Regulation of Cytoskeleton Organization by Sphingosine in a Mouse Cell Model of Progressive Ovarian Cancer

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
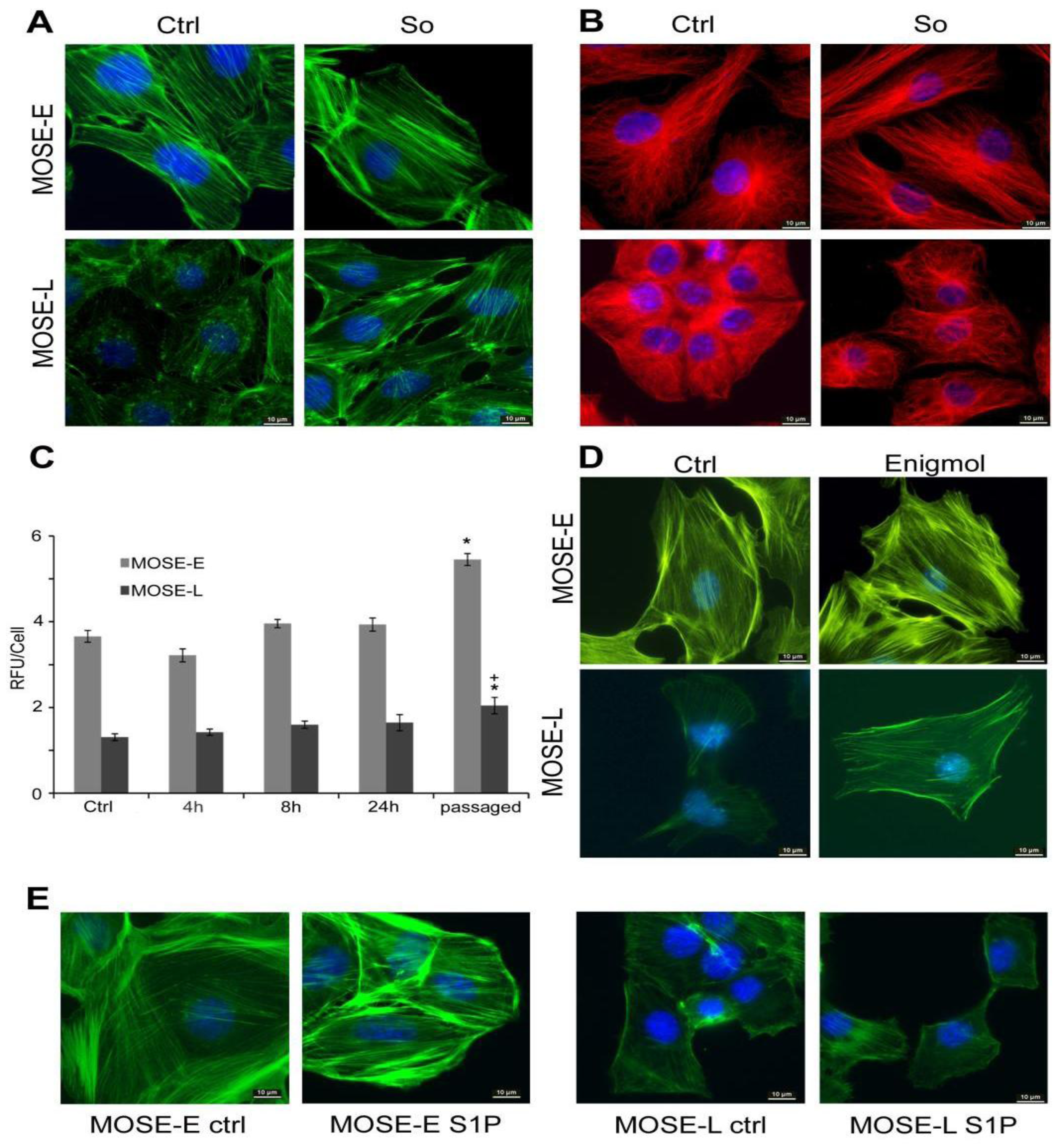
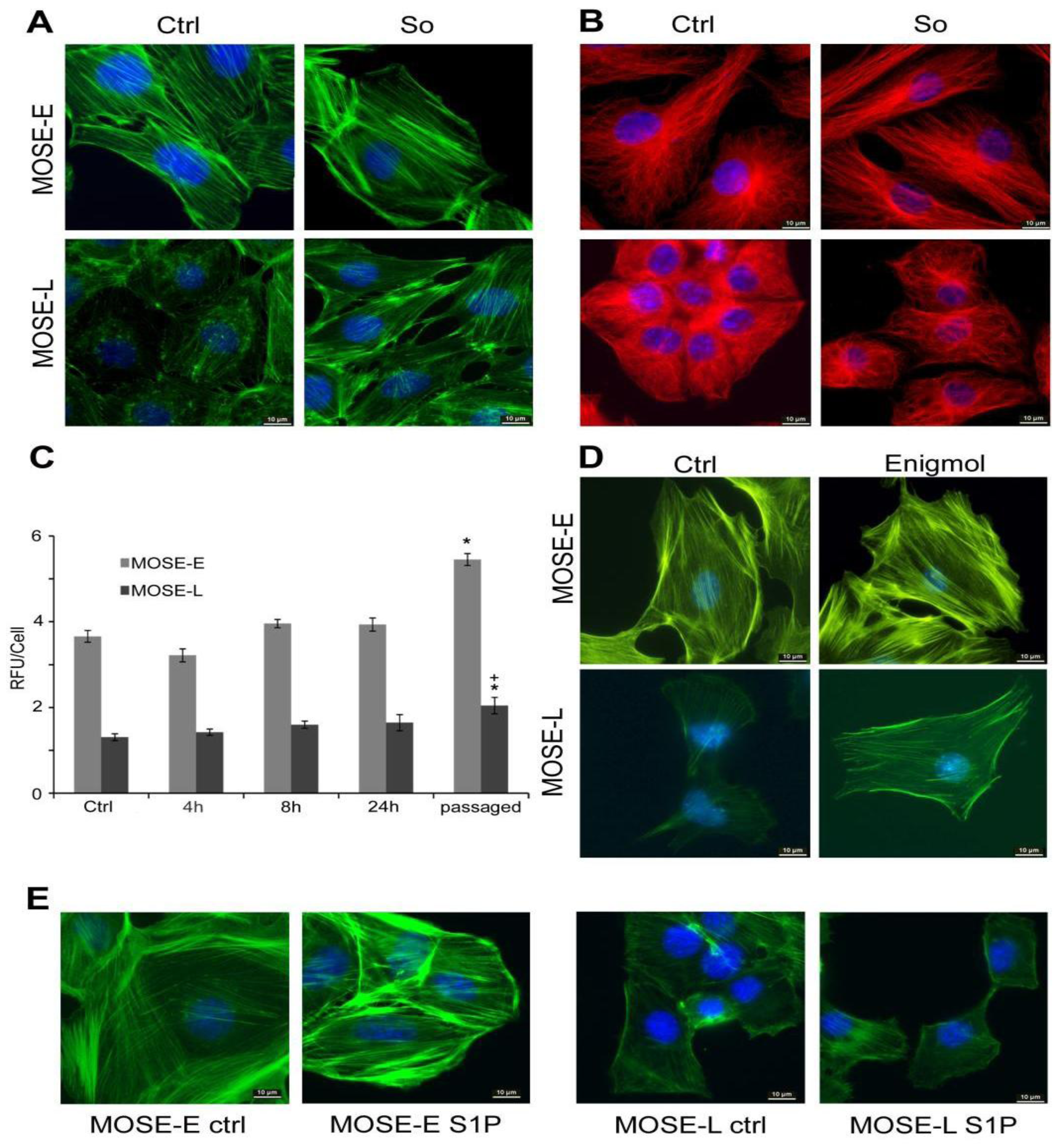
2.1. Sph Treatment Increased Cytoskeleton Organization
2.2. Sph Treatment Increased Actin Polymerization Levels
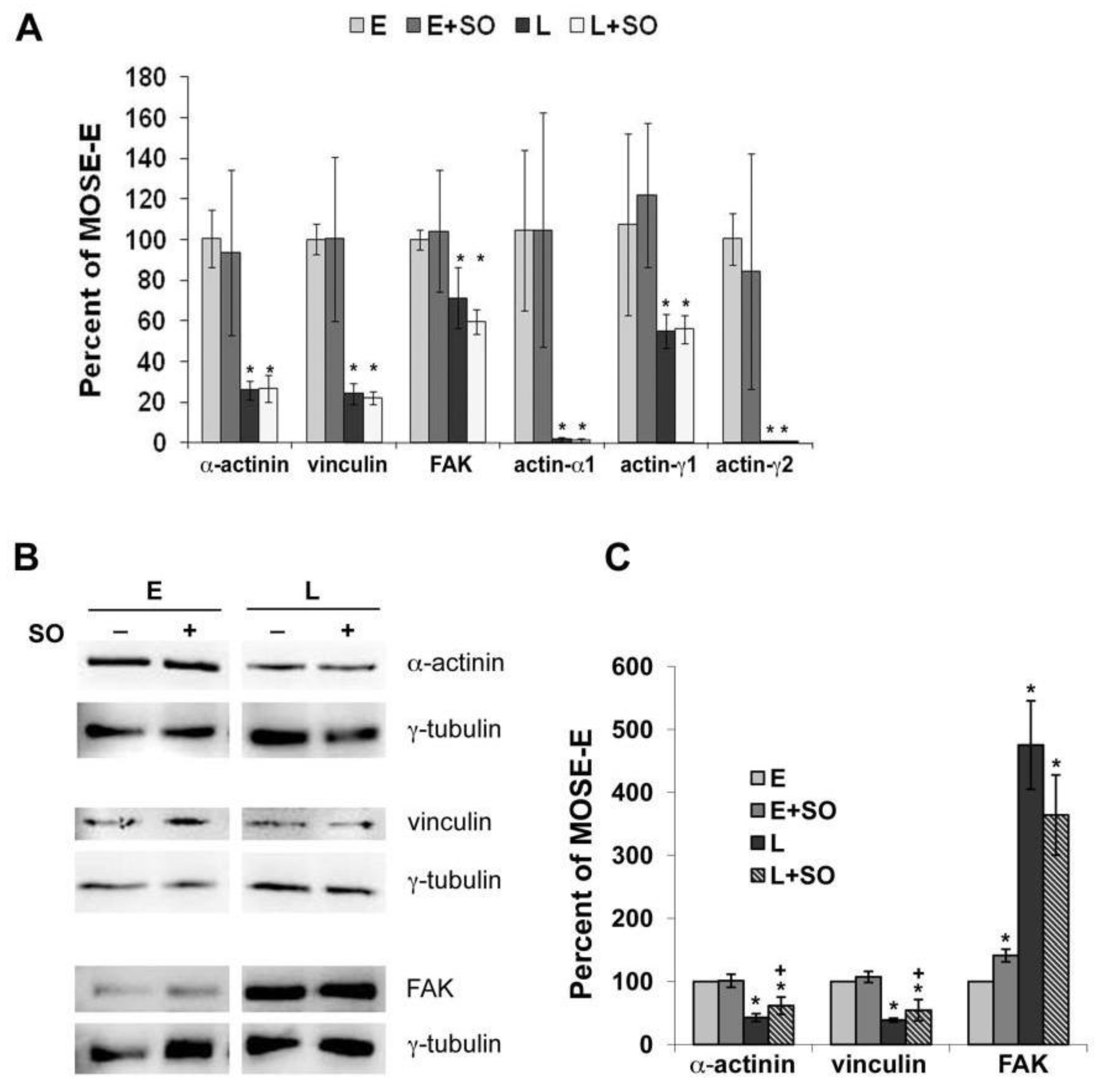
2.3. So Increased the Expression of Actin Regulatory Proteins

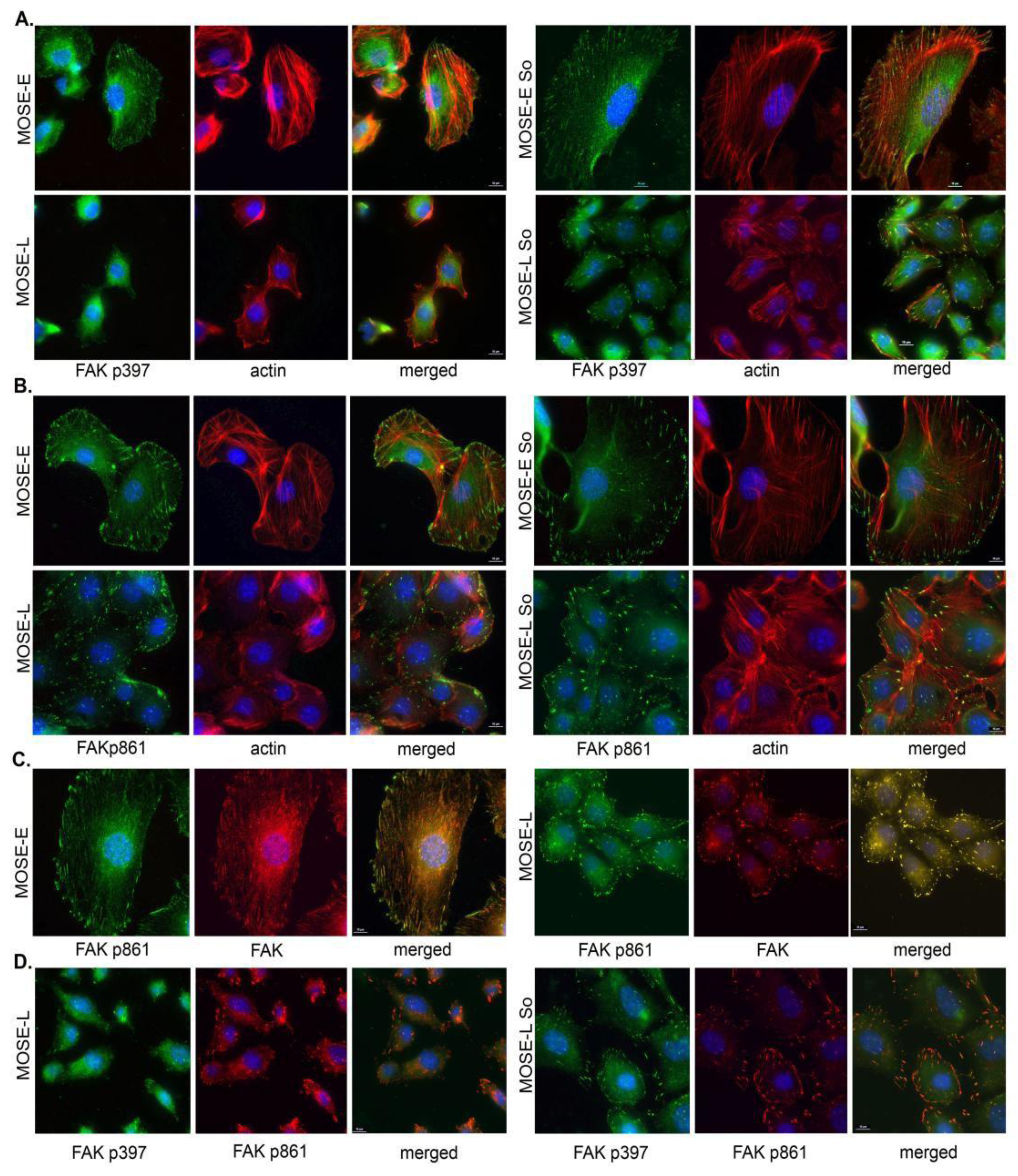
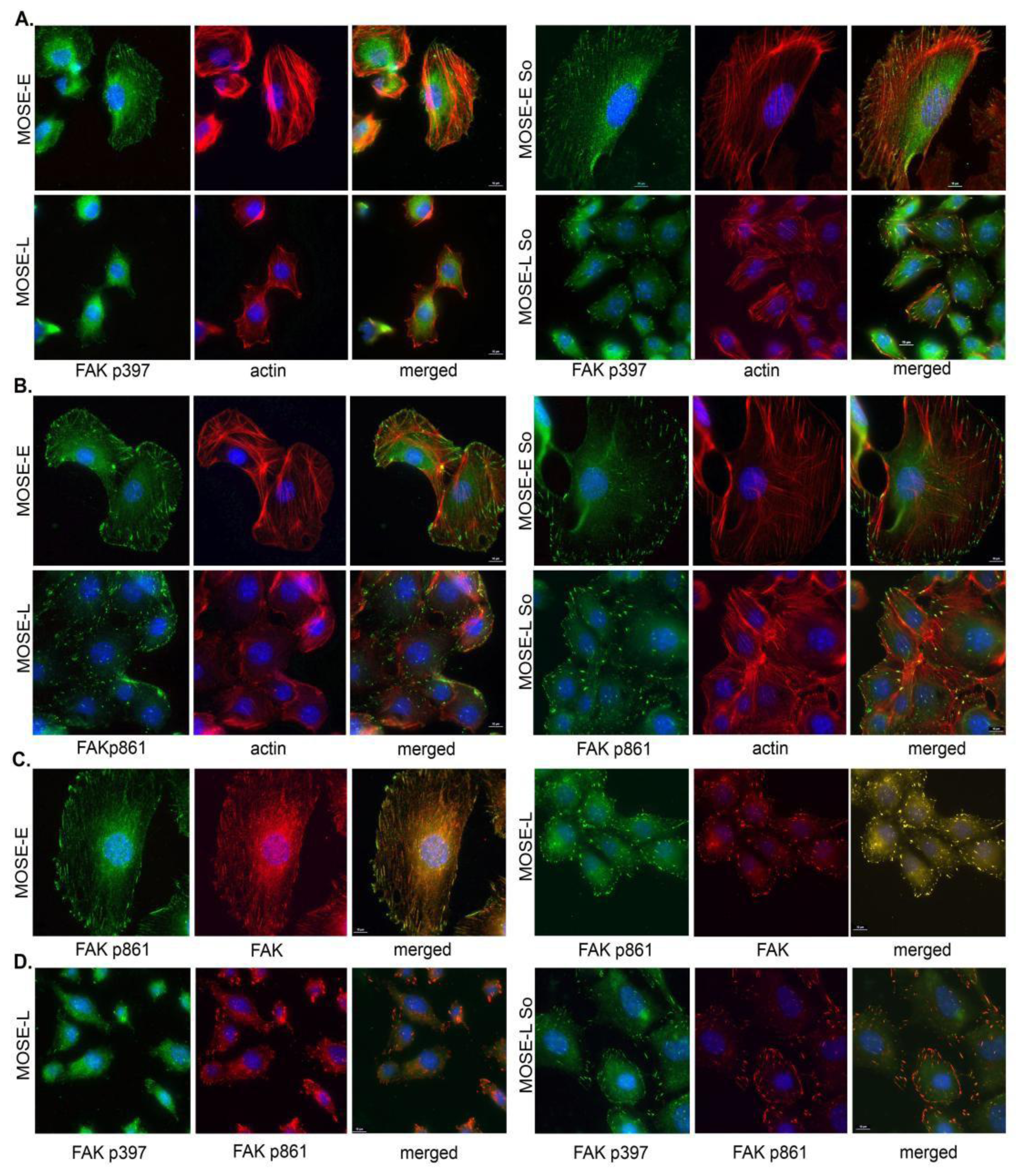
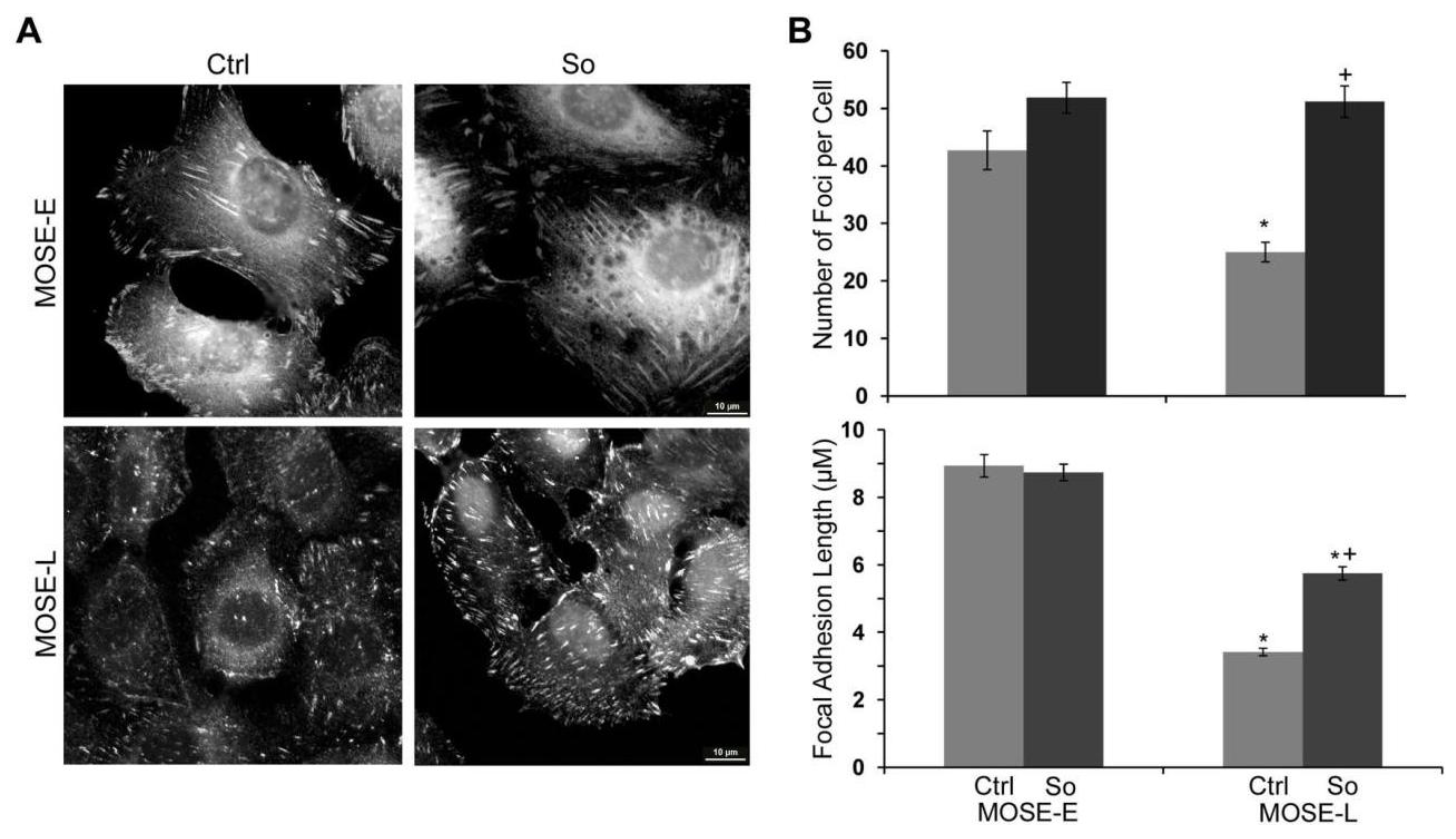
2.4. Sph Treatment Enhanced Focal Adhesion (FA) Formation and Maturation

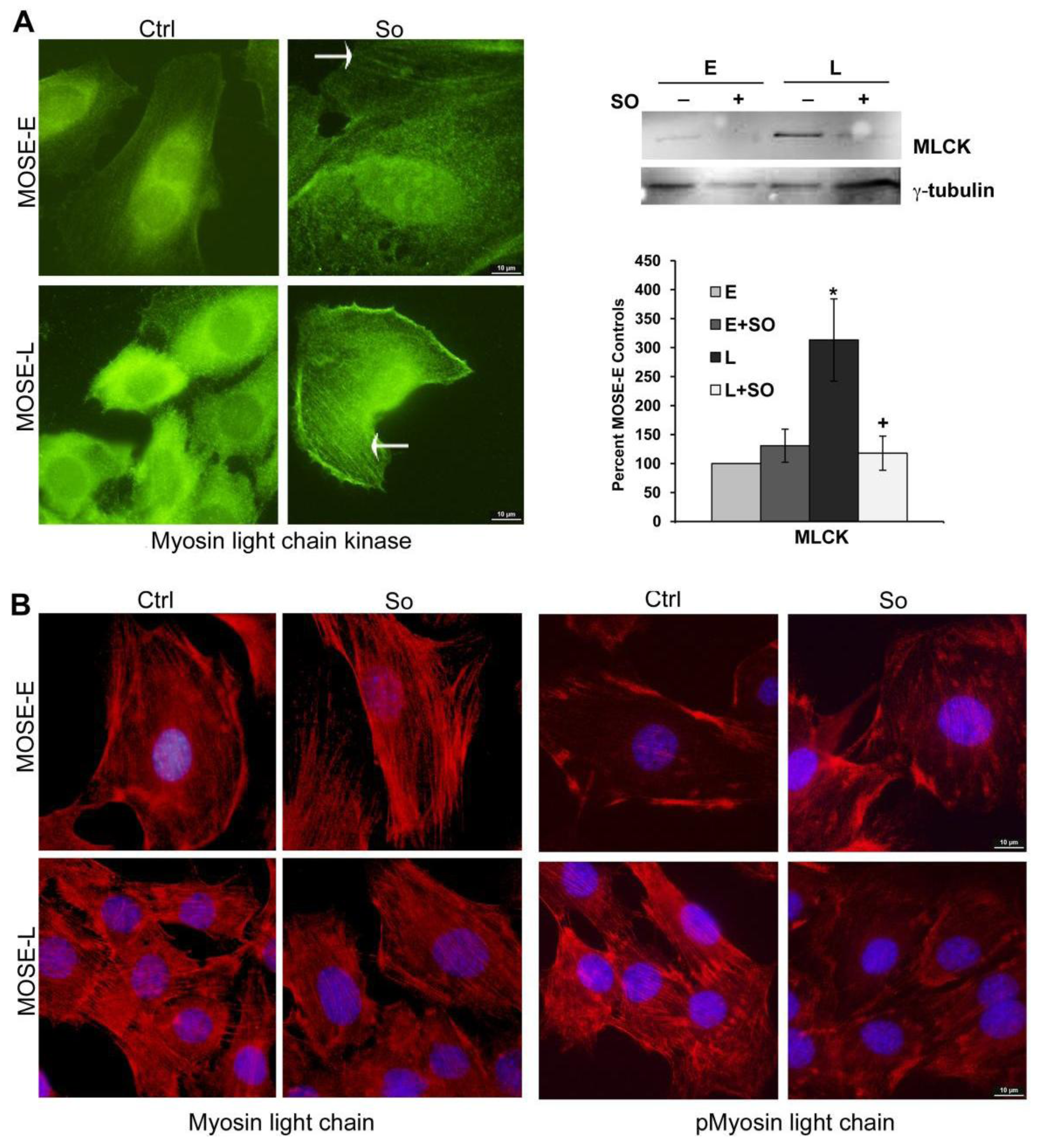
2.5. Regulation of FA and Actin Bundling
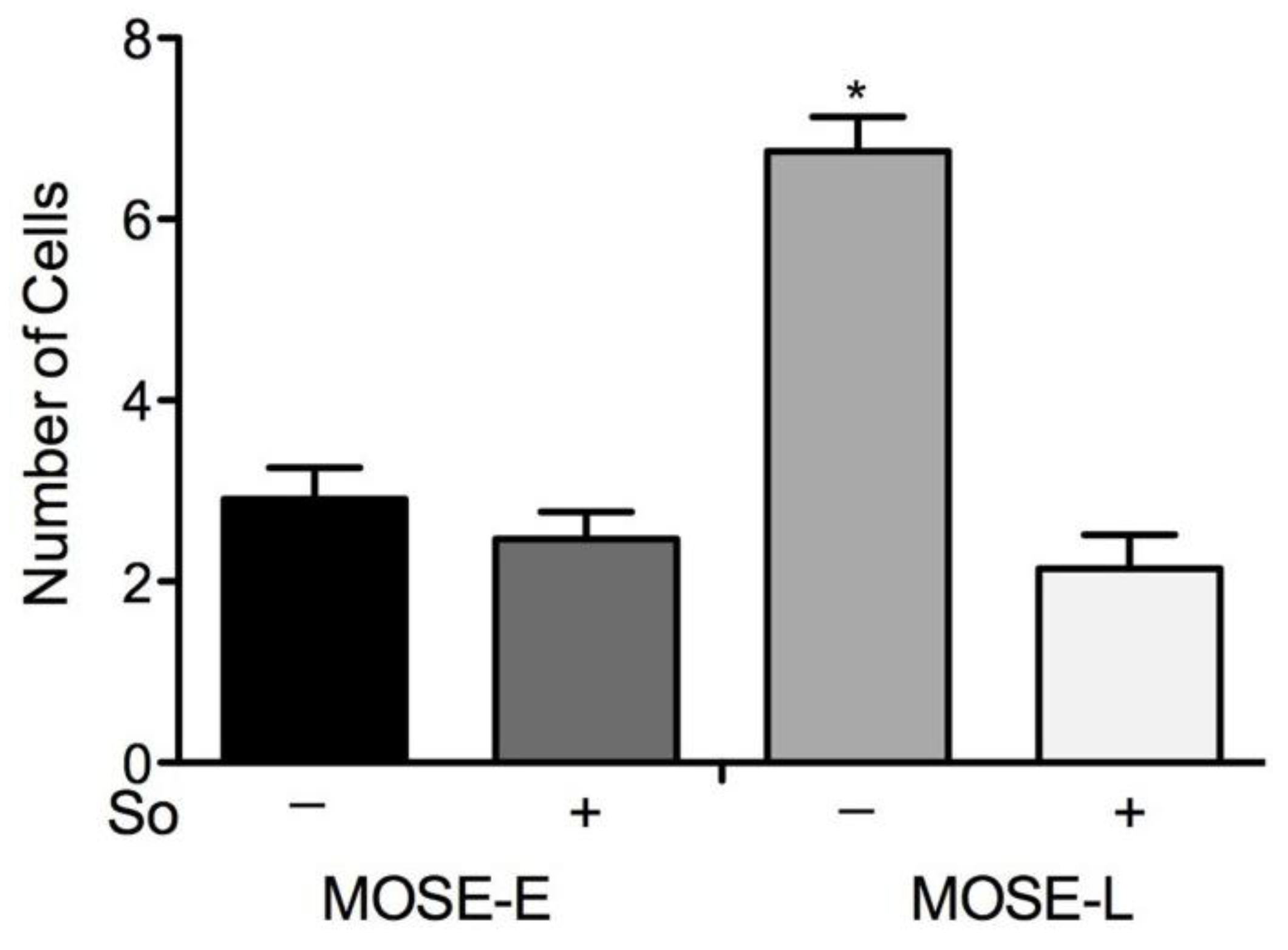
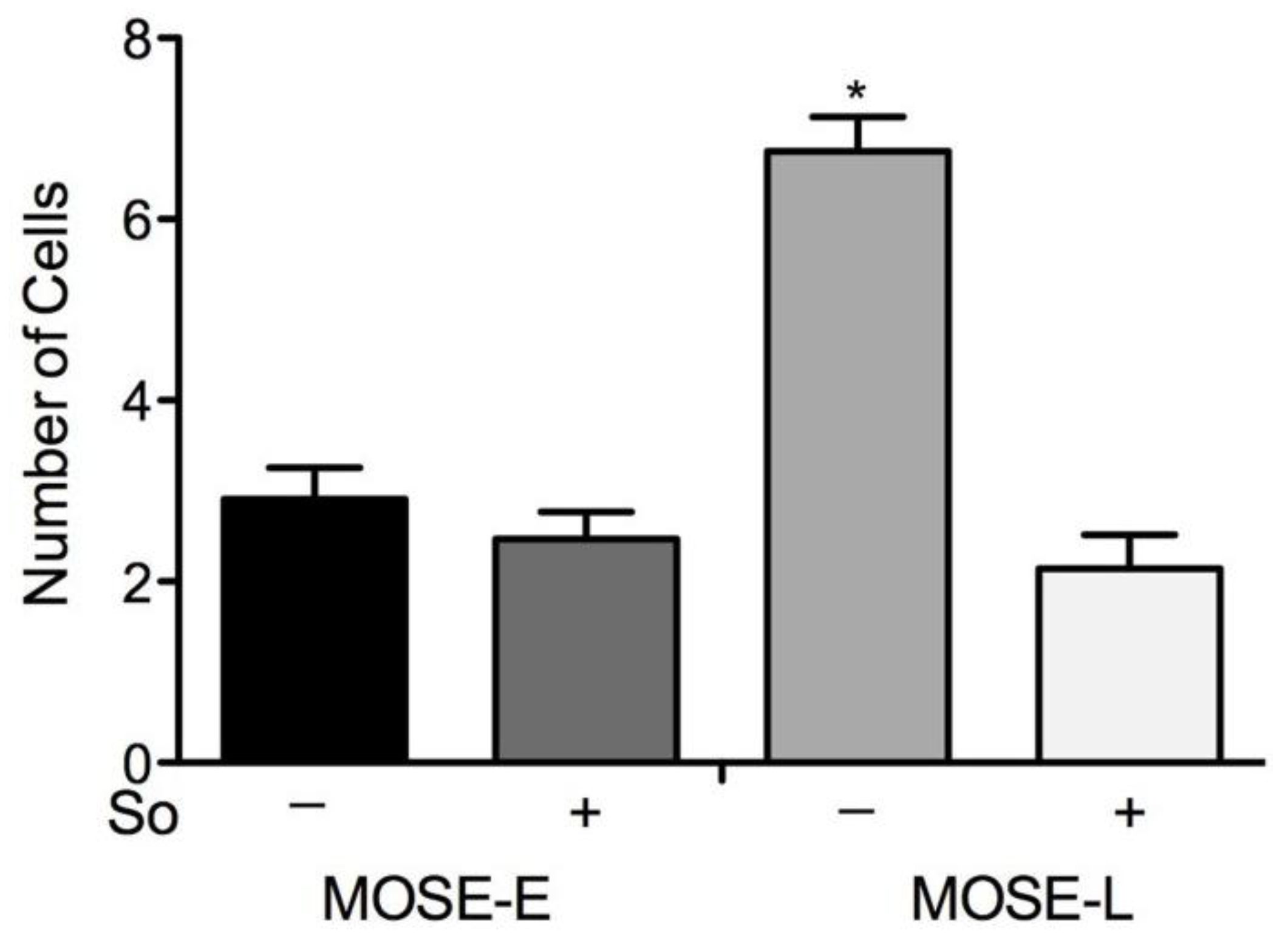
2.6. Suppression of Invasion in vitro
2.7. Discussion
3. Experimental Section
3.1. Cell Culture
3.2. Immunofluorescent Staining
3.3. Quantitation of Filamentous Actin
3.4. Western Blot Analysis
3.5. Quantitative Real-Time Polymerase Chain Reaction PCR (qRT-PCR)
3.6. Cell Invasion Assay
3.7. Statistics
4. Conclusions
Supplementary Materials
Supplementary File 1Acknowledgments
Conflict of Interest
References
- Howlader, N.; Noone, A.M.; Krapho, M.; Neyman, N.; Aminou, R.; Waldron, W.; Altekruse, S.F.; Kosary, C.L.; Ruhl, J.; Tatalovich, Z.; et al. SEER Cancer Statistics Review, 1975–2008. National Cancer Institute: Bethesda, MD, USA, 2011. Available online: http://seer.cancer.gov/csr/1975_2008/ (accessed on July 2, 2013). [Google Scholar]
- Jemal, A.; Siegel, R.; Xu, J.; Ward, E. Cancer statistics. CA Cancer J. Clin. 2010, 60, 277–300. [Google Scholar] [CrossRef]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; et al. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef]
- Pawlak, G.; Helfman, D.M. Cytoskeletal changes in cell transformation and tumorigenesis. Curr. Opin. Genet. Dev. 2001, 11, 41–47. [Google Scholar]
- Provenzano, P.P.; Keely, P.J. Mechanical signaling through the cytoskeleton regulates cell proliferation by coordinated focal adhesion and Rho GTPase signaling. J. Cell Sci. 2011, 124, 1195–1205. [Google Scholar] [CrossRef]
- Ayollo, D.V.; Zhitnyak, I.Y.; Vasiliev, J.M.; Gloushankova, N.A. Rearrangements of the actin cytoskeleton and E-cadherin-based adherens junctions caused by neoplasic transformation change cell-cell interactions. PLoS One 2009, 4, e8027. [Google Scholar]
- Lee, J.M.; Dedhar, S.; Kalluri, R.; Thompson, E.W. The epithelial-mesenchymal transition: new insights in signaling, development, and disease. J. Cell Biol. 2006, 172, 973–981. [Google Scholar]
- Roberts, P.C.; Mottillo, E.P.; Baxa, A.C.; Heng, H.H.; Doyon-Reale, N.; Gregoire, L.; Lancaster, W.D.; Rabah, R.; Schmelz, E.M. Sequential molecular and cellular events during neoplastic progression: a mouse syngeneic ovarian cancer model. Neoplasia 2005, 7, 944–956. [Google Scholar] [CrossRef]
- Creekmore, A.L.; Silkworth, W.T.; Cimini, D.; Jensen, R.V.; Roberts, P.C.; Schmelz, E.M. Changes in gene expression and cellular architecture in an ovarian cancer progression model. PLoS One 2011, 6, e17676. [Google Scholar]
- Anderson, A.S.; Roberts, P.C.; Frisard, M.I.; McMillan, R.P.; Brown, T.J.; Lawless, M.H.; Hulver, M.W.; Schmelz, E.M. Metabolic Changes During Ovarian Cancer Progression as Targets for Sphingosine Treatment. Exp. Cell Res. 2013, 319, 1431–1442. [Google Scholar] [CrossRef]
- Ketene, A.N.; Schmelz, E.M.; Roberts, P.C.; Agah, M. The effects of cancer progression on the viscoelasticity of ovarian cell cytoskeleton structures. Nanomedicine 2012, 8, 93–102. [Google Scholar]
- Ketene, A.N.; Roberts, P.C.; Shea, A.A.; Schmelz, E.M.; Agah, M. Actin filaments play a primary role for structural integrity and viscoelastic response in cells. Integrative Biology 2012, 4, 540–549. [Google Scholar]
- Salmanzadeh, A.; Kittur, H.; Sano, M.B.; Roberts, P.C.; Schmelz, E.M.; Davalos, R.V. Dielectrophoretic differentiation of mouse ovarian surface epithelial cells, macrophages, and fibroblasts using contactless dielectrophoresis. Biomicrofluidics 2012, 6, 24104–24113. [Google Scholar]
- Salmanzadeh, A.; Sano, M.B.; Gallo-Villanueva, R.C.; Roberts, P.C.; Schmelz, E.M.; Davalos, R.V. Investigating dielectric properties of different stages of syngeneic murine ovarian cancer cells. Biomicrofluidics 2013, 7. epublished online. [Google Scholar] [CrossRef]
- Gangoiti, P.; Camacho, L.; Arana, L.; Ouro, A.; Granado, M.H.; Brizuela, L.; Casas, J.; Fabrias, G.; Abad, J.L.; Delgado, A.; Gomez-Munoz, A. Control of metabolism and signaling of simple bioactive sphingolipids: Implications in disease. Prog. Lipid Res. 2010, 49, 316–334. [Google Scholar]
- Gault, C.R.; Obeid, L.M.; Hannun, Y.A. An overview of sphingolipid metabolism: from synthesis to breakdown. Adv. Exp. Med. Biol 2010, 688, 1–23. [Google Scholar]
- Ryland, L.K.; Fox, T.E.; Liu, X.; Loughran, T.P.; Kester, M. Dysregulation of sphingolipid metabolism in cancer. Cancer Biol. Ther. 2011, 11, 138–149. [Google Scholar] [CrossRef]
- Dillehay, D.L.; Webb, S.K.; Schmelz, E.M.; Merrill, A.H., Jr. Dietary sphingomyelin inhibits 1,2- dimethylhydrazine-induced colon cancer in CF1 mice. J. Nutr. 1994, 124, 615–620. [Google Scholar]
- Schmelz, E.M.; Dillehay, D.L.; Webb, S.K.; Reiter, A.; Adams, J.; Merrill, A.H., Jr. Sphingomyelin consumption suppresses aberrant colonic crypt foci and increases the proportion of adenomas vs. adenocarcinomas in CF1 mice treated with 1,2-dimethylhydrazine: implications for dietary sphingolipids and colon carcinogenesis. Cancer Res. 1996, 56, 4936–4941. [Google Scholar]
- Schmelz, E.M.; Sullards, M.C.; Dillehay, D.L.; Merrill, A.H., Jr. Colonic cell proliferation and aberrant crypt foci formation are inhibited by dairy glycosphingolipids in 1, 2-dimethylhydrazine-treated CF1 mice. J. Nutr. 2000, 130, 522–527. [Google Scholar]
- Schmelz, E.M.; Roberts, P.C.; Kustin, E.M.; Lemonnier, L.A.; Sullards, M.C.; Dillehay, D.L.; Merrill, A.H., Jr. Modulation of intracellular beta-catenin localization and intestinal tumorigenesis in vivo and in vitro by sphingolipids. Cancer Res. 2001, 61, 6723–6729. [Google Scholar]
- Lemonnier, L.A.; Dillehay, D.L.; Vespremi, M.J.; Abrams, J.; Brody, E.; Schmelz, E.M. Sphingomyelin in the suppression of colon tumors: Prevention vs. intervention. Arch. Biochem. Biophys. 2003, 419, 129–138. [Google Scholar] [CrossRef]
- Mazzei, J.C.; Zhou, H.; Brayfield, B.P.; Hontecillas, R.; Bassaganya-Riera, J.; Schmelz, E.M. Suppression of intestinal inflammation and inflammation-driven colon cancer in mice by dietary sphingomyelin: importance of peroxisome proliferator-activated receptor gamma expression. J. Nutr. Biochem. 2011, 22, 1160–1171. [Google Scholar] [CrossRef]
- Schmelz, E.M.; Crall, K.J.; Larocque, R.; Dillehay, D.L.; Merrill, A.H., Jr. Uptake and metabolism of sphingolipids in isolated intestinal loops of mice. J. Nutr. 1994, 124, 702–712. [Google Scholar]
- Ohlsson, L.; Hertervig, E.; Jonsson, B.A.; Duan, R.D.; Nyberg, L.; Svernlov, R.; Nilsson, A. Sphingolipids in human ileostomy content after meals containing milk sphingomyelin. Am. J. Clin. Nutr. 2010, 91, 672–678. [Google Scholar] [CrossRef]
- Silins, I.; Nordstrand, M.; Hogberg, J.; Stenius, U. Sphingolipids suppress preneoplastic rat hepatocytes in vitro and in vivo. Carcinogenesis 2003, 24, 1077–1083. [Google Scholar] [CrossRef]
- Simon, K.W.; Tait, L.; Miller, F.; Cao, C.; Davy, K.P.; Leroith, T.; Schmelz, E.M. Suppression of breast xenograft growth and progression in nude mice: implications for the use of orally administered sphingolipids as chemopreventive agents against breast cancer. Food Funct. 2010, 1, 90–98. [Google Scholar]
- Lim, K.G.; Sun, C.; Bittman, R.; Pyne, N.J.; Pyne, S. (R)-FTY720 methyl ether is a specific sphingosine kinase 2 inhibitor: Effect on sphingosine kinase 2 expression in HEK 293 cells and actin rearrangement and survival of MCF-7 breast cancer cells. Cell Signal. 2011, 23, 1590–1595. [Google Scholar] [CrossRef]
- Donati, C.; Bruni, P. Sphingosine 1-phosphate regulates cytoskeleton dynamics: implications in its biological response. Biochim. Biophys. Acta 2006, 1758, 2037–2048. [Google Scholar]
- Zeidan, Y.H.; Jenkins, R.W.; Hannun, Y.A. Remodeling of cellular cytoskeleton by the acid sphingomyelinase/ceramide pathway. J. Cell. Biol. 2008, 181, 335–350. [Google Scholar] [CrossRef]
- Feldhaus, M.J.; Weyrich, A.S.; Zimmerman, G.A.; McIntyre, T.M. Ceramide generation in situ alters leukocyte cytoskeletal organization and beta 2-integrin function and causes complete degranulation. J. Biol. Chem. 2002, 277, 4285–4293. [Google Scholar]
- Zheng, W.; Kollmeyer, J.; Symolon, H.; Momin, A.; Munter, E.; Wang, E.; Kelly, S., Allegood; Liu, Y.; Peng, Q.; Ramaraju, H.; Sullards, M.C.; Cabot, M.; Merrill, A.H., Jr. Ceramides and other bioactive sphingolipid backbones in health and disease: lipidomic analysis, metabolism and roles in membrane structure, dynamics, signaling and autophagy. Biochim. Biophys. Acta 2006, 1758, 1864–1884. [Google Scholar]
- Liotta, D.C.; Bushnev, A.S.; Baillie, M.T.; Holt, J.J.; Menaldino, D.S.; Merrill, A.H. An efficient asymmetric synthesis of Enigmols (1-deoxy-5-hydroxysphingoid bases), an important class of bioactive lipid modulators. Arkivoc 2010, viii, 263–277. [Google Scholar]
- Formigli, L.; Meacci, E.; Sassoli, C.; Chellini, F.; Giannini, R.; Quercioli, F.; Tiribilli, B.; Squecco, R.; Bruni, P.; Francini, F.; Zecchi-Orlandini, S. Sphingosine 1-phosphate induces cytoskeletal reorganization in C2C12 myoblasts: physiological relevance for stress fibres in the modulation of ion current through stretch-activated channels. J. Cell Sci. 2005, 118, 1161–1171. [Google Scholar]
- Serrels, B.; Serrels, A.; Brunton, V.G.; Holt, M.; McLean, G.W.; Gray, C.H.; Jones, G.E.; Frame, M.C. Focal adhesion kinase controls actin assembly via a FERM-mediated interaction with the Arp2/3 complex. Nat. Cell Biol. 2007, 9, 1046–1056. [Google Scholar] [CrossRef]
- Bai, C.Y.; Ohsugi, M.; Abe, Y.; Yamamoto, T. ZRP-1 controls Rho GTPase-mediated actin reorganization by localizing at cell-matrix and cell-cell adhesions. J. Cell Sci. 2007, 120, 2828–2837. [Google Scholar] [CrossRef]
- Tan, J.L.; Ravid, S.; Spudich, J.A. Control of nonmuscle myosins by phosphorylation. Annu. Rev. Biochem. 1992, 61, 721–759. [Google Scholar]
- Jefferson, A.B.; Schulman, H. Sphingosine inhibits calmodulin-dependent enzymes. J. Biol. Chem. 1988, 263, 15241–15244. [Google Scholar]
- Hanna, A.N.; Berthiaume, L.G.; Kikuchi, Y.; Begg, D.; Bourgoin, S.; Brindley, D.N. Tumor necrosis factor-alpha induces stress fiber formation through ceramide production: role of sphingosine kinase. Mol. Biol. Cell 2001, 12, 3618–3630. [Google Scholar]
- Fillet, M.; Cren-Olive, C.; Renert, A.F.; Piette, J.; Vandermoere, F.; Rolando, C.; Merville, M.P. Differential expression of proteins in response to ceramide-mediated stress signal in colon cancer cells by 2-D gel electrophoresis and MALDI-TOF-MS. J. Proteome Res. 2005, 4, 870–880. [Google Scholar]
- Singleton, P.A.; Dudek, S.M.; Chiang, E.T.; Garcia, J.G. Regulation of sphingosine 1-phosphate induced endothelial cytoskeletal rearrangement and barrier enhancement by S1P1 receptor, PI3 kinase, Tiam1/Rac1, and alpha-actinin. FASEB J. 2005, 19, 1646–1656. [Google Scholar]
- Wang, F.; Nobes, C.D.; Hall, A.; Spiegel, S. Sphingosine 1-phosphate stimulates rho-mediated tyrosine phosphorylation of focal adhesion kinase and paxillin in Swiss 3T3 fibroblasts. Biochem. J. 1997, 324, 481–488. [Google Scholar]
- Strub, G.M.; Maceyka, M.; Hait, N.C.; Milstien, S.; Spiegel, S. Extracellular and intracellular actions of sphingosine-1-phosphate. Adv. Exp. Med. Biol. 2010, 688, 141–155. [Google Scholar]
- Wang, D.; Zhao, Z.; Caperell-Grant, A.; Yang, G.; Mok, S.C.; Liu, J.; Bigsby, R.M.; Xu, Y. S1P differentially regulates migration of human ovarian cancer and human ovarian surface epithelial cells. Mol. Cancer Ther. 2008, 7, 1993–2002. [Google Scholar]
- Hong, G.; Baudhuin, L.M.; Xu, Y. Sphingosine-1-phosphate modulates growth and adhesion of ovarian cancer cells. FEBS Lett. 1999, 460, 513–518. [Google Scholar] [CrossRef]
- Canals, D.; Jenkins, R.W.; Roddy, P.; Hernandez-Corbacho, M.J.; Obeid, L.M.; Hannun, Y.A. Differential effects of ceramide and sphingosine 1-phosphate on ERM phosphorylation: probing sphingolipid signaling at the outer plasma membrane. J. Biol. Chem. 2010, 285, 32476–32485. [Google Scholar]
- Seufferlein, T.; Rozengurt, E. Sphingosine induces p125FAK and paxillin tyrosine phosphorylation, actin stress fiber formation, and focal contact assembly in Swiss 3T3 cells. J. Biol. Chem. 1994, 269, 27610–27617. [Google Scholar]
- Hannun, Y.A.; Loomis, C.R.; Merrill, A.H., Jr.; Bell, R.M. Sphingosine inhibition of protein kinase C activity and of phorbol dibutyrate binding in vitro and in human platelets. J. Biol. Chem. 1986, 261, 12604–12609. [Google Scholar]
- Symolon, H.; Bushnev, A.; Peng, Q.; Ramaraju, H.; Mays, S.G.; Allegood, J.C.; Pruett, S T.; Sullards, M.C.; Dillehay, D.L.; Liotta, D.C.; Merrill, A.H., Jr. Enigmol: a novel sphingolipid analogue with anticancer activity against cancer cell lines and in vivo models for intestinal and prostate cancer. Mol. Cancer Ther. 2011, 10, 648–657. [Google Scholar] [CrossRef]
- Tsai, M.A.; Waugh, R.E.; Keng, P.C. Passive mechanical behavior of human neutrophils: effects of colchicine and paclitaxel. Biophys. J. 1998, 74, 3282–3291. [Google Scholar]
- Suresh, S. Biomechanics and biophysics of cancer cells. ActaBiomater 2007, 3, 413–438. [Google Scholar]
- Rajfur, Z.; Roy, P.; Otey, C.; Romer, L.; Jacobson, K. Dissecting the link between stress fibres and focal adhesions by CALI with EGFP fusion proteins. Nat. Cell Biol. 2002, 4, 286–293. [Google Scholar]
- Salmanzadeh, A.; Elvington, E.S.; Roberts, P.C.; Schmelz, E.M.; Davalos, R.V. Sphingolipid metabolites modulate dielectric characteristics of cells in a mouse ovarian cancer progression model. Integr. Biol. 2013, 5, 843–852. [Google Scholar] [CrossRef]
- Ilic, D.; Furuta, Y.; Kanazawa, S.; Takeda, N.; Sobue, K.; Nakatsuji, N.; Nomura, S.; Fujimoto, J.; Okada, M.; Yamamoto, T. Reduced cell motility and enhanced focal adhesion contact formation in cells from FAK-deficient mice. Nature 1995, 377, 539–544. [Google Scholar]
- Grabel, L.B.; Hong, T. Migration of F9 parietal endoderm cells is regulated by the ERK pathway. J. Cell. Biochem. 2006, 97, 1339–1349. [Google Scholar]
- Sjoblom, B.; Salmazo, A.; Djinovic-Carugo, K. Alpha-actinin structure and regulation. Cell. Mol. Life Sci. 2008, 65, 2688–26701. [Google Scholar]
- Sood, A.K.; Armaiz-Pena, G.N.; Halder, J.; Nick, A.M.; Stone, R.L.; Hu, W.; Carroll, A.R.; Spannuth, W.A.; Deavers, M.T.; Allen, J.K.; et al. Adrenergic modulation of focal adhesion kinase protects human ovarian cancer cells from anoikis. J. Clin. Invest. 2010, 120, 1515–1523. [Google Scholar] [CrossRef]
- Schlaepfer, D.D.; Mitra, S.K.; Ilic, D. Control of motile and invasive cell phenotypes by focal adhesion kinase. Biochim. Biophys. Acta 2004, 1692, 77–102. [Google Scholar] [CrossRef]
- Schaller, M.D. Cellular functions of FAK kinases: insight into molecular mechanisms and novel functions. J. Cell. Sci. 2010, 123, 1007–1013. [Google Scholar] [CrossRef]
- Cui, W.J.; Liu, Y.; Zhou, X.L.; Wang, F.Z.; Zhang, X.D.; Ye, L.H. Myosin light chain kinase is responsible for high proliferative ability of breast cancer cells via anti-apoptosis involving p38 pathway. Acta Pharmacol. Sin. 2010, 31, 725–732. [Google Scholar]
- Gutjahr, M.C.; Rossy, J.; Niggli, V. Role of Rho, Rac, and Rho-kinase in phosphorylation of myosin light chain, development of polarity, and spontaneous migration of Walker 256 carcinosarcoma cells. Exp. Cell Res. 2005, 308, 422–438. [Google Scholar]
- Fazal, F.; Gu, L.; Ihnatovych, I.; Han, Y.; Hu, W.; Antic, N.; Carreira, F.; Blomquist, J.F.; Hope, T.J.; Ucker, D.S.; de Lanerolle, P. Inhibiting myosin light chain kinase induces apoptosis in vitro and in vivo. Mol. Cell Bio. 2005, 25, 6259–6266. [Google Scholar]
- Schmelz, E.M.; Bushnev, A.S.; Dillehay, D.L.; Liotta, D.C.; Merrill, A.H., Jr. Suppression of aberrant colonic crypt foci by synthetic sphingomyelins with saturated or unsaturated sphingoid base backbones. Nutr. Cancer 1997, 28, 81–85. [Google Scholar]
- Schmelz, E.M.; Dombrink-Kurtzman, M.A.; Roberts, P.C.; Kozutsumi, Y.; Kawasaki, T.; Merrill, A.H., Jr. Induction of apoptosis by fumonisin B1 in HT29 cells is mediated by the accumulation of endogenous free sphingoid bases. Toxicol. Appl. Pharmacol. 1998; 148, 252–260. [Google Scholar]
- Livak, K.J.; Schmittgen, T.D. Analysis of relative gene expression data using real-time quantitative PCR and the 2(-Delta Delta C(T)) Method. Methods, 2001; 25, 402–408. [Google Scholar]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Creekmore, A.L.; Heffron, C.L.; Brayfield, B.P.; Roberts, P.C.; Schmelz, E.M. Regulation of Cytoskeleton Organization by Sphingosine in a Mouse Cell Model of Progressive Ovarian Cancer. Biomolecules 2013, 3, 386-407. https://doi.org/10.3390/biom3030386
Creekmore AL, Heffron CL, Brayfield BP, Roberts PC, Schmelz EM. Regulation of Cytoskeleton Organization by Sphingosine in a Mouse Cell Model of Progressive Ovarian Cancer. Biomolecules. 2013; 3(3):386-407. https://doi.org/10.3390/biom3030386
Chicago/Turabian StyleCreekmore, Amy L., C. Lynn Heffron, Bradley P. Brayfield, Paul C. Roberts, and Eva M. Schmelz. 2013. "Regulation of Cytoskeleton Organization by Sphingosine in a Mouse Cell Model of Progressive Ovarian Cancer" Biomolecules 3, no. 3: 386-407. https://doi.org/10.3390/biom3030386