Human DNA Glycosylase NEIL1’s Interactions with Downstream Repair Proteins Is Critical for Efficient Repair of Oxidized DNA Base Damage and Enhanced Cell Survival


Abstract
:
1. Introduction

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Interaction partner of NEIL1 | Binding region in NEIL1 | Reference |
|---|---|---|
| Polβ | aa 312-349 | [12]; present study |
| LigIIIα | aa 312-349 | [12]; present study |
| XRCC1 | aa 312-349 | [12]; present study |
| FEN-1 | aa 312-349 | [26] |
| PCNA | aa 289-349 | [21] |
| RPA | aa 312-349 | [22] |
| hnRNP-U | aa 312-349 | [18] |
| PARP-1 | aa 312-389 | unpublished observation |
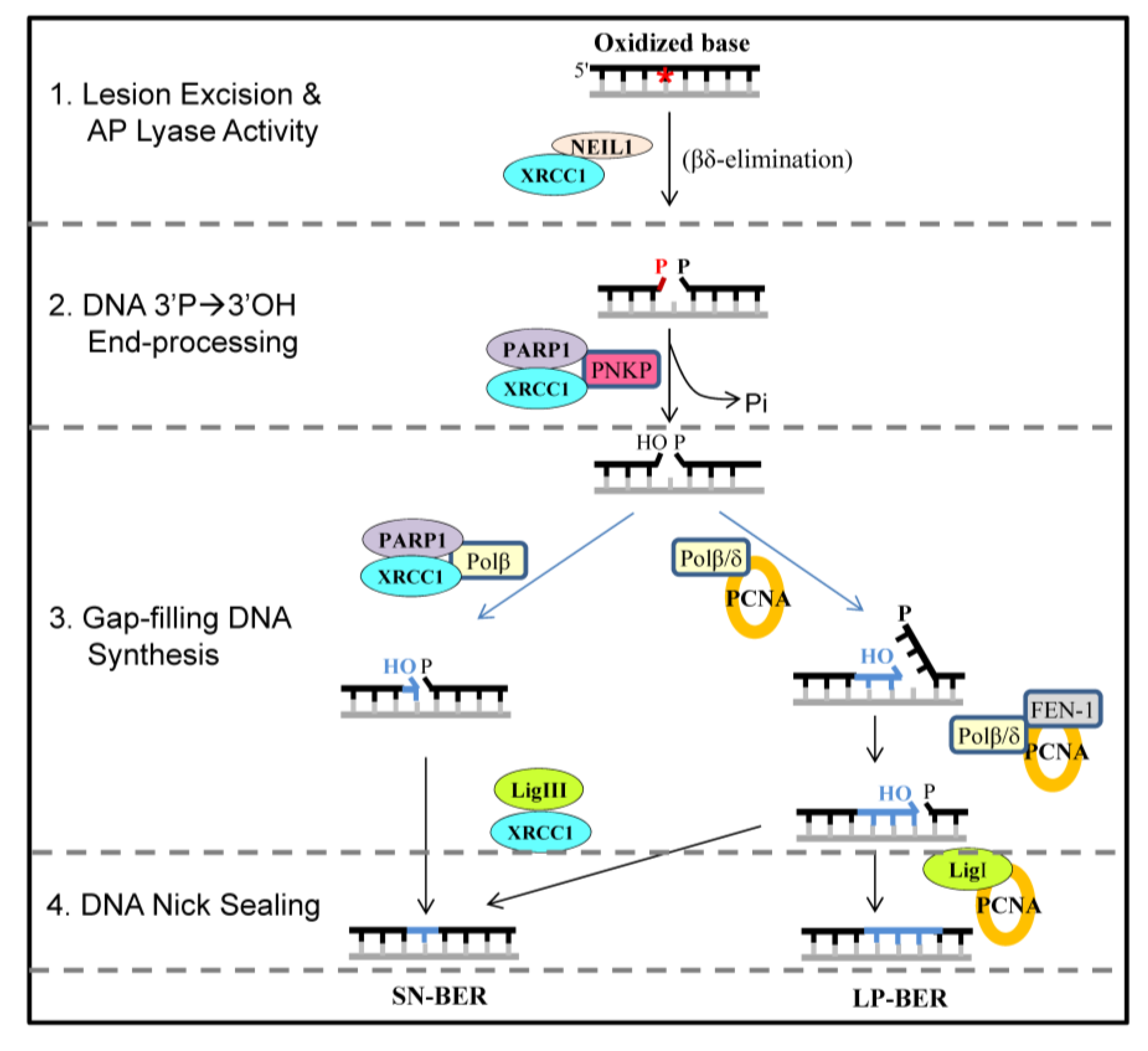
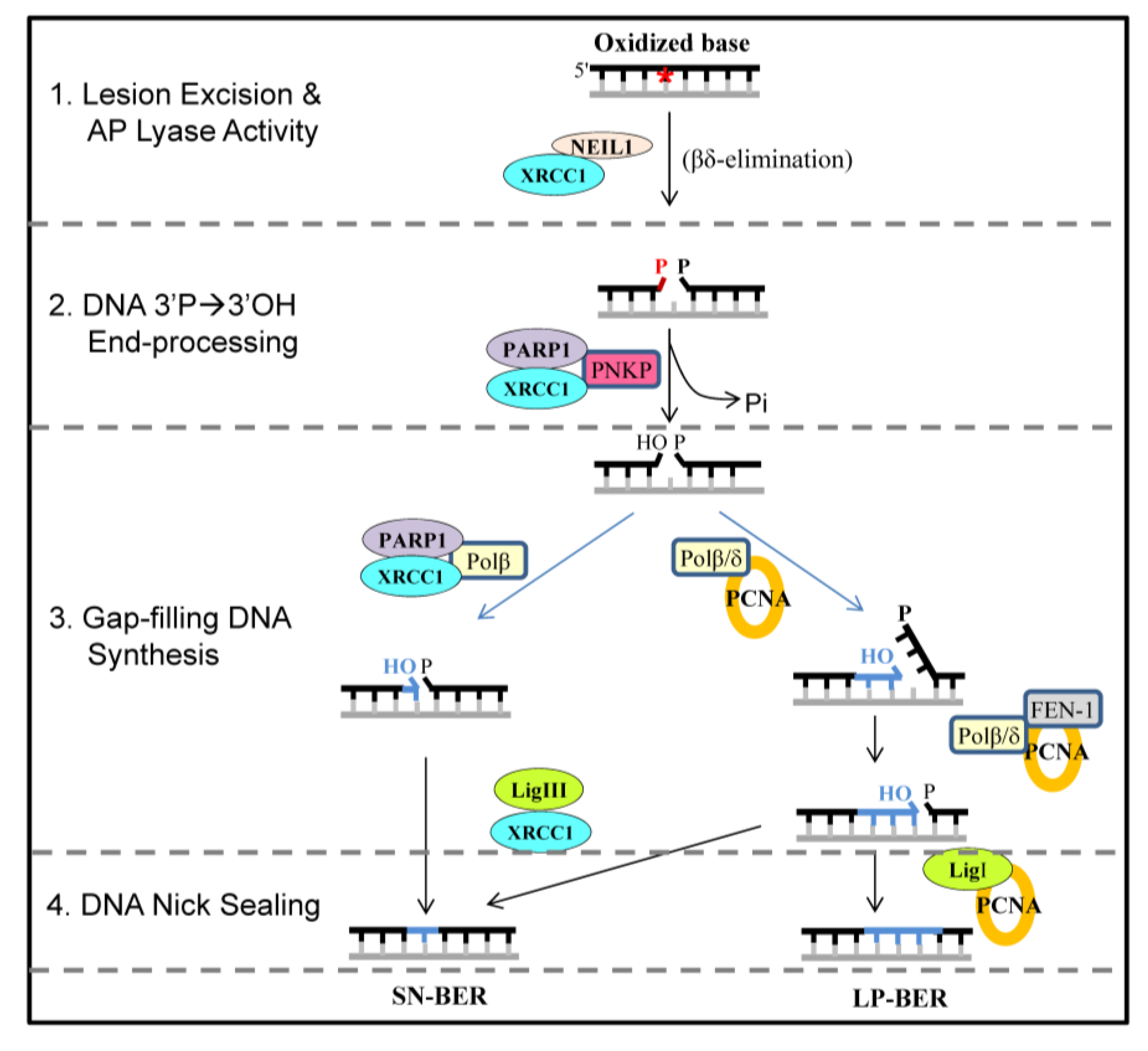
2. Results and Discussion
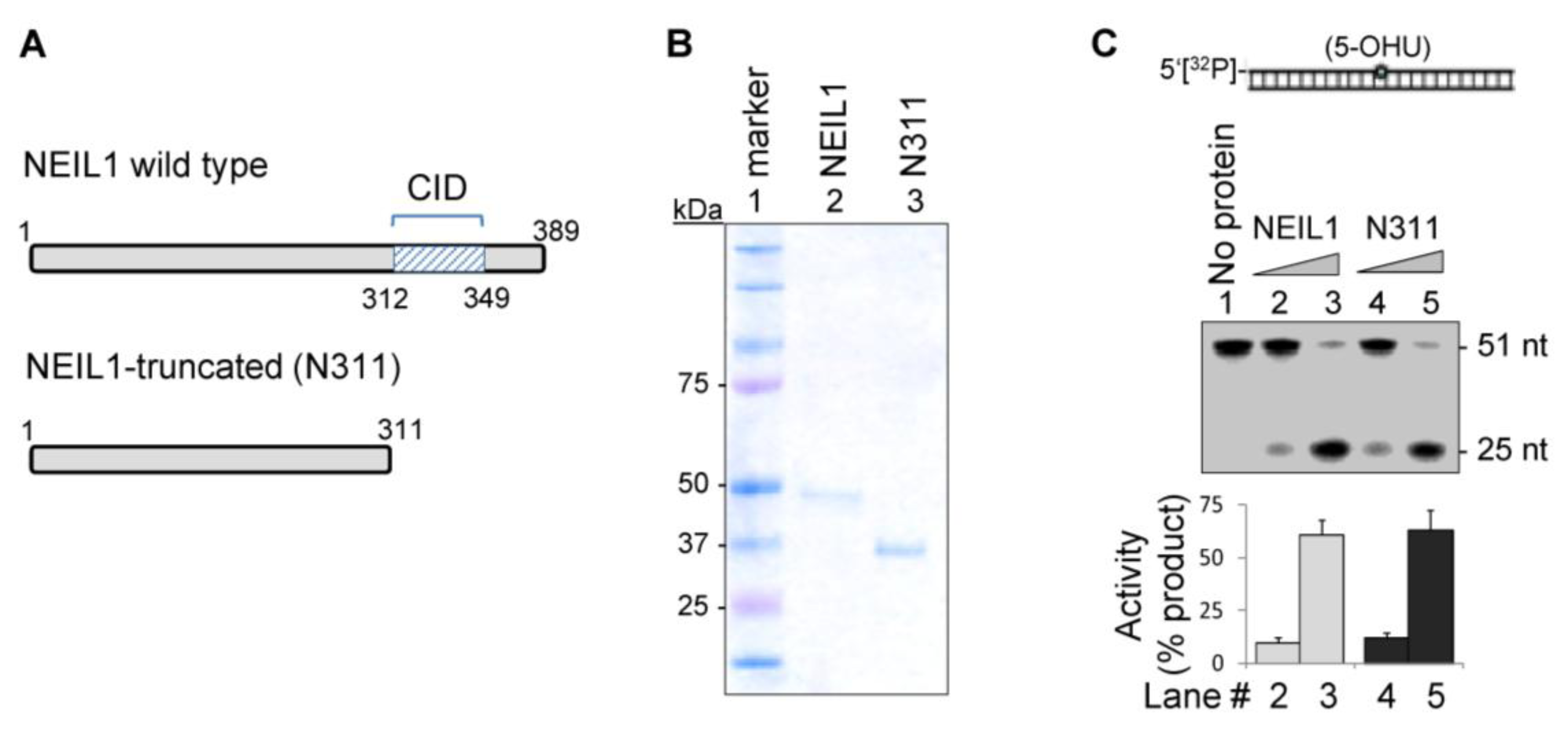
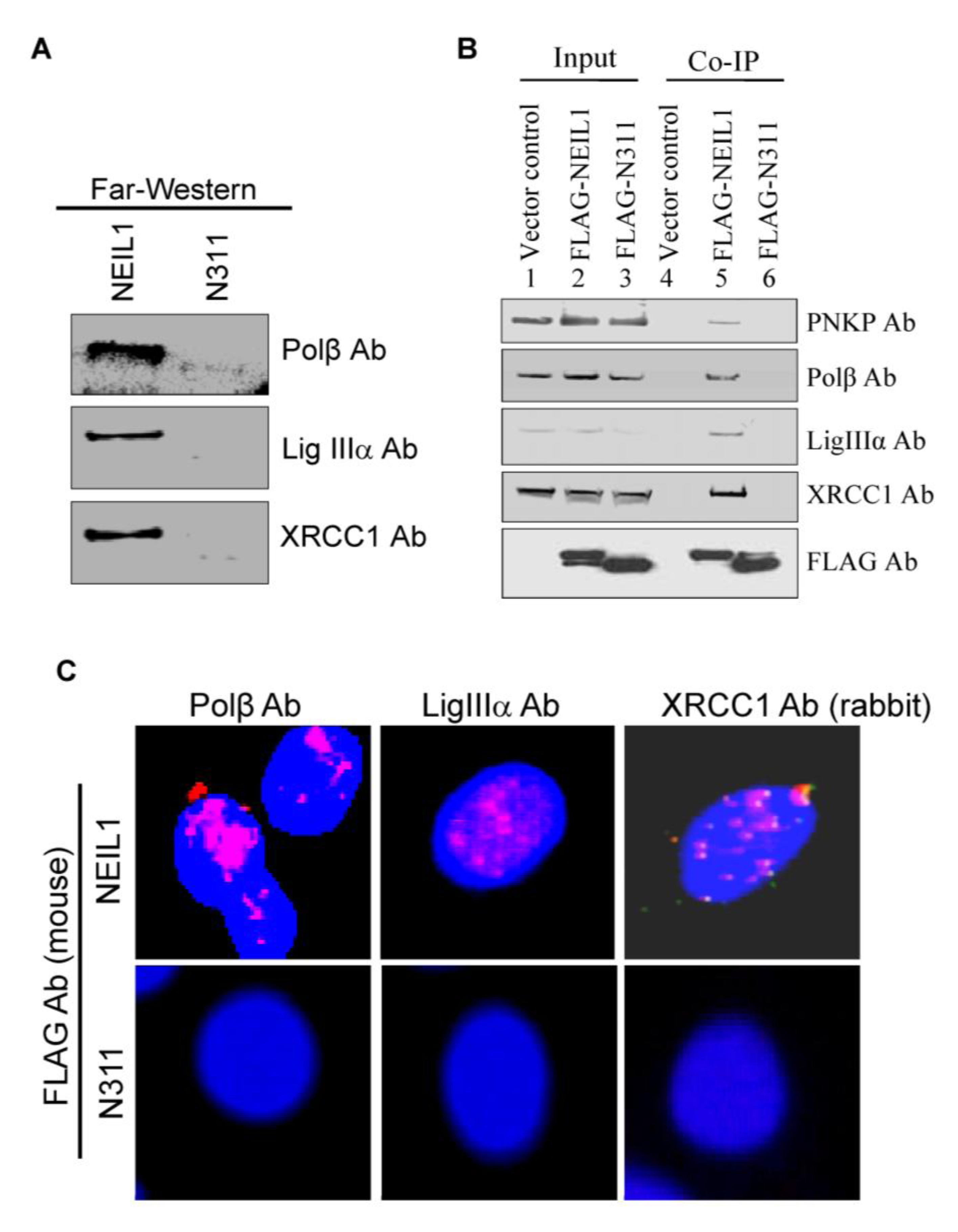
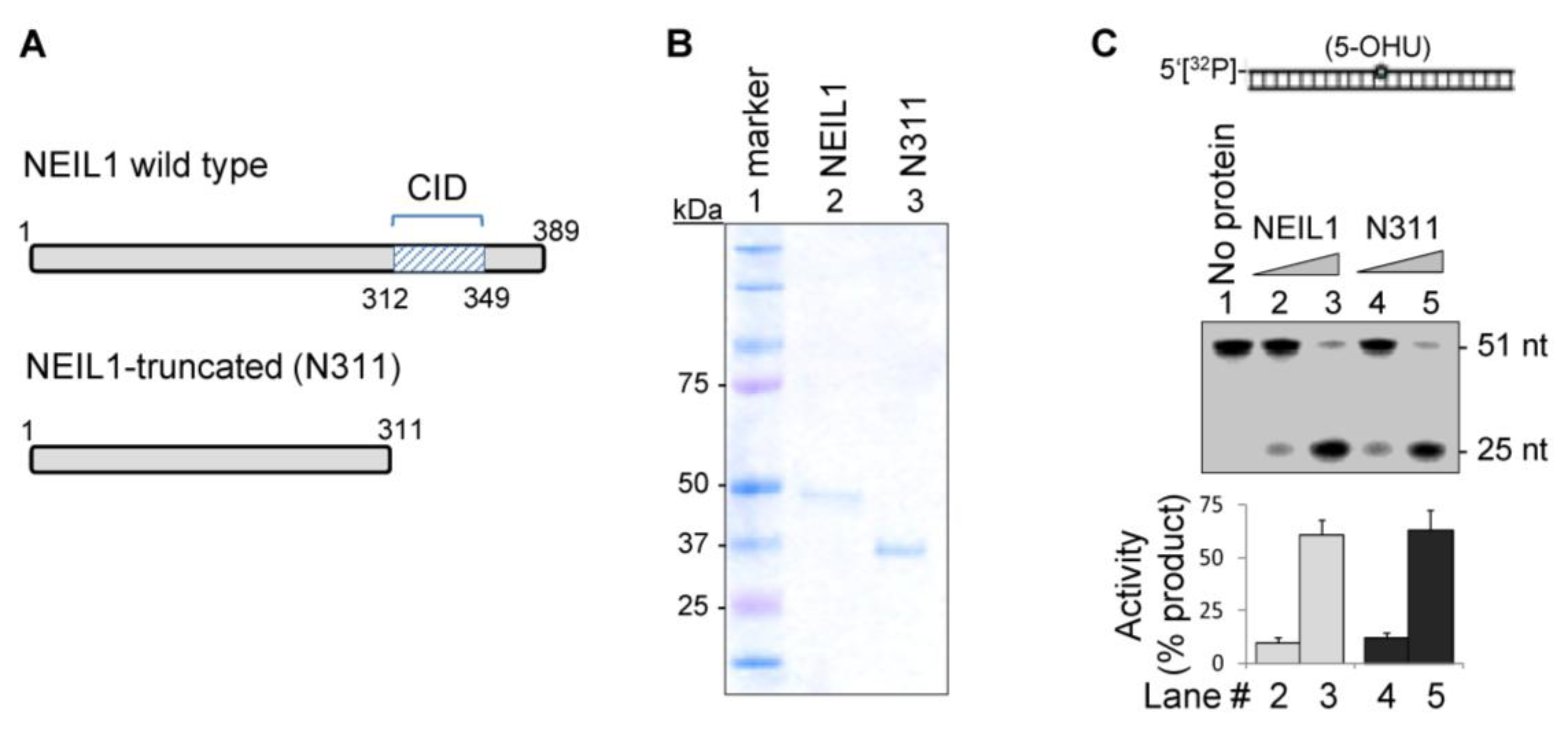
2.1. The CID Is Dispensable for NEIL1’s Glycosylase Activity in vitro, but Provides a Common Interaction Region for Protein-Protein Interactions

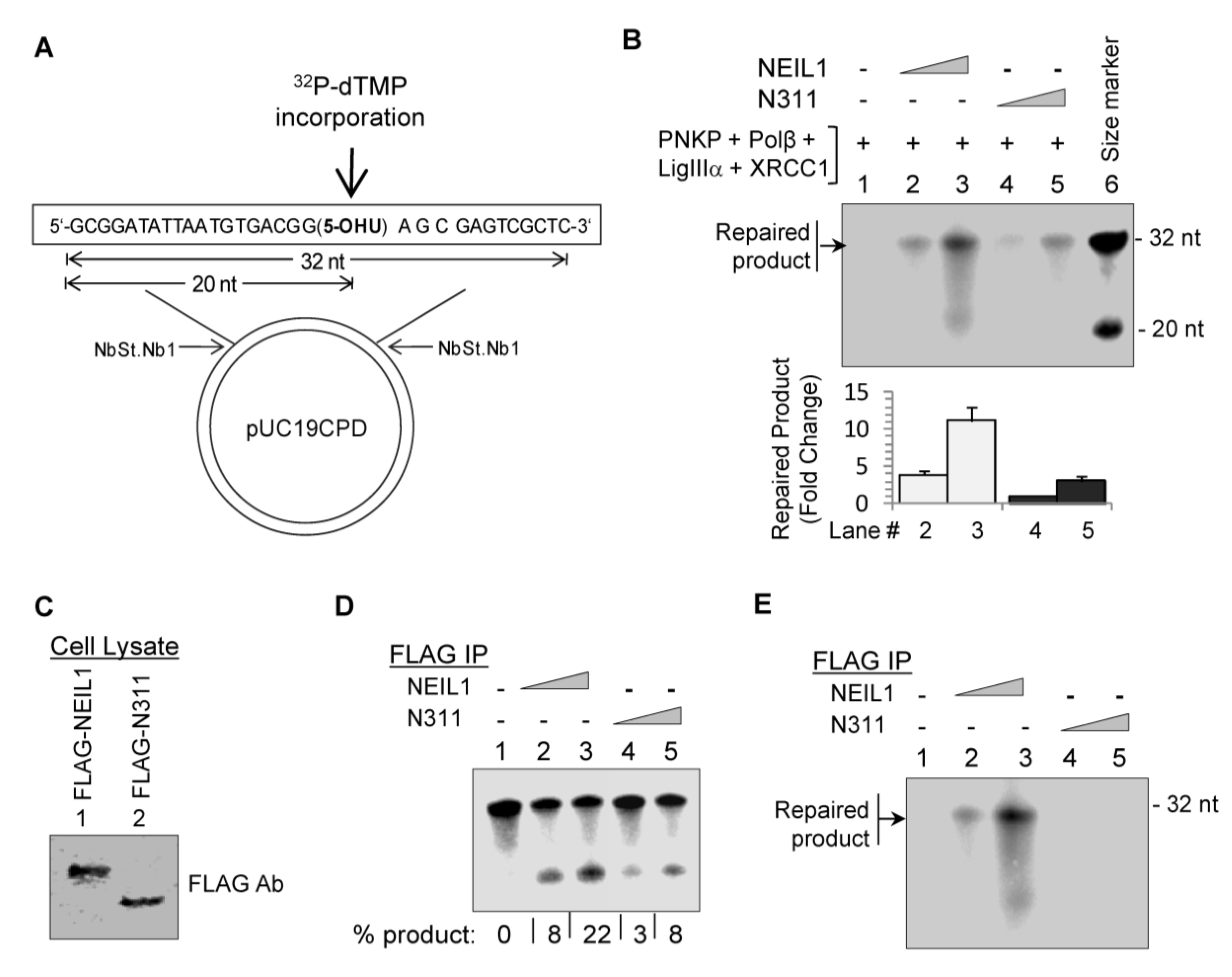
2.2. NEIL1’s CID Is Required for Efficient Repair of Oxidized DNA Bases
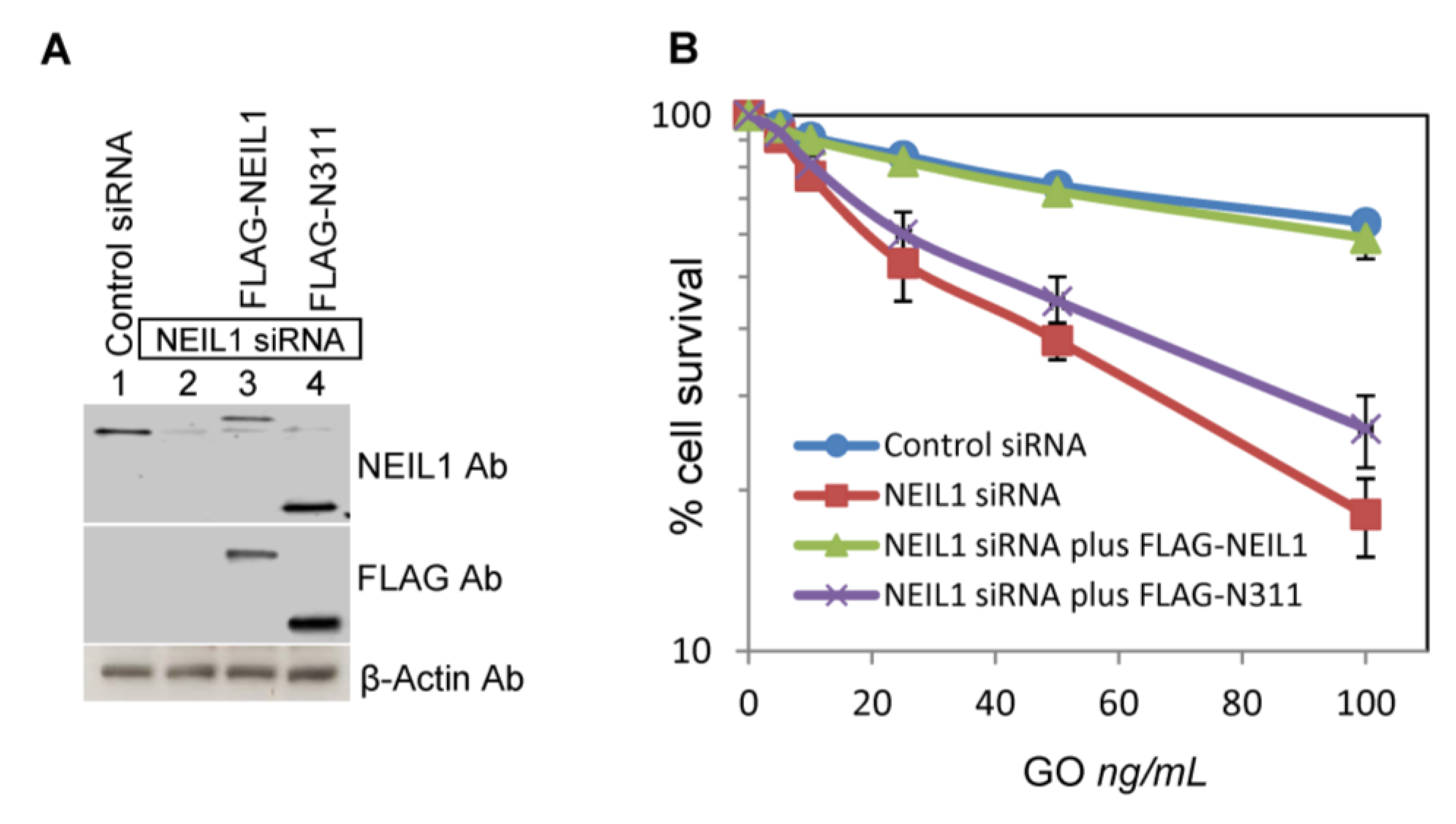
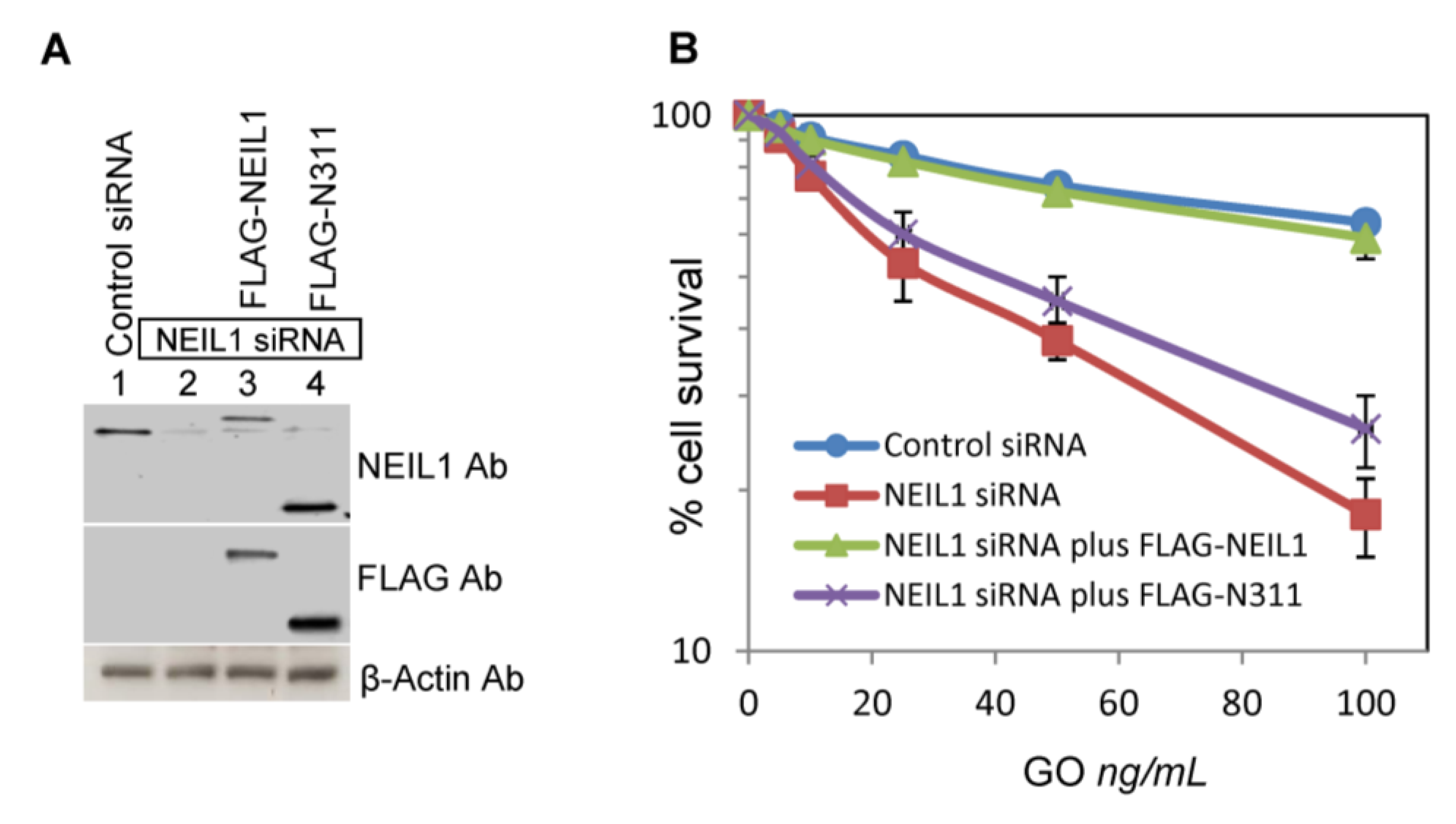
2.3. Ectopic Wild-Type but not Truncated NEIL1 Restores Resistance to ROS Toxicity in NEIL1-Depleted Cells
3. Experimental Section
3.1. Expression and Purification of Recombinant Proteins
3.2. Generation of FLAG-NEIL1 and FLAG-N311 Expression Plasmids and Their Stable Expression in HEK293 Cells
3.3. DNA Substrates for Repair Assay
3.4. Cell Culture and Co-Immunoprecipitation
3.5. Far Western Analysis
3.6. In Situ Proximity Ligation Assay (PLA)
3.7. DNA Glycosylase/AP Lyase Assay of NEIL1
3.8. Complete Repair Assay
3.9. Cell Survival Assay
4. Conclusions
Acknowledgments
Conflict of Interest
References
- Lavrovsky, Y.; Chatterjee, B.; Clark, R.A.; Roy, A.K. Role of redox-regulated transcription factors in inflammation, aging and age-related diseases. Exp. Gerontol. 2000, 35, 521–532. [Google Scholar] [CrossRef]
- Hegde, M.L.; Mantha, A.K.; Hazra, T.K.; Bhakat, K.K.; Mitra, S.; Szczesny, B. Oxidative genome damage and its repair: implications in aging and neurodegenerative diseases. Mech. Ageing Dev. 2012, 133, 157–168. [Google Scholar] [CrossRef]
- Hegde, M.L.; Izumi, T.; Mitra, S. Oxidized base damage and single-strand break repair in mammalian genomes: role of disordered regions and posttranslational modifications in early enzymes. Prog. Mol. Biol. Transl. Sci. 2012, 110, 123–153. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Rao, K.S.; Mitra, S. Oxidative genome damage and its repair in neurodegenerative diseases: function of transition metals as a double-edged sword. J. Alzheimers Dis. 2011, 24 Suppl. 2, 183–198. [Google Scholar]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; Cheadle, J.P. Inherited variants of MYH associated with somatic G:C-->T:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef]
- Ames, B.N.; Shigenaga, M.K.; Hagen, T.M. Oxidants, antioxidants, and the degenerative diseases of aging. Proc. Natl. Acad. Sci. USA 1993, 90, 7915–7922. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hegde, P.M.; Holthauzen, L.M.; Hazra, T.K.; Rao, K.S.; Mitra, S. Specific Inhibition of NEIL-initiated repair of oxidized base damage in human genome by copper and iron: potential etiological linkage to neurodegenerative diseases. J. Biol. Chem. 2010, 285, 28812–28825. [Google Scholar]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef]
- Liu, M.; Bandaru, V.; Bond, J.P.; Jaruga, P.; Zhao, X.; Christov, P.P.; Burrows, C.J.; Rizzo, C.J.; Dizdaroglu, M.; Wallace, S.S. The mouse ortholog of NEIL3 is a functional DNA glycosylase in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2010, 107, 4925–4930. [Google Scholar]
- Hazra, T.K.; Izumi, T.; Boldogh, I.; Imhoff, B.; Kow, Y.W.; Jaruga, P.; Dizdaroglu, M.; Mitra, S. Identification and characterization of a human DNA glycosylase for repair of modified bases in oxidatively damaged DNA. Proc. Natl. Acad. Sci. USA 2002, 99, 3523–3528. [Google Scholar]
- Hazra, T.K.; Kow, Y.W.; Hatahet, Z.; Imhoff, B.; Boldogh, I.; Mokkapati, S.K.; Mitra, S.; Izumi, T. Identification and characterization of a novel human DNA glycosylase for repair of cytosine-derived lesions. J. Biol. Chem. 2002, 277, 30417–30420. [Google Scholar]
- Wiederhold, L.; Leppard, J.B.; Kedar, P.; Karimi-Busheri, F.; Rasouli-Nia, A.; Weinfeld, M.; Tomkinson, A.E.; Izumi, T.; Prasad, R.; Wilson, S.H.; Mitra, S.; Hazra, T.K. AP endonuclease-independent DNA base excision repair in human cells. Mol. Cell 2004, 15, 209–220. [Google Scholar]
- Matsumoto, Y. Molecular mechanism of PCNA-dependent base excision repair. Prog. Nucleic Acid Res. Mol. Biol. 2001, 68, 129–138. [Google Scholar] [CrossRef]
- Klungland, A.; Lindahl, T. Second pathway for completion of human DNA base excision-repair: reconstitution with purified proteins and requirement for DNase IV (FEN1). EMBO J. 1997, 16, 3341–3348. [Google Scholar]
- Della-Maria, J.; Hegde, M.L.; McNeill, D.R.; Matsumoto, Y.; Tsai, M.S.; Ellenberger, T.; Wilson, D.M.; Mitra, S.; Tomkinson, A.E. The interaction between Polynucleotide Kinase Phosphatase and the DNA Repair Protein XRCC1 is Critical for Repair of DNA Alkylation Damage and Stable Association at DNA Damage Sites. J. Biol. Chem. 2012, 287, 39233–39244. [Google Scholar]
- Odell, I.D.; Barbour, J.E.; Murphy, D.L.; Della-Maria, J.A.; Sweasy, J.B.; Tomkinson, A.E.; Wallace, S.S.; Pederson, D.S. Nucleosome disruption by DNA ligase III-XRCC1 promotes efficient base excision repair. Mol. Cell Biol. 2011, 31, 4623–4632. [Google Scholar]
- Noren Hooten, N.; Kompaniez, K.; Barnes, J.; Lohani, A.; Evans, M.K. Poly(ADP-ribose) polymerase 1 (PARP-1) binds to 8-oxoguanine-DNA glycosylase (OGG1). J. Biol. Chem. 2011, 286, 44679–44690. [Google Scholar]
- Hegde, M.L.; Banerjee, S.; Hegde, P.M.; Bellot, L.A.; Hazra, T.K.; Boldogh, I.; Mitra, S. Enhancement of NEIL1-initiated oxidized DNA base excision repair by heterogeneous nuclear Ribonucleoprotein U (hnRNP-U) via direct interaction. J. Biol. Chem. 2012, 287, 34202–34211. [Google Scholar]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential repair of oxidized base damage in the transcribed genes of mammalian cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar]
- Das, A.; Boldogh, I.; Lee, J.W.; Harrigan, J.A.; Hegde, M.L.; Piotrowski, J.; de Souza Pinto, N.; Ramos, W.; Greenberg, M.M.; Hazra, T.K.; Mitra, S.; Bohr, V.A. The human Werner syndrome protein stimulates repair of oxidative DNA base damage by the DNA glycosylase NEIL1. J. Biol. Chem. 2007, 282, 26591–26602. [Google Scholar]
- Dou, H.; Theriot, C.A.; Das, A.; Hegde, M.L.; Matsumoto, Y.; Boldogh, I.; Hazra, T.K.; Bhakat, K.K.; Mitra, S. Interaction of the human DNA glycosylase NEIL1 with proliferating cell nuclear antigen. The potential for replication-associated repair of oxidized bases in mammalian genomes. J. Biol. Chem. 2008, 283, 3130–3140. [Google Scholar]
- Theriot, C.A.; Hegde, M.L.; Hazra, T.K.; Mitra, S. RPA physically interacts with the human DNA glycosylase NEIL1 to regulate excision of oxidative DNA base damage in primer-template structures. DNA Repair (Amst) 2010, 9, 643–652. [Google Scholar] [CrossRef]
- Bandaru, V.; Sunkara, S.; Wallace, S.S.; Bond, J.P. A novel human DNA glycosylase that removes oxidative DNA damage and is homologous to Escherichia coli endonuclease VIII. DNA Repair (Amst) 2002, 1, 517–529. [Google Scholar] [CrossRef]
- Takao, M.; Kanno, S.; Shiromoto, T.; Hasegawa, R.; Ide, H.; Ikeda, S.; Sarker, A.H.; Seki, S.; Xing, J.Z.; Le, X.C.; Weinfeld, M.; Kobayashi, K.; Miyazaki, J.; Muijtjens, M.; Hoeijmakers, J.H.; van der Horst, G.; Yasui, A. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. Embo. J. 2002, 21, 3486–3493. [Google Scholar]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of oxidized bases in DNA bubble structures by human DNA glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar]
- Hegde, M.L.; Theriot, C.A.; Das, A.; Hegde, P.M.; Guo, Z.; Gary, R.K.; Hazra, T.K.; Shen, B.; Mitra, S. Physical and functional interaction between human oxidized base-specific DNA glycosylase NEIL1 and flap endonuclease 1. J. Biol. Chem. 2008, 283, 27028–27037. [Google Scholar]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Functions of disordered regions in mammalian early base excision repair proteins. Cell Mol. Life Sci. 2010, 67, 3573–3587. [Google Scholar]
- Johansson, H.; Svensson, F.; Runnberg, R.; Simonsson, T.; Simonsson, S. Phosphorylated nucleolin interacts with translationally controlled tumor protein during mitosis and with Oct4 during interphase in ES cells. PLoS One 2010, 5, e13678. [Google Scholar]
- Fredriksson, S.; Gullberg, M.; Jarvius, J.; Olsson, C.; Pietras, K.; Gustafsdottir, S.M.; Ostman, A.; Landegren, U. Protein detection using proximity-dependent DNA ligation assays. Nat. Biotechnol. 2002, 20, 473–477. [Google Scholar]
- Mandal, S.M.; Hegde, M.L.; Chatterjee, A.; Hegde, P.M.; Szczesny, B.; Banerjee, D.; Boldogh, I.; Gao, R.; Falkenberg, M.; Gustafsson, C.M.; Sarkar, P.S.; Hazra, T.K. Role of human DNA glycosylase Nei-like 2 (NEIL2) and single strand break repair protein polynucleotide kinase 3'-phosphatase in maintenance of mitochondrial genome. J. Biol. Chem. 2012, 287, 2819–2829. [Google Scholar]
- Izumi, T.; Mitra, S. Deletion analysis of human AP-endonuclease: minimum sequence required for the endonuclease activity. Carcinogenesis 1998, 19, 525–527. [Google Scholar] [CrossRef]
- Izumi, T.; Wiederhold, L.R.; Roy, G.; Roy, R.; Jaiswal, A.; Bhakat, K.K.; Mitra, S.; Hazra, T.K. Mammalian DNA base excision repair proteins: their interactions and role in repair of oxidative DNA damage. Toxicology 2003, 193, 43–65. [Google Scholar] [CrossRef]
- Bhakat, K.K.; Izumi, T.; Yang, S.H.; Hazra, T.K.; Mitra, S. Role of acetylated human AP-endonuclease (APE1/Ref-1) in regulation of the parathyroid hormone gene. EMBO J. 2003, 22, 6299–6309. [Google Scholar] [CrossRef]
- Fuxreiter, M.; Tompa, P.; Simon, I.; Uversky, V.N.; Hansen, J.C.; Asturias, F.J. Malleable machines take shape in eukaryotic transcriptional regulation. Nat. Chem. Biol. 2008, 4, 728–737. [Google Scholar] [CrossRef]
- Tompa, P.; Fuxreiter, M. Fuzzy complexes: polymorphism and structural disorder in protein-protein interactions. Trends Biochem. Sci. 2008, 33, 2–8. [Google Scholar]
- Fan, J.; Wilson, D.M., 3rd. Protein-protein interactions and posttranslational modifications in mammalian base excision repair. Free Radic. Biol. Med. 2005, 38, 1121–1138. [Google Scholar] [CrossRef]
- Mitra, S.; Hegde, M.L.; Theriot, C.A.; Das, A.; Hegde, P.M.; Hazra, T.K. Complexity in repair of oxidative genome damage and its regulation. In Proceedings of Princess Takamatsu Symposium, Tokyo, Japan, 10–12 November 2009.
- Das, A.; Wiederhold, L.; Leppard, J.B.; Kedar, P.; Prasad, R.; Wang, H.; Boldogh, I.; Karimi-Busheri, F.; Weinfeld, M.; Tomkinson, A.E.; Wilson, S.H.; Mitra, S.; Hazra, T.K. NEIL2-initiated, APE-independent repair of oxidized bases in DNA: Evidence for a repair complex in human cells. DNA Repair (Amst) 2006, 5, 1439–1448. [Google Scholar] [CrossRef]
- Chattopadhyay, R.; Das, S.; Maiti, A.K.; Boldogh, I.; Xie, J.; Hazra, T.K.; Kohno, K.; Mitra, S.; Bhakat, K.K. Regulatory role of human AP-endonuclease (APE1/Ref-1) in YB-1-mediated activation of the multidrug resistance gene MDR1. Mol. Cell Biol. 2008, 28, 7066–7080. [Google Scholar] [CrossRef]
- Lindahl, T. Instability and decay of the primary structure of DNA. Nature 1993, 362, 709–715. [Google Scholar]
- Jaiswal, A.S.; Banerjee, S.; Panda, H.; Bulkin, C.D.; Izumi, T.; Sarkar, F.H.; Ostrov, D.A.; Narayan, S. A novel inhibitor of DNA polymerase beta enhances the ability of temozolomide to impair the growth of colon cancer cells. Molecular Cancer Res. MCR 2009, 7, 1973–1983. [Google Scholar] [CrossRef]
- Wilson, D.M., 3rd; Simeonov, A. Small molecule inhibitors of DNA repair nuclease activities of APE1. Cell Mol. Life Sci. 2010, 67, 3621–3631. [Google Scholar] [CrossRef]
- Bapat, A.; Glass, L.S.; Luo, M.; Fishel, M.L.; Long, E.C.; Georgiadis, M.M.; Kelley, M.R. Novel small-molecule inhibitor of apurinic/apyrimidinic endonuclease 1 blocks proliferation and reduces viability of glioblastoma cells. J. Pharmacol Exp. Ther. 2010, 334, 988–998. [Google Scholar]
- Fishel, M.L.; Jiang, Y.; Rajeshkumar, N.V.; Scandura, G.; Sinn, A.L.; He, Y.; Shen, C.; Jones, D.R.; Pollok, K.; Ivan, M.; Maitra, A.; Kelley, M.R. Impact of APE1/Ref-1 Redox Inhibition on Pancreatic Tumor Growth. Mol. Cancer Ther. 2011, 10, 1698–1708. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license ( http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Hegde, M.L.; Hegde, P.M.; Arijit, D.; Boldogh, I.; Mitra, S. Human DNA Glycosylase NEIL1’s Interactions with Downstream Repair Proteins Is Critical for Efficient Repair of Oxidized DNA Base Damage and Enhanced Cell Survival. Biomolecules 2012, 2, 564-578. https://doi.org/10.3390/biom2040564
Hegde ML, Hegde PM, Arijit D, Boldogh I, Mitra S. Human DNA Glycosylase NEIL1’s Interactions with Downstream Repair Proteins Is Critical for Efficient Repair of Oxidized DNA Base Damage and Enhanced Cell Survival. Biomolecules. 2012; 2(4):564-578. https://doi.org/10.3390/biom2040564
Chicago/Turabian StyleHegde, Muralidhar L., Pavana M. Hegde, Dutta Arijit, Istvan Boldogh, and Sankar Mitra. 2012. "Human DNA Glycosylase NEIL1’s Interactions with Downstream Repair Proteins Is Critical for Efficient Repair of Oxidized DNA Base Damage and Enhanced Cell Survival" Biomolecules 2, no. 4: 564-578. https://doi.org/10.3390/biom2040564