, 50, 1852–1862. [Google Scholar]
- Hu, W.M.; Li, L.; Jing, B.Q.; Zhao, Y.S.; Wang, C.L.; Feng, L.; Xie, Y.E. Effect of s1p5 on proliferation and migration of human esophageal cancer cells. World J. Gastroenterol.: WJG 2010, 16, 1859–1866. [Google Scholar] [CrossRef]
- Hengst, J.A.; Guilford, J.M.; Fox, T.E.; Wang, X.; Conroy, E.J.; Yun, J.K. Sphingosine kinase 1 localized to the plasma membrane lipid raft microdomain overcomes serum deprivation induced growth inhibition. Arch. Biochem. Biophys. 2009, 492, 62–73. [Google Scholar] [CrossRef]
- Pitson, S.M.; Moretti, P.A.; Zebol, J.R.; Lynn, H.E.; Xia, P.; Vadas, M.A.; Wattenberg, B.W. Activation of sphingosine kinase 1 by erk1/2-mediated phosphorylation. EMBO J. 2003, 22, 5491–5500. [Google Scholar] [CrossRef]
- Tepper, A.D.; Diks, S.H.; van Blitterswijk, W.J.; Borst, J. Glucosylceramide synthase does not attenuate the ceramide pool accumulating during apoptosis induced by cd95 or anti-cancer regimens. J. Biol. Chem. 2000, 275, 34810–34817. [Google Scholar]
- Licitra, L.; Felip, E. Squamous cell carcinoma of the head and neck: Esmo clinical recommendations for diagnosis, treatment and follow-up. Ann. Oncol. 2009, 20, 121–122. [Google Scholar]
- Parkin, D.M.; Bray, F.; Ferlay, J.; Pisani, P. Global cancer statistics, 2002. CA 2005, 55, 74–108. [Google Scholar]
- Siegel, R.; DeSantis, C.; Virgo, K.; Stein, K.; Mariotto, A.; Smith, T.; Cooper, D.; Gansler, T.; Lerro, C.; Fedewa, S.; et al. Cancer treatment and survivorship statistics, 2012. CA 2012, 62, 220–241. [Google Scholar]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics, 2010. CA 2010, 60, 277–300. [Google Scholar]
- Znaor, A.; Brennan, P.; Gajalakshmi, V.; Mathew, A.; Shanta, V.; Varghese, C.; Boffetta, P. Independent and combined effects of tobacco smoking, chewing and alcohol drinking on the risk of oral, pharyngeal and esophageal cancers in indian men. Int. J. Cancer 2003, 105, 681–686. [Google Scholar] [CrossRef]
- Barnes, L.E.J.; Reichart, P.; Sidransky, D. Cancer Pathology and Genetics; WHO Press: Geneva, Switzerland, 2005; p. 430. [Google Scholar]
- D’Souza, G.; Kreimer, A.R.; Viscidi, R.; Pawlita, M.; Fakhry, C.; Koch, W.M.; Westra, W.H.; Gillison, M.L. Case-control study of human papillomavirus and oropharyngeal cancer. N. Engl. J. Med. 2007, 356, 1944–1956. [Google Scholar] [CrossRef]
- Marur, S.; D’Souza, G.; Westra, W.H.; Forastiere, A.A. Hpv-associated head and neck cancer: A virus-related cancer epidemic. Lancet Oncol. 2010, 11, 781–789. [Google Scholar] [CrossRef]
- Agrawal, N.; Frederick, M.J.; Pickering, C.R.; Bettegowda, C.; Chang, K.; Li, R.J.; Fakhry, C.; Xie, T.X.; Zhang, J.; Wang, J.; et al. Exome sequencing of head and neck squamous cell carcinoma reveals inactivating mutations in notch1. Science 2011, 333, 1154–1157. [Google Scholar] [CrossRef]
- Stransky, N.; Egloff, A.M.; Tward, A.D.; Kostic, A.D.; Cibulskis, K.; Sivachenko, A.; Kryukov, G.V.; Lawrence, M.S.; Sougnez, C.; McKenna, A.; et al. The mutational landscape of head and neck squamous cell carcinoma. Science 2011, 333, 1157–1160. [Google Scholar] [CrossRef] [Green Version]
- Leemans, C.R.; Braakhuis, B.J.; Brakenhoff, R.H. The molecular biology of head and neck cancer. Nat. Rev. Cancer 2011, 11, 9–22. [Google Scholar] [CrossRef]
- Lindenbergh-van der Plas, M.; Brakenhoff, R.H.; Kuik, D.J.; Buijze, M.; Bloemena, E.; Snijders, P.J.; Leemans, C.R.; Braakhuis, B.J. Prognostic significance of truncating tp53 mutations in head and neck squamous cell carcinoma. Clin. Cancer Res. 2011, 17, 3733–3741. [Google Scholar] [CrossRef]
- Facchinetti, M.M.; Gandini, N.A.; Fermento, M.E.; Sterin-Speziale, N.B.; Ji, Y.; Patel, V.; Gutkind, J.S.; Rivadulla, M.G.; Curino, A.C. The expression of sphingosine kinase-1 in head and neck carcinoma. Cells Tissues Organs 2010, 192, 314–324. [Google Scholar] [CrossRef]
- Pan, J.; Tao, Y.F.; Zhou, Z.; Cao, B.R.; Wu, S.Y.; Zhang, Y.L.; Hu, S.Y.; Zhao, W.L.; Wang, J.; Lou, G.L.; et al. An novel role of sphingosine kinase-1 (sphk1) in the invasion and metastasis of esophageal carcinoma. J. Transl. Med. 2011, 9, 157. [Google Scholar] [CrossRef]
- Guan, H.; Liu, L.; Cai, J.; Liu, J.; Ye, C.; Li, M.; Li, Y. Sphingosine kinase 1 is overexpressed and promotes proliferation in human thyroid cancer. Mol. Endocrinol. 2011, 25, 1858–1866. [Google Scholar] [CrossRef]
- Karahatay, S.; Thomas, K.; Koybasi, S.; Senkal, C.E.; Elojeimy, S.; Liu, X.; Bielawski, J.; Day, T.A.; Gillespie, M.B.; Sinha, D.; et al. Clinical relevance of ceramide metabolism in the pathogenesis of human head and neck squamous cell carcinoma (hnscc): Attenuation of c(18)-ceramide in hnscc tumors correlates with lymphovascular invasion and nodal metastasis. Cancer Lett. 2007, 256, 101–111. [Google Scholar] [CrossRef]
- Sinha, U.K.; Schorn, V.J.; Hochstim, C.; Chinn, S.B.; Zhu, S.; Masood, R. Increased radiation sensitivity of head and neck squamous cell carcinoma with sphingosine kinase 1 inhibition. Head Neck 2011, 33, 178–188. [Google Scholar] [CrossRef]
- Egloff, A.M.; Rothstein, M.E.; Seethala, R.; Siegfried, J.M.; Grandis, J.R.; Stabile, L.P. Cross-talk between estrogen receptor and epidermal growth factor receptor in head and neck squamous cell carcinoma. Clin. Cancer Res. 2009, 15, 6529–6540. [Google Scholar] [CrossRef]
- Sukocheva, O.; Wadham, C.; Holmes, A.; Albanese, N.; Verrier, E.; Feng, F.; Bernal, A.; Derian, C.K.; Ullrich, A.; Vadas, M.A.; et al. Estrogen transactivates egfr via the sphingosine 1-phosphate receptor edg-3: The role of sphingosine kinase-1. J. Cell Biol. 2006, 173, 301–310. [Google Scholar] [CrossRef]
- Ledgerwood, L.; Tinling, S.; Gandour-Edwards, R.; Farwell, D. Sphingosine-1-phosphate Receptor s1p1 Expression Predicts Cervical Metastasis in Oral Cavity Squamous Cell Carcinoma. American Head & Neck Society. In Proceedings of 8th International Conference on Head and Neck Cancer, Toronto, ON, Canada, 22–24 July 2012; UC Davis Medical Center : Toronto, ON, Canada, 2012. [Google Scholar]
- Miller, A.V.; Alvarez, S.E.; Spiegel, S.; Lebman, D.A. Sphingosine kinases and sphingosine-1-phosphate are critical for transforming growth factor beta-induced extracellular signal-regulated kinase 1 and 2 activation and promotion of migration and invasion of esophageal cancer cells. Mol. Cell. Biol. 2008, 28, 4142–4151. [Google Scholar] [CrossRef]
- Gallo, O.; Franchi, A.; Magnelli, L.; Sardi, I.; Vannacci, A.; Boddi, V.; Chiarugi, V.; Masini, E. Cyclooxygenase-2 pathway correlates with vegf expression in head and neck cancer. Implications for tumor angiogenesis and metastasis. Neoplasia 2001, 3, 53–61. [Google Scholar]
- Camacho, M.; Leon, X.; Fernandez-Figueras, M.T.; Quer, M.; Vila, L. Prostaglandin e(2) pathway in head and neck squamous cell carcinoma. Head Neck 2008, 30, 1175–1181. [Google Scholar] [CrossRef]
- Chan, G.; Boyle, J.O.; Yang, E.K.; Zhang, F.; Sacks, P.G.; Shah, J.P.; Edelstein, D.; Soslow, R.A.; Koki, A.T.; Woerner, B.M.; et al. Cyclooxygenase-2 expression is up-regulated in squamous cell carcinoma of the head and neck. Cancer Res. 1999, 59, 991–994. [Google Scholar]
- Shiotani, H.; Denda, A.; Yamamoto, K.; Kitayama, W.; Endoh, T.; Sasaki, Y.; Tsutsumi, N.; Sugimura, M.; Konishi, Y. Increased expression of cyclooxygenase-2 protein in 4-nitroquinoline-1-oxide-induced rat tongue carcinomas and chemopreventive efficacy of a specific inhibitor, nimesulide. Cancer Res. 2001, 61, 1451–1456. [Google Scholar]
- Vishwanatha, J.K.; Swinney, R.; Banerjee, A.G. Modulation of annexin i and cyclooxygenase-2 in smokeless tobacco-induced inflammation and oral cancer. Mol. Cell. Biochem. 2003, 248, 67–75. [Google Scholar] [CrossRef]
- Liu, C.H.; Chang, S.H.; Narko, K.; Trifan, O.C.; Wu, M.T.; Smith, E.; Haudenschild, C.; Lane, T.F.; Hla, T. Overexpression of cyclooxygenase-2 is sufficient to induce tumorigenesis in transgenic mice. J. Biol. Chem. 2001, 276, 18563–18569. [Google Scholar] [CrossRef]
- Kawamori, T.; Kitamura, T.; Watanabe, K.; Uchiya, N.; Maruyama, T.; Narumiya, S.; Sugimura, T.; Wakabayashi, K. Prostaglandin e receptor subtype ep(1) deficiency inhibits colon cancer development. Carcinogenesis 2005, 26, 353–357. [Google Scholar]
- Mutoh, M.; Watanabe, K.; Kitamura, T.; Shoji, Y.; Takahashi, M.; Kawamori, T.; Tani, K.; Kobayashi, M.; Maruyama, T.; Kobayashi, K.; et al. Involvement of prostaglandin e receptor subtype ep(4) in colon carcinogenesis. Cancer Res. 2002, 62, 28–32. [Google Scholar]
- Kawamori, T.; Osta, W.; Johnson, K.R.; Pettus, B.J.; Bielawski, J.; Tanaka, T.; Wargovich, M.J.; Reddy, B.S.; Hannun, Y.A.; Obeid, L.M.; et al. Sphingosine kinase 1 is up-regulated in colon carcinogenesis. FASEB J. 2006, 20, 386–388. [Google Scholar]
- Balthasar, S.; Bergelin, N.; Lof, C.; Vainio, M.; Andersson, S.; Tornquist, K. Interactions between sphingosine-1-phosphate and vascular endothelial growth factor signalling in ml-1 follicular thyroid carcinoma cells. Endocr.-Relat. Cancer 2008, 15, 521–534. [Google Scholar] [CrossRef]
- Windh, R.T.; Lee, M.J.; Hla, T.; An, S.; Barr, A.J.; Manning, D.R. Differential coupling of the sphingosine 1-phosphate receptors edg-1, edg-3, and h218/edg-5 to the g(i), g(q), and g(12) families of heterotrimeric g proteins. J. Biol. Chem. 1999, 274, 27351–27358. [Google Scholar]
- Yamazaki, Y.; Kon, J.; Sato, K.; Tomura, H.; Sato, M.; Yoneya, T.; Okazaki, H.; Okajima, F.; Ohta, H. Edg-6 as a putative sphingosine 1-phosphate receptor coupling to ca(2+) signaling pathway. Biochem. Biophys. Res. Commun. 2000, 268, 583–589. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Graler, M.H.; Bernhardt, G.; Hobson, J.P.; Lipp, M.; Spiegel, S. Sphingosine-1-phosphate is a ligand for the g protein-coupled receptor edg-6. Blood 2000, 95, 2624–2629. [Google Scholar]
- Graler, M.H.; Grosse, R.; Kusch, A.; Kremmer, E.; Gudermann, T.; Lipp, M. The sphingosine 1-phosphate receptor s1p4 regulates cell shape and motility via coupling to gi and g12/13. J. Cell. Biochem. 2003, 89, 507–519. [Google Scholar] [CrossRef]
- Im, D.S.; Heise, C.E.; Ancellin, N.; O’Dowd, B.F.; Shei, G.J.; Heavens, R.P.; Rigby, M.R.; Hla, T.; Mandala, S.; McAllister, G.; et al. Characterization of a novel sphingosine 1-phosphate receptor, edg-8. J. Biol. Chem. 2000, 275, 14281–14286. [Google Scholar] [CrossRef]
- Taha, T.A.; Argraves, K.M.; Obeid, L.M. Sphingosine-1-phosphate receptors: Receptor specificity versus functional redundancy. Biochim. Biophys. Acta 2004, 1682, 48–55. [Google Scholar] [CrossRef]
- Long, J.S.; Fujiwara, Y.; Edwards, J.; Tannahill, C.L.; Tigyi, G.; Pyne, S.; Pyne, N.J. Sphingosine 1-phosphate receptor 4 uses her2 (erbb2) to regulate extracellular signal regulated kinase-1/2 in mda-mb-453 breast cancer cells. J. Biol. Chem. 2010, 285, 35957–35966. [Google Scholar]
- Yamashita, H.; Kitayama, J.; Shida, D.; Yamaguchi, H.; Mori, K.; Osada, M.; Aoki, S.; Yatomi, Y.; Takuwa, Y.; Nagawa, H. Sphingosine 1-phosphate receptor expression profile in human gastric cancer cells: Differential regulation on the migration and proliferation. J. Surg. Res. 2006, 130, 80–87. [Google Scholar] [CrossRef]
- Bergelin, N.; Lof, C.; Balthasar, S.; Kalhori, V.; Tornquist, K. S1p1 and vegfr-2 form a signaling complex with extracellularly regulated kinase 1/2 and protein kinase c-alpha regulating ml-1 thyroid carcinoma cell migration. Endocrinology 2010, 151, 2994–3005. [Google Scholar] [CrossRef]
- Arikawa, K.; Takuwa, N.; Yamaguchi, H.; Sugimoto, N.; Kitayama, J.; Nagawa, H.; Takehara, K.; Takuwa, Y. Ligand-dependent inhibition of b16 melanoma cell migration and invasion via endogenous s1p2 g protein-coupled receptor. Requirement of inhibition of cellular rac activity. J. Biol. Chem. 2003, 278, 32841–32851. [Google Scholar] [CrossRef]
- Van Brocklyn, J.R.; Young, N.; Roof, R. Sphingosine-1-phosphate stimulates motility and invasiveness of human glioblastoma multiforme cells. Cancer Lett. 2003, 199, 53–60. [Google Scholar] [CrossRef]
- Brocklyn, J.R. Regulation of cancer cell migration and invasion by sphingosine-1-phosphate. World J. Biol. Chem. 2010, 1, 307–312. [Google Scholar] [CrossRef]
- Young, N.; van Brocklyn, J.R. Roles of sphingosine-1-phosphate (s1p) receptors in malignant behavior of glioma cells. Differential effects of s1p2 on cell migration and invasiveness. Exp. Cell Res. 2007, 313, 1615–1627. [Google Scholar] [CrossRef]
- Ang, K.K.; Berkey, B.A.; Tu, X.; Zhang, H.Z.; Katz, R.; Hammond, E.H.; Fu, K.K.; Milas, L. Impact of epidermal growth factor receptor expression on survival and pattern of relapse in patients with advanced head and neck carcinoma. Cancer Res. 2002, 62, 7350–7356. [Google Scholar]
- Kalyankrishna, S.; Grandis, J.R. Epidermal growth factor receptor biology in head and neck cancer. J. Clin. Oncol. 2006, 24, 2666–2672. [Google Scholar] [CrossRef]
- Sok, J.C.; Coppelli, F.M.; Thomas, S.M.; Lango, M.N.; Xi, S.; Hunt, J.L.; Freilino, M.L.; Graner, M.W.; Wikstrand, C.J.; Bigner, D.D.; et al. Mutant epidermal growth factor receptor (egfrviii) contributes to head and neck cancer growth and resistance to egfr targeting. Clin. Cancer Res. 2006, 12, 5064–5073. [Google Scholar] [CrossRef]
- Grandis, J.R.; Tweardy, D.J. Elevated levels of transforming growth factor alpha and epidermal growth factor receptor messenger rna are early markers of carcinogenesis in head and neck cancer. Cancer Res. 1993, 53, 3579–3584. [Google Scholar]
- Ongkeko, W.M.; Altuna, X.; Weisman, R.A.; Wang-Rodriguez, J. Expression of protein tyrosine kinases in head and neck squamous cell carcinomas. Am. J. Clin. Pathol. 2005, 124, 71–76. [Google Scholar] [CrossRef]
- Iihara, K.; Shiozaki, H.; Tahara, H.; Kobayashi, K.; Inoue, M.; Tamura, S.; Miyata, M.; Oka, H.; Doki, Y.; Mori, T. Prognostic significance of transforming growth factor-alpha in human esophageal carcinoma. Implication for the autocrine proliferation. Cancer 1993, 71, 2902–2909. [Google Scholar] [CrossRef]
- Molinolo, A.A.; Hewitt, S.M.; Amornphimoltham, P.; Keelawat, S.; Rangdaeng, S.; Meneses Garcia, A.; Raimondi, A.R.; Jufe, R.; Itoiz, M.; Gao, Y.; et al. Dissecting the akt/mammalian target of rapamycin signaling network: Emerging results from the head and neck cancer tissue array initiative. Clini. Cancer Res. 2007, 13, 4964–4973. [Google Scholar] [CrossRef]
- Yu, Z.; Weinberger, P.M.; Sasaki, C.; Egleston, B.L.; Speier, W.F., 4th.; Haffty, B.; Kowalski, D.; Camp, R.; Rimm, D.; Vairaktaris, E.; et al. Phosphorylation of akt (ser473) predicts poor clinical outcome in oropharyngeal squamous cell cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 553–558. [Google Scholar] [CrossRef]
- Massarelli, E.; Liu, D.D.; Lee, J.J.; El-Naggar, A.K.; Lo Muzio, L.; Staibano, S.; De Placido, S.; Myers, J.N.; Papadimitrakopoulou, V.A. Akt activation correlates with adverse outcome in tongue cancer. Cancer 2005, 104, 2430–2436. [Google Scholar]
- Czerninski, R.; Amornphimoltham, P.; Patel, V.; Molinolo, A.A.; Gutkind, J.S. Targeting mammalian target of rapamycin by rapamycin prevents tumor progression in an oral-specific chemical carcinogenesis model. Cancer Prev. Res. 2009, 2, 27–36. [Google Scholar] [CrossRef]
- Vitale-Cross, L.; Molinolo, A.A.; Martin, D.; Younis, R.H.; Maruyama, T.; Patel, V.; Chen, W.; Schneider, A.; Gutkind, J.S. Metformin prevents the development of oral squamous cell carcinomas from carcinogen-induced premalignant lesions. Cancer Prev. Res. 2012, 5, 562–573. [Google Scholar] [CrossRef]
- Lin, D.T.; Subbaramaiah, K.; Shah, J.P.; Dannenberg, A.J.; Boyle, J.O. Cyclooxygenase-2: A novel molecular target for the prevention and treatment of head and neck cancer. Head Neck 2002, 24, 792–799. [Google Scholar] [CrossRef]
- Kelley, D.J.; Mestre, J.R.; Subbaramaiah, K.; Sacks, P.G.; Schantz, S.P.; Tanabe, T.; Inoue, H.; Ramonetti, J.T.; Dannenberg, A.J. Benzo[a]pyrene up-regulates cyclooxygenase-2 gene expression in oral epithelial cells. Carcinogenesis 1997, 18, 795–799. [Google Scholar] [CrossRef]
- Greenhough, A.; Smartt, H.J.; Moore, A.E.; Roberts, H.R.; Williams, A.C.; Paraskeva, C.; Kaidi, A. The cox-2/pge2 pathway: Key roles in the hallmarks of cancer and adaptation to the tumour microenvironment. Carcinogenesis 2009, 30, 377–386. [Google Scholar] [CrossRef]
- Yoshida, K.; Tanaka, T.; Kohno, H.; Sakata, K.; Kawamori, T.; Mori, H.; Wakabayashi, K. A cox-2 inhibitor, nimesulide, inhibits chemically-induced rat tongue carcinogenesis through suppression of cell proliferation activity and cox-2 and inos expression. Histol. Histopathol. 2003, 18, 39–48. [Google Scholar]
- Kawamori, T.; Kaneshiro, T.; Okumura, M.; Maalouf, S.; Uflacker, A.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Role for sphingosine kinase 1 in colon carcinogenesis. FASEB J. 2009, 23, 405–414. [Google Scholar]
- Abrahao, A.C.; Castilho, R.M.; Squarize, C.H.; Molinolo, A.A.; dos Santos-Pinto, D., Jr.; Gutkind, J.S. A role for cox2-derived pge2 and pge2-receptor subtypes in head and neck squamous carcinoma cell proliferation. Oral Oncol. 2010, 46, 880–887. [Google Scholar] [CrossRef]
- Papadimitrakopoulou, V.A.; William, W.N., Jr.; Dannenberg, A.J.; Lippman, S.M.; Lee, J.J.; Ondrey, F.G.; Peterson, D.E.; Feng, L.; Atwell, A.; El-Naggar, A.K.; et al. Pilot randomized phase ii study of celecoxib in oral premalignant lesions. Clin. Cancer Res. 2008, 14, 2095–2101. [Google Scholar] [CrossRef]
- Mulshine, J.L.; Atkinson, J.C.; Greer, R.O.; Papadimitrakopoulou, V.A.; van Waes, C.; Rudy, S.; Martin, J.W.; Steinberg, S.M.; Liewehr, D.J.; Avis, I.; et al. Randomized, double-blind, placebo-controlled phase iib trial of the cyclooxygenase inhibitor ketorolac as an oral rinse in oropharyngeal leukoplakia. Clin. Cancer Res. 2004, 10, 1565–1573. [Google Scholar] [CrossRef]
- Gillespie, M.B.; Moody, M.W.; Lee, F.S.; Poole, L.J.; Hornig, J.D.; Lathers, D.; Young, M.R.; Day, T.A. Head and neck cancer recurrence and mortality in nonselective cyclooxygenase inhibitor users. Arch. Otolaryngol. Head Neck Surg. 2007, 133, 28–31. [Google Scholar] [CrossRef]
- Fu, S.; Rivera, M.; Ko, E.C.; Sikora, A.G.; Chen, C.T.; Vu, H.L.; Cannan, D.; Eisenstein, S.; Rosenstein, B.S.; Aguirre-Ghiso, J.; et al. Combined inhibition of epidermal growth factor receptor and cyclooxygenase-2 as a novel approach to enhance radiotherapy. J. Cell Sci. Ther. 2011, 1. S1-002. [Google Scholar]
- Zimmermann, K.C.; Sarbia, M.; Weber, A.A.; Borchard, F.; Gabbert, H.E.; Schror, K. Cyclooxygenase-2 expression in human esophageal carcinoma. Cancer Res. 1999, 59, 198–204. [Google Scholar]
- Wilson, K.T.; Fu, S.; Ramanujam, K.S.; Meltzer, S.J. Increased expression of inducible nitric oxide synthase and cyclooxygenase-2 in barrett's esophagus and associated adenocarcinomas. Cancer Res. 1998, 58, 2929–2934. [Google Scholar]
- Akhurst, R.J.; Derynck, R. Tgf-beta signaling in cancer--a double-edged sword. Trends Cell Biol. 2001, 11, S44–S51. [Google Scholar]
- Roberts, A.B.; Wakefield, L.M. The two faces of transforming growth factor beta in carcinogenesis. Proc. Natl. Acad. Sci. USA 2003, 100, 8621–8623. [Google Scholar] [CrossRef]
- Massague, J.; Chen, Y.G. Controlling tgf-beta signaling. Genes Dev. 2000, 14, 627–644. [Google Scholar]
- Javelaud, D.; Mauviel, A. Crosstalk mechanisms between the mitogen-activated protein kinase pathways and smad signaling downstream of tgf-beta: Implications for carcinogenesis. Oncogene 2005, 24, 5742–5750. [Google Scholar] [CrossRef]
- Radeke, H.H.; von Wenckstern, H.; Stoidtner, K.; Sauer, B.; Hammer, S.; Kleuser, B. Overlapping signaling pathways of sphingosine 1-phosphate and tgf-beta in the murine langerhans cell line xs52. J. Immunol. 2005, 174, 2778–2786. [Google Scholar]
- Gellings Lowe, N.; Swaney, J.S.; Moreno, K.M.; Sabbadini, R.A. Sphingosine-1-phosphate and sphingosine kinase are critical for transforming growth factor-beta-stimulated collagen production by cardiac fibroblasts. Cardiovasc. Res. 2009, 82, 303–312. [Google Scholar]
- Sauer, B.; Vogler, R.; von Wenckstern, H.; Fujii, M.; Anzano, M.B.; Glick, A.B.; Schafer-Korting, M.; Roberts, A.B.; Kleuser, B. Involvement of smad signaling in sphingosine 1-phosphate-mediated biological responses of keratinocytes. J. Biol. Chem. 2004, 279, 38471–38479. [Google Scholar] [CrossRef]
- Xin, C.; Ren, S.; Kleuser, B.; Shabahang, S.; Eberhardt, W.; Radeke, H.; Schafer-Korting, M.; Pfeilschifter, J.; Huwiler, A. Sphingosine 1-phosphate cross-activates the smad signaling cascade and mimics transforming growth factor-beta-induced cell responses. J. Biol. Chem. 2004, 279, 35255–35262. [Google Scholar] [CrossRef]
- Keller, C.D.; Rivera Gil, P.; Tolle, M.; van der Giet, M.; Chun, J.; Radeke, H.H.; Schafer-Korting, M.; Kleuser, B. Immunomodulator fty720 induces myofibroblast differentiation via the lysophospholipid receptor s1p3 and smad3 signaling. Am. J. Pathol. 2007, 170, 281–292. [Google Scholar] [CrossRef]
- Sugimoto, N.; Takuwa, N.; Okamoto, H.; Sakurada, S.; Takuwa, Y. Inhibitory and stimulatory regulation of rac and cell motility by the g12/13-rho and gi pathways integrated downstream of a single g protein-coupled sphingosine-1-phosphate receptor isoform. Mol. Cell. Biol. 2003, 23, 1534–1545. [Google Scholar] [CrossRef]
- Goparaju, S.K.; Jolly, P.S.; Watterson, K.R.; Bektas, M.; Alvarez, S.; Sarkar, S.; Mel, L.; Ishii, I.; Chun, J.; Milstien, S.; et al. The s1p2 receptor negatively regulates platelet-derived growth factor-induced motility and proliferation. Mol. Cell. Biol. 2005, 25, 4237–4249. [Google Scholar] [CrossRef]
- Bakin, A.V.; Rinehart, C.; Tomlinson, A.K.; Arteaga, C.L. P38 mitogen-activated protein kinase is required for tgfbeta-mediated fibroblastic transdifferentiation and cell migration. J. Cell Sci. 2002, 115, 3193–3206. [Google Scholar]
- Greco, A.; Borrello, M.G.; Miranda, C.; Degl’Innocenti, D.; Pierotti, M.A. Molecular pathology of differentiated thyroid cancer. Q. J. Nucl. Med. Mol. Imaging 2009, 53, 440–453. [Google Scholar]
- Scopa, C.D. Histopathology of thyroid tumors. An overview. Hormones (Athens) 2004, 3, 100–110. [Google Scholar]
- Guo, R.J.; Huang, E.; Ezaki, T.; Patel, N.; Sinclair, K.; Wu, J.; Klein, P.; Suh, E.R.; Lynch, J.P. Cdx1 inhibits human colon cancer cell proliferation by reducing beta-catenin/t-cell factor transcriptional activity. J. Biol. Chem. 2004, 279, 36865–36875. [Google Scholar]
- Morin, P.J. Beta-catenin signaling and cancer. BioEssays 1999, 21, 1021–1030. [Google Scholar] [CrossRef]
- Bergelin, N.; Blom, T.; Heikkila, J.; Lof, C.; Alam, C.; Balthasar, S.; Slotte, J.P.; Hinkkanen, A.; Tornquist, K. Sphingosine kinase as an oncogene: Autocrine sphingosine 1-phosphate modulates ml-1 thyroid carcinoma cell migration by a mechanism dependent on protein kinase c-alpha and erk1/2. Endocrinology 2009, 150, 2055–2063. [Google Scholar]
- Liu, G.; Zheng, H.; Zhang, Z.; Wu, Z.; Xiong, H.; Li, J.; Song, L. Overexpression of sphingosine kinase 1 is associated with salivary gland carcinoma progression and might be a novel predictive marker for adjuvant therapy. BMC Cancer 2010, 10, 495. [Google Scholar] [CrossRef]
- Senkal, C.E.; Ponnusamy, S.; Rossi, M.J.; Bialewski, J.; Sinha, D.; Jiang, J.C.; Jazwinski, S.M.; Hannun, Y.A.; Ogretmen, B. Role of human longevity assurance gene 1 and c18-ceramide in chemotherapy-induced cell death in human head and neck squamous cell carcinomas. Mol. Cancer Ther. 2007, 6, 712–722. [Google Scholar]
- Saddoughi, S.A.; Garrett-Mayer, E.; Chaudhary, U.; O’Brien, P.E.; Afrin, L.B.; Day, T.A.; Gillespie, M.B.; Sharma, A.K.; Wilhoit, C.S.; Bostick, R.; et al. Results of a phase ii trial of gemcitabine plus doxorubicin in patients with recurrent head and neck cancers: Serum c(1)(8)-ceramide as a novel biomarker for monitoring response. Clin. Cancer Res. 2011, 17, 6097–6105. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Ceramide glycosylation potentiates cellular multidrug resistance. FASEB J. 2001, 15, 719–730. [Google Scholar] [CrossRef]
- Bleicher, R.J.; Cabot, M.C. Glucosylceramide synthase and apoptosis. Biochim. Biophys. Acta 2002, 1585, 172–178. [Google Scholar] [CrossRef]
- Marques Filho, M.F.; Walder, F.; Takahashi, H.K.; Guimaraes, L.L.; Tanaka, A.K.; Cervantes, O.; Straus, A.H. Glycosphingolipid expression in squamous cell carcinoma of the upper aerodigestive tract. Braz. J. Otorhinolaryngol. 2006, 72, 25–30. [Google Scholar]
- Neubauer, H.A.; Pitson, S.M. Roles, regulation and inhibitors of sphingosine kinase 2. FEBS J. 2013. [Google Scholar] [CrossRef]
- Wallington-Beddoe, C.T.; Bradstock, K.F.; Bendall, L.J. Oncogenic properties of sphingosine kinases in haematological malignancies. Br. J. Haematol. 2013, 161, 623–638. [Google Scholar]
- French, K.J.; Zhuang, Y.; Maines, L.W.; Gao, P.; Wang, W.; Beljanski, V.; Upson, J.J.; Green, C.L.; Keller, S.N.; Smith, C.D. Pharmacology and antitumor activity of abc294640, a selective inhibitor of sphingosine kinase-2. J. Pharmacol. Exp. Ther. 2010, 333, 129–139. [Google Scholar] [CrossRef]
- Beljanski, V.; Knaak, C.; Smith, C.D. A novel sphingosine kinase inhibitor induces autophagy in tumor cells. J. Pharmacol. Exp. Ther. 2010, 333, 454–464. [Google Scholar] [CrossRef]
- Beljanski, V.; Knaak, C.; Zhuang, Y.; Smith, C.D. Combined anticancer effects of sphingosine kinase inhibitors and sorafenib. Investig. New Drugs 2011, 29, 1132–1142. [Google Scholar] [CrossRef]
- Antoon, J.W.; Meacham, W.D.; Bratton, M.R.; Slaughter, E.M.; Rhodes, L.V.; Ashe, H.B.; Wiese, T.E.; Burow, M.E.; Beckman, B.S. Pharmacological inhibition of sphingosine kinase isoforms alters estrogen receptor signaling in human breast cancer. J. Mol. Endocrinol. 2011, 46, 205–216. [Google Scholar] [CrossRef]
- Antoon, J.W.; White, M.D.; Slaughter, E.M.; Driver, J.L.; Khalili, H.S.; Elliott, S.; Smith, C.D.; Burow, M.E.; Beckman, B.S. Targeting nfkb mediated breast cancer chemoresistance through selective inhibition of sphingosine kinase-2. Cancer Biol. Ther. 2011, 11, 678–689. [Google Scholar] [CrossRef]
- Liu, K.; Guo, T.L.; Hait, N.C.; Allegood, J.; Parikh, H.I.; Xu, W.; Kellogg, G.E.; Grant, S.; Spiegel, S.; Zhang, S. Biological characterization of 3-(2-amino-ethyl)-5-[3-(4-butoxyl-phenyl)-propylidene]-thiazolidine-2,4-dione (k145) as a selective sphingosine kinase-2 inhibitor and anticancer agent. PLoS One 2013, 8, e56471. [Google Scholar]
- Kharel, Y.; Raje, M.; Gao, M.; Gellett, A.M.; Tomsig, J.L.; Lynch, K.R.; Santos, W.L. Sphingosine kinase type 2 inhibition elevates circulating sphingosine 1-phosphate. Biochem. J. 2012, 447, 149–157. [Google Scholar] [CrossRef]
- Taha, T.A.; Kitatani, K.; El-Alwani, M.; Bielawski, J.; Hannun, Y.A.; Obeid, L.M. Loss of sphingosine kinase-1 activates the intrinsic pathway of programmed cell death: Modulation of sphingolipid levels and the induction of apoptosis. FASEB J. 2006, 20, 482–484. [Google Scholar]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef]
- Huang, W.C.; Chen, C.L.; Lin, Y.S.; Lin, C.F. Apoptotic sphingolipid ceramide in cancer therapy. J. Lipids 2011, 2011, 565316. [Google Scholar]
- Venkataraman, K.; Riebeling, C.; Bodennec, J.; Riezman, H.; Allegood, J.C.; Sullards, M.C.; Merrill, A.H., Jr.; Futerman, A.H. Upstream of growth and differentiation factor 1 (uog1), a mammalian homolog of the yeast longevity assurance gene 1 (lag1), regulates n-stearoyl-sphinganine (c18-(dihydro)ceramide) synthesis in a fumonisin b1-independent manner in mammalian cells. J. Biol. Chem. 2002, 277, 35642–35649. [Google Scholar] [CrossRef]
- Agostinis, P.; Berg, K.; Cengel, K.A.; Foster, T.H.; Girotti, A.W.; Gollnick, S.O.; Hahn, S.M.; Hamblin, M.R.; Juzeniene, A.; Kessel, D.; et al. Photodynamic therapy of cancer: An update. CA 2011, 61, 250–281. [Google Scholar]
- Dolgachev, V.; Farooqui, M.S.; Kulaeva, O.I.; Tainsky, M.A.; Nagy, B.; Hanada, K.; Separovic, D. De novo ceramide accumulation due to inhibition of its conversion to complex sphingolipids in apoptotic photosensitized cells. J. Biol. Chem. 2004, 279, 23238–23249. [Google Scholar]
- Separovic, D.; Breen, P.; Joseph, N.; Bielawski, J.; Pierce, J.S.; van Buren, E.; Gudz, T.I. Sirna-mediated down-regulation of ceramide synthase 1 leads to apoptotic resistance in human head and neck squamous carcinoma cells after photodynamic therapy. Anticancer Res. 2012, 32, 2479–2485. [Google Scholar]
- Separovic, D.; Breen, P.; Joseph, N.; Bielawski, J.; Pierce, J.S.; van Buren, E.; Gudz, T.I. Ceramide synthase 6 knockdown suppresses apoptosis after photodynamic therapy in human head and neck squamous carcinoma cells. Anticancer Res. 2012, 32, 753–760. [Google Scholar]
- Senkal, C.E.; Ponnusamy, S.; Bielawski, J.; Hannun, Y.A.; Ogretmen, B. Antiapoptotic roles of ceramide-synthase-6-generated c16-ceramide via selective regulation of the atf6/chop arm of er-stress-response pathways. FASEB J. 2010, 24, 296–308. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Giuliano, A.E.; Cabot, M.C. Expression of glucosylceramide synthase, converting ceramide to glucosylceramide, confers adriamycin resistance in human breast cancer cells. J. Biol. Chem. 1999, 274, 1140–1146. [Google Scholar]
- Baran, Y.; Bielawski, J.; Gunduz, U.; Ogretmen, B. Targeting glucosylceramide synthase sensitizes imatinib-resistant chronic myeloid leukemia cells via endogenous ceramide accumulation. J. Cancer Res. Clin. Oncol. 2011, 137, 1535–1544. [Google Scholar] [CrossRef]
- Liu, Y.Y.; Han, T.Y.; Yu, J.Y.; Bitterman, A.; Le, A.; Giuliano, A.E.; Cabot, M.C. Oligonucleotides blocking glucosylceramide synthase expression selectively reverse drug resistance in cancer cells. J. Lipid Res. 2004, 45, 933–940. [Google Scholar] [CrossRef]
- Gouaze, V.; Yu, J.Y.; Bleicher, R.J.; Han, T.Y.; Liu, Y.Y.; Wang, H.; Gottesman, M.M.; Bitterman, A.; Giuliano, A.E.; Cabot, M.C. Overexpression of glucosylceramide synthase and p-glycoprotein in cancer cells selected for resistance to natural product chemotherapy. Mol. Cancer Ther. 2004, 3, 633–639. [Google Scholar]
- De Rosa, M.F.; Sillence, D.; Ackerley, C.; Lingwood, C. Role of multiple drug resistance protein 1 in neutral but not acidic glycosphingolipid biosynthesis. J. Biol. Chem. 2004, 279, 7867–7876. [Google Scholar]
- Lucci, A.; Cho, W.I.; Han, T.Y.; Giuliano, A.E.; Morton, D.L.; Cabot, M.C. Glucosylceramide: A marker for multiple-drug resistant cancers. Anticancer Res. 1998, 18, 475–480. [Google Scholar]
- Perales, M.; Cervantes, F.; Cobo, F.; Montserrat, E. Non-hodgkin’s lymphoma associated with gaucher’s disease. Leuk. Lymphoma 1998, 31, 609–612. [Google Scholar]
- Marsh, N.L.; Elias, P.M.; Holleran, W.M. Glucosylceramides stimulate murine epidermal hyperproliferation. J. Clin. Investig. 1995, 95, 2903–2909. [Google Scholar] [CrossRef]
- Marchell, N.L.; Uchida, Y.; Brown, B.E.; Elias, P.M.; Holleran, W.M. Glucosylceramides stimulate mitogenesis in aged murine epidermis. J. Investig. Dermatol. 1998, 110, 383–387. [Google Scholar] [CrossRef]
- Fujiwara, K.; Kitatani, K.; Fukushima, K.; Yazama, H.; Umehara, H.; Kikuchi, M.; Igarashi, Y.; Kitano, H.; Okazaki, T. Inhibitory effects of dietary glucosylceramides on squamous cell carcinoma of the head and neck in nod/scid mice. Int. J. Clin. Oncol. 2011, 16, 133–140. [Google Scholar] [CrossRef]
- Inamine, M.; Suzui, M.; Morioka, T.; Kinjo, T.; Kaneshiro, T.; Sugishita, T.; Okada, T.; Yoshimi, N. Inhibitory effect of dietary monoglucosylceramide L-o-beta-glucosyl-N-2'-hydroxyarachidoyl-4,8-sphingadienine on two different categories of colon preneoplastic lesions induced by 1,2-dimethylhydrazine in f344 rats. Cancer Sci. 2005, 96, 876–881. [Google Scholar] [CrossRef]
- Lee, J.; Moon, C. Current status of experimental therapeutics for head and neck cancer. Exp. Biol. Med. 2011, 236, 375–389. [Google Scholar] [CrossRef]
- Schnute, M.E.; McReynolds, M.D.; Kasten, T.; Yates, M.; Jerome, G.; Rains, J.W.; Hall, T.; Chrencik, J.; Kraus, M.; Cronin, C.N.; et al. Modulation of cellular s1p levels with a novel, potent and specific inhibitor of sphingosine kinase-1. Biochem. J. 2012, 444, 79–88. [Google Scholar] [CrossRef]
- French, K.J.; Upson, J.J.; Keller, S.N.; Zhuang, Y.; Yun, J.K.; Smith, C.D. Antitumor activity of sphingosine kinase inhibitors. J. Pharmacol. Exp. Ther. 2006, 318, 596–603. [Google Scholar] [CrossRef]
- Beckham, T.H.; Elojeimy, S.; Cheng, J.C.; Turner, L.S.; Hoffman, S.R.; Norris, J.S.; Liu, X. Targeting sphingolipid metabolism in head and neck cancer: Rational therapeutic potentials. Expert Opin. Ther. Targets 2010, 14, 529–539. [Google Scholar] [CrossRef]
- Pyne, S.; Bittman, R.; Pyne, N.J. Sphingosine kinase inhibitors and cancer: Seeking the golden sword of hercules. Cancer Res. 2011, 71, 6576–6582. [Google Scholar] [CrossRef]
- Visentin, B.; Vekich, J.A.; Sibbald, B.J.; Cavalli, A.L.; Moreno, K.M.; Matteo, R.G.; Garland, W.A.; Lu, Y.; Yu, S.; Hall, H.S.; et al. Validation of an anti-sphingosine-1-phosphate antibody as a potential therapeutic in reducing growth, invasion, and angiogenesis in multiple tumor lineages. Cancer Cell 2006, 9, 225–238. [Google Scholar] [CrossRef]
- O’Brien, N.; Jones, S.T.; Williams, D.G.; Cunningham, H.B.; Moreno, K.; Visentin, B.; Gentile, A.; Vekich, J.; Shestowsky, W.; Hiraiwa, M.; et al. Production and characterization of monoclonal anti-sphingosine-1-phosphate antibodies. J. Lipid Res. 2009, 50, 2245–2257. [Google Scholar] [CrossRef]
- Coward, J.; Ambrosini, G.; Musi, E.; Truman, J.P.; Haimovitz-Friedman, A.; Allegood, J.C.; Wang, E.; Merrill, A.H., Jr.; Schwartz, G.K. Safingol (l-threo-sphinganine) induces autophagy in solid tumor cells through inhibition of pkc and the pi3-kinase pathway. Autophagy 2009, 5, 184–193. [Google Scholar] [CrossRef]
- Noda, T.; Iwai, S.; Hamada, M.; Fujita, Y.; Yura, Y. Induction of apoptosis of detached oral squamous cell carcinoma cells by safingol. Possible role of bim, focal adhesion kinase and endonuclease G. Apoptosis 2009, 14, 287–297. [Google Scholar] [CrossRef]
- Hamada, M.; Sumi, T.; Iwai, S.; Nakazawa, M.; Yura, Y. Induction of endonuclease g-mediated apopotosis in human oral squamous cell carcinoma cells by protein kinase c inhibitor safingol. Apoptosis 2006, 11, 47–56. [Google Scholar] [CrossRef]
- Senkal, C.E.; Ponnusamy, S.; Rossi, M.J.; Sundararaj, K.; Szulc, Z.; Bielawski, J.; Bielawska, A.; Meyer, M.; Cobanoglu, B.; Koybasi, S.; et al. Potent antitumor activity of a novel cationic pyridinium-ceramide alone or in combination with gemcitabine against human head and neck squamous cell carcinomas in vitro and in vivo. J. Pharmacol. Exp. Ther. 2006, 317, 1188–1199. [Google Scholar] [CrossRef]
- Norris, J.S.; Bielawska, A.; Day, T.; El-Zawahri, A.; ElOjeimy, S.; Hannun, Y.; Holman, D.; Hyer, M.; Landon, C.; Lowe, S.; et al. Combined therapeutic use of adgfpfasl and small molecule inhibitors of ceramide metabolism in prostate and head and neck cancers: A status report. Cancer Gene Ther. 2006, 13, 1045–1051. [Google Scholar] [CrossRef]
- Elojeimy, S.; Liu, X.; McKillop, J.C.; El-Zawahry, A.M.; Holman, D.H.; Cheng, J.Y.; Meacham, W.D.; Mahdy, A.E.; Saad, A.F.; Turner, L.S.; et al. Role of acid ceramidase in resistance to fasl: Therapeutic approaches based on acid ceramidase inhibitors and fasl gene therapy. Mol. Ther. 2007, 15, 1259–1263. [Google Scholar] [CrossRef]
- Brizuela, L.; Dayon, A.; Doumerc, N.; Ader, I.; Golzio, M.; Izard, J.C.; Hara, Y.; Malavaud, B.; Cuvillier, O. The sphingosine kinase-1 survival pathway is a molecular target for the tumor-suppressive tea and wine polyphenols in prostate cancer. FASEB J. 2010, 24, 3882–3894. [Google Scholar] [CrossRef]
- Signorelli, P.; Ghidoni, R. Resveratrol as an anticancer nutrient: Molecular basis, open questions and promises. J. Nutr. Biochem. 2005, 16, 449–466. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Tamashiro, P.M.; Furuya, H.; Shimizu, Y.; Iino, K.; Kawamori, T. The Impact of Sphingosine Kinase-1 in Head and Neck Cancer. Biomolecules 2013, 3, 481-513. https://doi.org/10.3390/biom3030481
Tamashiro PM, Furuya H, Shimizu Y, Iino K, Kawamori T. The Impact of Sphingosine Kinase-1 in Head and Neck Cancer. Biomolecules. 2013; 3(3):481-513. https://doi.org/10.3390/biom3030481
Chicago/Turabian StyleTamashiro, Paulette M., Hideki Furuya, Yoshiko Shimizu, Kayoko Iino, and Toshihiko Kawamori. 2013. "The Impact of Sphingosine Kinase-1 in Head and Neck Cancer" Biomolecules 3, no. 3: 481-513. https://doi.org/10.3390/biom3030481
APA StyleTamashiro, P. M., Furuya, H., Shimizu, Y., Iino, K., & Kawamori, T. (2013). The Impact of Sphingosine Kinase-1 in Head and Neck Cancer. Biomolecules, 3(3), 481-513. https://doi.org/10.3390/biom3030481