Rafts, Nanoparticles and Neural Disease

{kind=link}
{kind=link}
Abstract
:1. Introduction
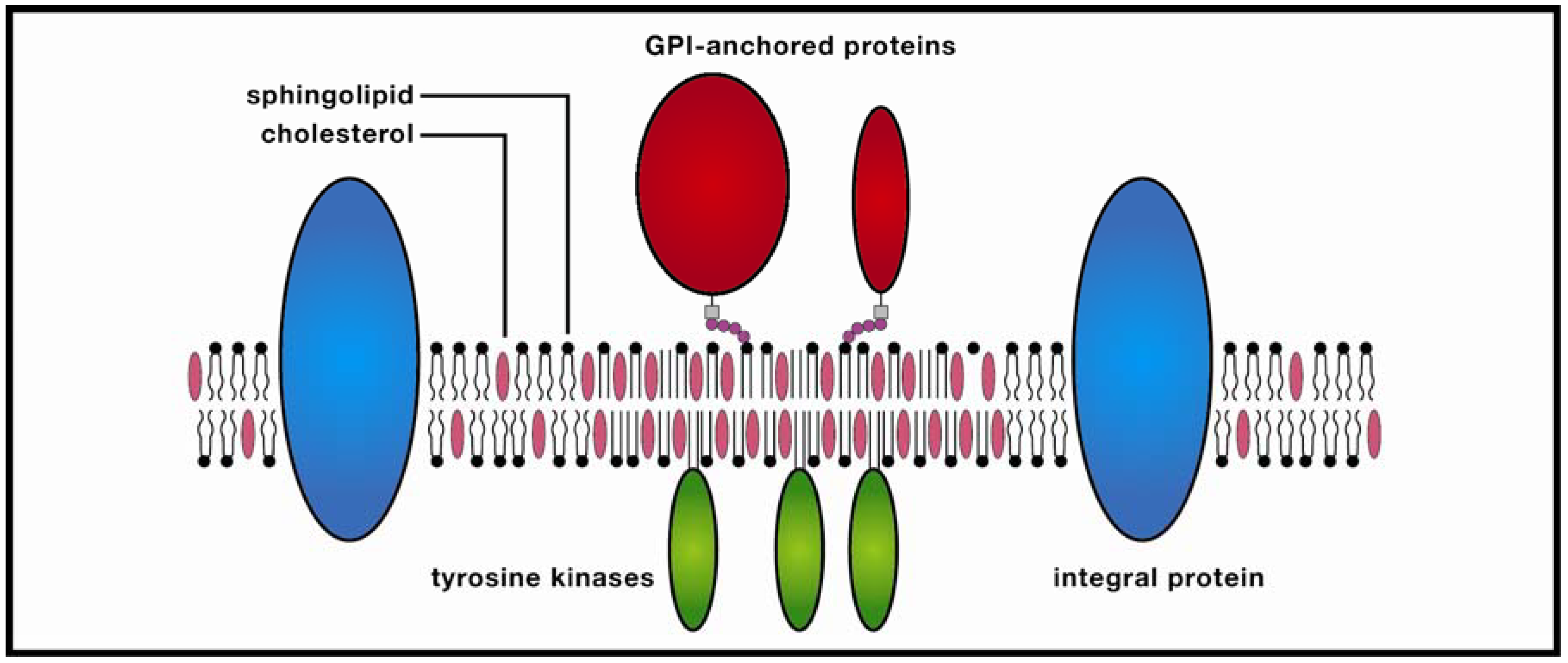
2. Methodological Issues: Do Membrane Rafts Exist?
2.1. Detergent Extraction and Cholesterol Depletion
2.2. Biophysical Methods
2.3. Raft Sizes and Lifetimes: Toward a Rapprochement

3. Rafts and Neural Disease
3.1. Epilepsy
3.2. Parkinson’s Disease
3.3. Alzheimer’s Disease
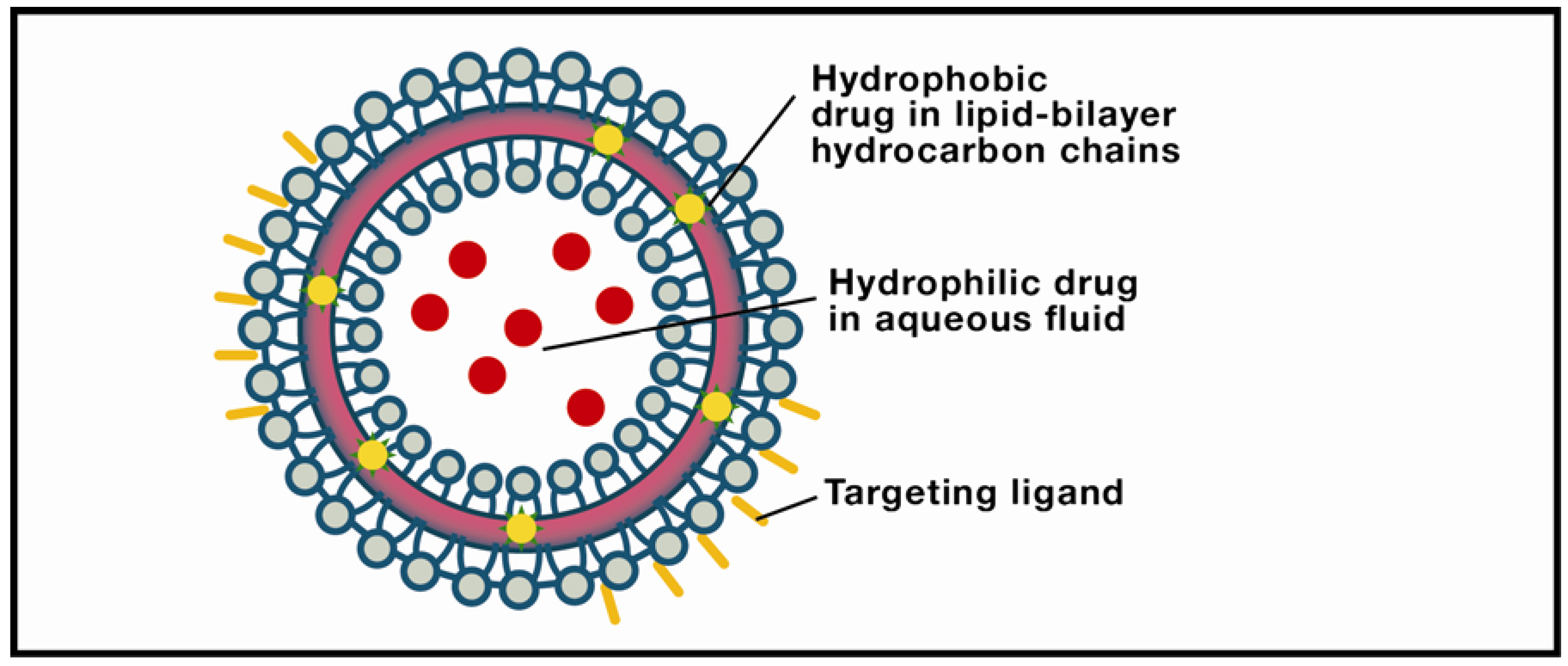
4. Lipid-based Nanocarriers: Liposomes and Solid Lipid Nanoparticles
4.1. Liposomes: Overview
4.2. Solid Lipid Nanoparticles: Overview
5. LBNs and Raft Targeting in Epilepsy, PD, and AD
5.1. Epilepsy
5.2. Parkinson’s Disease
5.3. Alzheimer’s Disease
6. Conclusions
References
- Bowman, W.C. Neuromuscular block. Br. J. Pharmacol. 2006, 147, 277–286. [Google Scholar] [CrossRef]
- Bronswijk, P.; Cohen, A.F. The first recordings of pharmacological effects. Br. J. Clin. Pharmacol. 2008, 66, 588–593. [Google Scholar]
- Rajendran, L.; Knölker, H.-J.; Simons, K. Subcellular targeting strategies for drug design and delivery. Nat. Rev. Drug Discov. 2010, 9, 29–42. [Google Scholar]
- Breunig, M.; Bauer, S.; Goepferich, A. Polymers and nanoparticles: Intelligent tools for intracellular targeting? Eur. J. Pharm. Biopharm. 2008, 68, 112–128. [Google Scholar] [CrossRef]
- Sengupta, P.; Baird, B.; Holowka, D. Lipid rafts, fluid/fluid phase separation, and their relevance to plasma membrane structure and function. Semin. Cell Dev. Biol. 2007, 18, 583–590. [Google Scholar] [CrossRef]
- Pike, L. The challenge of lipid rafts. J. Lipid Res. 2009, 50, 323–328. [Google Scholar] [CrossRef]
- Escribá, P.V. Membrane-lipid therapy: A new approach in molecular medicine. Trends Mol. Med. 2006, 12, 34–43. [Google Scholar] [CrossRef]
- Michel, V.; Bakovic, M. Lipid rafts in health and disease. Biol. Cell 2007, 99, 129–140. [Google Scholar] [CrossRef]
- Sandhiya, S.; Dkhar, S.A.; Surendiran, A. Emerging trends of nanomedicine: An overview. Fundam. Clin. Pharmacol. 2009, 23, 263–269. [Google Scholar] [CrossRef]
- Juliano, R.L.; Alam, R.; Dixit, V.; Min Kang, H. Cell-targeting and cell-penetrating peptides for delivery of therapeutic and imaging agents. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 324–335. [Google Scholar] [CrossRef]
- Head, B.P.; Hu, Y.; Finley, J.C.; Saldana, M.D.; Bonds, J.A.; Mivanohara, A.; Niesman, I.R.; Ali, S.S.; Murray, F.; Insel, P.A.; Roth, D.M.; Patel, H.H.; Patel, P.M. Neuron-targeted caveolin-1 protein enhances signaling and promotes arborization of primary neurons. J. Biol. Chem. 2011, 286, 33310–33321. [Google Scholar]
- Berta, A.I.; Boesze-Battaglia, K.; Magyar, A.; Szél, A.; Kiss, A.I. Localization of caveolin-1 and c-src in mature and differentiating photoreceptors: Raft proteins co-distribute with rhodopsin during development. J. Mol. Histol. 2011, 42, 523–533. [Google Scholar] [CrossRef]
- Nuñez, E.; Alonso-Torres, P.; Fornés, A.; Aragón, C.; López-Corcuera, B. The neuronal glycine transporter GLYT2 associates with membrane rafts: Functional modulation by lipid environment. J. Neurochem. 2008, 105, 2080–2090. [Google Scholar] [CrossRef]
- Cai, M.; Zhao, W.; Shang, X.; Jiang, J.; Ji, H.; Tang, Z.; Wang, H. Direct evidence of lipid rafts by in situ atomic force microscopy. Small 2012, 8, 1243–1250. [Google Scholar] [CrossRef]
- Lai, E.C. Lipid rafts make for slippery platforms. J. Cell Biol. 2003, 162, 365–370. [Google Scholar] [CrossRef]
- Calder, P.C.; Yaqoob, P. Lipid rafts—Composition, characterization, and controversie. J. Nutr. 2007, 137, 545–547. [Google Scholar]
- Fan, J.; Sammalkorpi, M.; Haataja, M. Formation and regulation of lipid microdomains in cell membranes: Theory, modeling, and speculation. FEBS Lett. 2009, 584, 1678–1684. [Google Scholar]
- Brown, D.A.; Rose, J.K. Sorting of GPI-anchored proteins to glycolipid-enriched membrane subdomains during transport to the apical cell surface. Cell 1992, 68, 533–544. [Google Scholar] [CrossRef]
- Pike, L.J. Lipid rafts: Bringing order to chaos. J. Lipid. Res. 2003, 44, 655–667. [Google Scholar] [CrossRef]
- Brown, D.A. Lipid rafts, detergent-resistant membranes, and raft targeting signals. Physiology 2006, 21, 430–439. [Google Scholar] [CrossRef]
- Day, C.A.; Kenworthy, A.K. Tracking microdomain dynamics in cell membranes. Biochim. Biophys. Acta 2009, 1788, 245–253. [Google Scholar] [CrossRef]
- Korade, Z.; Kenworthy, A.K. Lipid rafts, cholesterol, and the brain. Neuropharmacology 2008, 55, 1265–1273. [Google Scholar] [CrossRef]
- Kenworthy, A.K.; Edidin, M. Distribution of a glycoslphosphatidylinositol-anchored protein at the apical surface of MDCK cells examined at a resolution of < 100 Å using imaging fluorescence resonance energy transfer. J. Cell Biol. 1998, 142, 69–84. [Google Scholar] [CrossRef]
- Varma, R.; Mayor, S. GPI-anchored proteins are organized in submicron domains at the cell surface. Nature 1998, 394, 798–801. [Google Scholar]
- Scheiffle, P.; Roth, M.G.; Simons, K. Interactions of influenza virus haemagglutinin with sphingolipid-cholesterol membrane domains via its transmembrane domain. EMBO J. 1997, 16, 5501–5508. [Google Scholar] [CrossRef]
- Shvartsman, D.E.; Kotler, M.; Tall, R.D.; Roth, M.G.; Henis, Y.I. Differently anchored influenza hemagglutinin mutants display distinct interaction dynamics with mutual rafts. J. Cell Biol. 2003, 163, 879–888. [Google Scholar] [CrossRef]
- Niv, H.; Gutman, O.; Kloog, Y.; Henis, Y.I. Activated K-Ras and H-Ras display different interactions with saturable nonraft sites at the surface of live cells. J. Cell Biol. 2002, 157, 865–872. [Google Scholar]
- Eisenberg, S.; Schvartsman, D.E.; Ehrlich, M.; Henis, Y.I. Clustering of raft-associated proteins in the external membrane leaflet modulates internal leaflet H-Ras diffusion and signaling. Mol. Cell. Biol. 2006, 19, 7190–7200. [Google Scholar]
- Zhou, X.; Wang, L. Uses of single-particle tracking in living cells. Drug Discov. Ther. 2010, 4, 62–69. [Google Scholar]
- Kusumi, A.; Nakada, C.; Ritchie, K.; Murase, K.; Suzuki, K.; Murakoshi, H.; Kasai, R.S.; Kondo, J.; Fujiwara, T. Paradigm shift of the plasma membrane concept from the two-dimensional continuum fluid to the partitioned fluid: High-speed single-molecule tracking of membrane molecules. Ann. Rev. Biophys. Biomol. Struct. 2005, 34, 351–378. [Google Scholar] [CrossRef]
- Tsuji, A.; Ohnishi, S. Restriction of the lateral motion of band 3 in the erythrocyte membrane by the cytoskeletal network: Dependence on spectrin association state. Biochemistry 1986, 25, 133–139. [Google Scholar]
- Kusumi, A.; Koyama-Honda, I.; Suzuki, K. Molecular dynamics and interactions for creation of stimulation-induced stabilized rafts from small unstable steady-state rafts. Traffic 2004, 5, 213–230. [Google Scholar] [CrossRef]
- Anderson, R.G.W.; Jacobson, K. A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domain. Science 2002, 296, 1821–1824. [Google Scholar]
- Fantini, J.; Barrantes, F.J. Sphingolipid/cholesterol regulation of neurotransmitter receptor conformation and function. Biochim. Biophys. Acta. 2009, 1788, 2345–2361. [Google Scholar]
- Fantini, J.; Garmy, N.; Mahfoud, R.; Yahi, N. Lipid rafts: Structure, function, and role in HIV, Alzheimer’s, and prion diseases. Expert Rev. Mol. Med. 2002, 4, 1–22. [Google Scholar]
- Simons, K.; Ehehalt, R. Cholesterol, lipid rafts, and diseases. J. Clin. Invest. 2002, 110, 597–603. [Google Scholar]
- Escribá, P.V.; Gonzalez-Ros, J.M.; Gonñi, F.M.; Kinnunen, P.K.J.; Vigh, L.; Sánchez-Magraner, L.; Fernández, A.M.; Busquets, X.; Horváth, I.; Barceló-Coblijn, G. Membranes: A meeting point for lipids, proteins, and therapies. J. Cell. Mol. Med. 2008, 12, 829–875. [Google Scholar]
- Schengrund, C.-L. Lipid rafts: Keys to neurodegeneration. Brain Res. Bull. 2010, 82, 7–12. [Google Scholar] [CrossRef]
- Wallace, R. Neural membrane signaling platforms. Int. J. Mol. Sci. 2010, 11, 2421–2442. [Google Scholar] [CrossRef]
- Kwan, P.; Sander, J.W. The natural history of epilepsy: An epidemiological view. J. Neurol. Neurosurg. Psychiatry 2004, 75, 1376–1381. [Google Scholar] [CrossRef]
- Kirsch, H.E.; Grossman, M. Tracing the roots and routes of cognitive dysfunction in epilepsy. Neurology 2008, 71, 1854–1855. [Google Scholar] [CrossRef]
- Schachter, S.C. Seizure disorders. Med. Clin. North Am. 2009, 93, 343–351. [Google Scholar] [CrossRef]
- Ngugi, A.K.; Kariuki, S.M.; Bottomley, C.; Kleinschmidt, I.; Sander, J.W.; Newton, C.R. Incidence of epilepsy: A systematic review and meta-analysis. Neurology 2011, 77, 1005–1012. [Google Scholar]
- Rodin, E. The Prognosis of Patients with Epilepsy; Charles C. Thomas: Springfield, IL, USA, 1968. [Google Scholar]
- Hering, H.; Lin, C.-C.; Sheng, M. Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci. 2003, 23, 3262–3271. [Google Scholar]
- Schrattenholz, A.; Soskic, V. NMDA receptors are not alone: Dynamic regulation of NMDA receptor structure and function by neuregulins and transient cholesterol-rich membrane domains leads to disease-specific nuances of glutamate-signaling. Curr. Top. Med. Chem. 2006, 6, 663–686. [Google Scholar] [CrossRef]
- Hou, Q.; Huang, Y.; Amato, S.; Snyder, S.H.; Huganir, R.L.; Man, H.-Y. Regulation of AMPA receptor localization in lipid rafts. Mol. Cell. Neurosci. 2008, 38, 213–223. [Google Scholar] [CrossRef]
- Bowie, D. Ionotropic glutamate receptors & CNS disorders. CNS Neurol. Disord. Drug Targets 2008, 7, 129–143. [Google Scholar] [CrossRef]
- Kalia, L.V.; Kalia, S.K.; Salter, M.W. NMDA receptors in clinical neurology: Excitatory times ahead. Lancet Neurol. 2008, 7, 742–755. [Google Scholar] [CrossRef]
- Lonze, B.E.; Ginty, D.D. Function and regulation of CREB family transcription factors in the nervous system. Neuron 2002, 35, 605–623. [Google Scholar] [CrossRef]
- McEachem, J.C.; Shaw, C.A. An alternative to the LTP orthodoxy: A plasticity-pathology continuum model. Brain Res. Rev. 1996, 1, 51–92. [Google Scholar]
- McEachem, J.C.; Shaw, C.A. The plasticity-pathology continuum: Defining a role for the LTP phenomenon. J. Neurosci. Res. 1999, 58, 42–61. [Google Scholar] [CrossRef]
- Cooke, S.F.; Bliss, T.V.P. Plasticity in the human nervous system. Brain 2006, 129, 1659–1673. [Google Scholar]
- Meador, K.J. The basic science of memory as it applies to epilepsy. Epilepsia 2007, 48, 23–25. [Google Scholar] [CrossRef]
- Naegele, J. Epilepsy and the plastic mind. Epilepsy Curr. 2009, 9, 166–169. [Google Scholar] [CrossRef]
- Ghasemi, M.; Schachter, S.C. The NMDA receptor complex as a therapeutic target in epilepsy: A review. Epilepsy Behav. 2011, 22, 617–640. [Google Scholar] [CrossRef]
- Zhang, Y.; Su, P.; Liang, P.; Liu, T.; Liu, X.; Liu, X.-Y.; Zhang, B.; Han, T.; Zhu, Y.-B.; Yin, D.-M.; Li, J.; Zhou, Z.; Wang, K.-W.; Wang, Y. The DREAM protein negatively regulates the NMDA receptor through interaction with the NR1 subunit. J. Neurosci. 2010, 30, 7575–7586. [Google Scholar]
- Gomez-Villafuertes, R.; Torres, B.; Barrio, J.; Savignac, M.; Gabellini, N.; Rizzato, F.; Pintado, B.; Gutierrez-Adan, A.; Mellström, B.; Carafoli, E.; Naranjo, J.R. Downstream regulatory element antagonist modulator regulates Ca2+ homeostasis and viability in cerebellar neurons. J. Neurosci. 2005, 25, 10822–10830. [Google Scholar]
- Parkinson, J. An essay on the shaking palsy. J. Neuropsychiatry Clin. Neurosci. 2002, 14, 223–236. [Google Scholar] [CrossRef]
- Thomas, B.; Beal, M.F. Parkinson’s disease. Hum. Mol. Genet. 2007, 16, 183–194. [Google Scholar] [CrossRef]
- Morley, J.F.; Hurtig, H.J. Current understanding and management of Parkinson’s disease: Five new things. Neurology 2010, 75, S9–S15. [Google Scholar] [CrossRef]
- Cookson, M.R.; Hardy, J.; Lewis, P.A. Genetic neuropathology of Parkinson’s Disease. Int. J. Clin. Exp. Pathol. 2008, 1, 217–231. [Google Scholar]
- Cookson, M.R. Unraveling the role of defective genes. Prog. Brain Res. 2010, 183, 43–57. [Google Scholar] [CrossRef]
- Hardy, J. Genetic analysis of pathways to Parkinson disease. Neuron 2010, 68, 201–206. [Google Scholar] [CrossRef]
- Cicchetti, F.; Drouin-Ouellet, J.; Gross, R.E. Environmental toxins and Parkinson’s disease: What have we learned from pesticide-induced animal models? Trends Pharmacol. Sci. 2009, 30, 475–483. [Google Scholar] [CrossRef]
- Satake, W.; Nakabayashi, Y.; Mizuta, I.; Hirota, Y.; Ito, C.; Kubo, M. Genome-wide association study identifies common variants at four loci as genetic risk factors for Parkinson’s disease. Nat. Genet. 2009, 41, 1303–1307. [Google Scholar] [CrossRef]
- Simon-Sanchez, J.; Schulte, C.; Bras, J.M.; Sharma, M.; Gibbs, J.R.; Berg, D. Genome-wide association study reveals genetic risk underlying Parkinson’s disease. Nat. Genet. 2009, 41, 1308–1312. [Google Scholar] [CrossRef]
- Goetz, C.G. The history of Parkinson’s disease: Early clinical descriptions and neurological therapies. Cold Spring Harb. Perspect. Med. 2011, 1, a008862. [Google Scholar]
- Maguire-Zeiss, K.A. α-Synuclein: A therapeutic target for Parkinson’s disease. Pharmacol. Res. 2008, 58, 271–280. [Google Scholar] [CrossRef]
- Auluck, P.K.; Caraveo, G.; Lindquist, S. α-Synuclein: Membrane interactions and toxicity in Parkinson’s disease. Ann. Rev. Cell Dev. Biol. 2010, 26, 211–233. [Google Scholar]
- DeSantis, M.E.; Dersh, D. Preventing Parkinson’s pathology. Dis. Model. Mech. 2010, 3, 399–400. [Google Scholar] [CrossRef]
- Fortin, D.L.; Nemani, V.M.; Nakamura, K.; Edwards, R.H. The behavior of α-synuclein in neurons. Mov. Disord. 2010, 25, S21–S26. [Google Scholar]
- Franssens, V.; Boelen, E.; Anandhakumar, J.; Vanhelmont, T.; Büttner, S.; Winderickx, J. Yeast unfolds the road map toward α-synuclein-induced cell death. Cell Death Differ. 2010, 17, 746–753. [Google Scholar] [CrossRef]
- Nemani, V.M.; Lu, W.; Berge, V.; Nakamura, K.; Onoa, B.; Lee, M.K.; Chaudhry, F.A.; Nicoli, R.A.; Edwards, R.H. Increased expression of α-synuclein reduces neurotransmitter release by inhibiting synaptic vesicle reclustering after endocytosis. Neuron 2010, 65, 66–79. [Google Scholar] [CrossRef]
- Scott, D.A.; Taberean, I.; Tang, Y.; Cartier, A.; Masliah, E.; Roy, S. A pathologic cascade leading to synaptic dysfunction in α-synuclein-induced neurodegeneration. J. Neurosci. 2010, 30, 8083–8095. [Google Scholar]
- Sharma, M.; Barré, J.; Südhof, T.C. CSP α-promotes SNARE-complex assembly by chaperoning SNAP-25 during synaptic activity. Nat. Cell Biol. 2011, 13, 30–39. [Google Scholar] [CrossRef]
- Kubo, S.; Nemani, V.M.; Chalkey, R.J.; Anthony, M.D.; Hattori, N.; Mizuno, Y.; Edwards, R.H.; Fortin, D.L. A combinatorial code for the interaction of α-synuclein with membranes. J. Biol. Chem. 2005, 280, 31664–31672. [Google Scholar]
- Outeiro, T.F.; Lindquist, S. Yeast cells provide insight into alpha-synuclein biology and pathobiology. Science 2003, 302, 1772–1775. [Google Scholar] [CrossRef]
- Bodner, C.R.; Dobson, C.M.; Bax, A. Multiple tight phospholipid-binding modes of alpha-synuclein revealed by solution NMR spectroscopy. J. Mol. Biol. 2009, 390, 775–790. [Google Scholar]
- Devi, L.; Raghavendran, V.; Prabhu, B.M.; Avadhani, N.G.; Anandatheerthavarada, H.K. Mitochondrial import and accumulation of alpha-synuclein impair complex I in human dopaminergic neuronal cultures and Parkinson disease brain. J. Biol. Chem. 2008, 283, 9089–9100. [Google Scholar]
- Esteves, A.R.; Arduino, D.M.; Silva, D.F.F.; Oliveira, C.R.; Cardoso, S.M. Mitochondrial dysfunction: The road to alpha-synuclein oligomerization in PD. Parkinson Dis. 2011, 1–20. [Google Scholar]
- Keane, P.C.; Kurzawa, M.; Blain, P.G.; Morris, C.M. Mitochondrial dysfunction in Parkinson’s disease. Parkinson Dis. 2011, 2011, 1–18. [Google Scholar]
- Fortin, D.L.; Troyer, M.D.; Nakamura, K.; Kubo, S.; Anthony, M.D.; Edwards, R.H. Lipid rafts mediate the synaptic localization of α-synuclein. J. Neurosci. 2004, 24, 6715–6723. [Google Scholar] [CrossRef]
- Dawkins, R. The Blind Watchmaker: Why the Evidence of Evolution Reveals a Universe without Design; W.W. Norton: New York, NY, USA, 1986. [Google Scholar]
- Abe, K.; Kobayashi, N.; Sode, K.; Ikebukuro, K. Peptide ligand screening of α-synuclein aggregation modulators by in silico panning. BMC Bioinforma. 2007, 8, 1–7. [Google Scholar] [CrossRef]
- El-Agnaf, O.; Paleologol, K.E.; Greer, B.; Abogrein, A.M.; King, J.E.; Salem, S.A.; Fullwood, N.G.; Benson, F.E.; Hewitt, R.; Ford, K.J.; Martin, F.L.; Harriott, P.; Cookson, M.R.; Allsop, D. A strategy for designing inhibitors of α-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB J. 2004, 18, 1315–1317. [Google Scholar]
- Möller, H.J.; Graeber, M.B. The case described by Alois Alzheimer in 1911. Historical and conceptual perspectives based on the clinical record and neurohistological sections. Eur. Arch. Psychiatry 1998, 248, 111–122. [Google Scholar]
- Alzheimer’s Association. 2012 Alzheimer’s disease facts and figures. Alzheimers Dement. 2012, 8, 131–168. [CrossRef]
- Selkoe, D.J. Alzheimer’s disease: Genes, proteins, and therapy. Physiol. Rev. 2001, 81, 741–766. [Google Scholar]
- Zheng, H.; Jiang, M.; Trumbauer, M.E.; Sirinathsinghji, D.J.S.; Hopkins, R.; Smith, D.W.; Heavens, R.P.; Dawson, G.R.; Boyce, S.; Conner, M.W.; Stevens, K.A.; Slunt, H.H.; Sisodia, S.S.; Chen, H.Y.; van der Ploeg, L.H.T. β-amyloid precursor protein-deficient mice show reactive gliosis and decreased locomotor activity. Cell 1995, 81, 525–531. [Google Scholar]
- Cole, S.T.; Vassar, R. The role of amyloid precursor protein processing by BACE1, the β secretase in Alzheimer’s disease pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. [Google Scholar]
- Deuss, M.; Reiss, K.; Hartmann, D. Part-time alpha-secretases: The functional biology of ADAM 9, 10, and 17. Curr. Alzheimer Res. 2008, 5, 187–201. [Google Scholar] [CrossRef]
- Harris, B.; Pereira, I.; Parkin, E. Targeting ADAM 10 to lipid rafts in neuroblastoma SH-SY5Y cells impairs amyloidogenic processing of the amyloid precursor protein. Brain Res. 2009, 1296, 203–215. [Google Scholar] [CrossRef]
- Fahrenholz, F. Alpha secretase as a therapeutic target. Curr. Alzheimer Res. 2007, 4, 412–417. [Google Scholar] [CrossRef]
- Skovronsky, D.M.; Lee, V.M.-Y.; Trojanowski, J.Q. Neurodegenerative diseases: New concepts of pathogenesis and their therapeutic implications. Ann. Rev. Pathol. 2006, 1, 151–170. [Google Scholar] [CrossRef]
- Selkoe, D.J. Alzheimer’s disease is a synaptic failure. Science 2002, 298, 789–791. [Google Scholar] [CrossRef]
- Hong, L.; Koelsch, G.; Lin, X.; Wu, S.; Terzyan, S.; Ghosh, A.K.; Zhang, X.C.; Tang, J. Structure of the protease domain of memapsin 2 (beta-secretase) complexed with inhibitor. Science 2000, 290, 150–153. [Google Scholar]
- Tomita, T. Secretase inhibitors and modulators of Alzheimer’s disease. Expert Rev. Neurother. 2009, 9, 661–679. [Google Scholar] [CrossRef]
- Cheng, H.; Vetrivel, K.S.; Gong, P.; Meckler, X.; Parent, A.; Thinakaran, G. Mechanisms of disease: New therapeutic strategies for Alzheimer’s disease—Targeting APP processing in lipid rafts. Nat. Clin. Pract. Neurol. 2007, 3, 374–382. [Google Scholar]
- Ehehalt, R.; Keller, P.; Haass, C.; Thiele, C.; Simons, K. Amyloidogenic processing of the Alzheimer’s β amyloid precursor protein depends on lipid rafts. J. Cell Biol. 2003, 160, 113–123. [Google Scholar] [CrossRef]
- Simons, M.; Keller, P.; de Strooper, B.; Bayreuther, K.; Dotti, C.G.; Simons, K. Cholesterol depletion inhibits the generation of beta-amyloid in hippocampal neurons. Proc. Natl. Acad. Sci. USA 1998, 95, 6460–6464. [Google Scholar]
- Lakshmana, M.K.; Roy, S.; Mi, K.; Kang, D.E. Amyloidogenic processing of APP in lipid rafts. Open Biol. J. 2010, 3, 21–31. [Google Scholar] [CrossRef]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid β-peptide: Isoform-specific effects and implications for late-onset Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar]
- Querfurth, H.W.; LaFerla, F.M. Mechanisms of disease: Alzheimer’s disease. N. Engl. J. Med. 2010, 362, 329–344. [Google Scholar] [CrossRef]
- Fassbender, K.; Simons, M.; Bergmann, C.; Stroick, M.; Lutjohann, D.; Keller, P.; Runz, H.; Kuhl, S.; Bertsch, T.; von Bergmann, K.; Hennerici, M.; Bayreuther, K.; Hartmann, T. Simvastatin strongly reduces levels of Alzheimer’s disease beta-amyloid peptides Abeta 42 and Abeta 40 in vitro and in vivo. Proc. Natl. Acad. Sci. USA 2001, 98, 5856–5861. [Google Scholar]
- Refolo, L.M.; Sambamurti, K.; Efthimiopoulos, S.; Pappolla, M.A.; Robakis, N.K. Evidence that secretase cleavage of cell surface amyloid precursor occurs after normal endocytic internalization. J. Neurosci. Res. 1995, 40, 694–706. [Google Scholar] [CrossRef]
- Harder, T.; Scheiffele, P.; Verkade, P.; Simons, K. Lipid domain structure of the plasma membrane revealed by patching of membrane components. J. Cell Biol. 1998, 141, 929–942. [Google Scholar] [CrossRef]
- Marquer, C.; Devauges, V.; Cossec, J.-C.; Liot, G.; Lécart, S.; Sadou, F.; Duyckaerts, C.; Lévêque-Fort, S.; Potier, M.C. Local cholesterol increase triggers amyloid precursorprotein-Bace1 clustering in lipid rafts and rapid endocytosis. FASEB J. 2011, 25, 1295–1305. [Google Scholar]
- Cole, S.L.; Vassar, R. The role of amyloid precursor protein processing by BACE1, the β-secretase, in Alzheimer disease pathophysiology. J. Biol. Chem. 2008, 283, 29621–29625. [Google Scholar] [CrossRef]
- Cataldo, A.M.; Petanceska, S.; Terio, N.B.; Peterhoff, C.M.; Durham, R.; Mercken, M.; Mehta, P.D.; Buxbaum, J.; Haroutunian, V.; Nixon, R.A. Abeta localization in abnormal endosomes: Association with earliest Abeta elevations in AD and Down syndrome. Neurobiol. Aging 2004, 25, 1263–1272. [Google Scholar]
- Jiang, Y.; Mullaney, K.A.; Peterhoff, C.M.; Che, S.; Schmidt, S.D.; Boyer-Boiteau, A.; Ginsberg, S.D.; Cataldo, A.M.; Mathews, P.M.; Nixon, R.A. Alzheimer’s-related endosome dysfunction in Down syndrome is Abeta-independent but requires APP and is reversed by BACE-1 inhibition. Proc. Natl. Acad. Sci. USA 2010, 107, 1630–1635. [Google Scholar]
- Savonenko, A.V.; Melnikova, T.; Laird, F.M.; Stewart, K.-A.; Price, D.L.; Wong, P.C. Alteration of BACE1-dependent NRG1/ErbB4 signaling and schizophrenia-like phenotypes in BACE1-null mice. Proc. Natl. Acad. Sci. USA 2008, 105, 5585–5590. [Google Scholar]
- Sankaranarayanan, S.; Price, E.A.; Wu, G.; Crouthamel, M.C.; Shi, X.P.; Tugusheva, K.; Tyler, K.X.; Kahana, J.; Ellis, J.; Jin, L.; Steele, T.; Stachel, S.; Coburn, C.; Simon, A.J. In vivo beta-secretase 1 inhibition leads to brain Abeta lowering and increased alpha-secretase processing of amyloid precursor protein without effect on neuregulin-1. J. Pharmacol. Exp. Ther. 2008, 324, 3431–3433. [Google Scholar]
- McConlogue, L.; Buttini, M.; Anderson, J.P.; Brigham, E.F.; Chen, K.S.; Freedman, S.B.; Games, D.; Johnson-Wood, K.; Lee, M.; Zeller, M.; Liu, W.; Motter, R.; Sinha, S. Partial reduction of BACE1 has dramatic effects on Alzheimer plaque and synaptic pathology in APP transgenic mice. J. Biol. Chem. 2007, 282, 26326–26334. [Google Scholar]
- Feynman, R.P. There’s plenty of room at the bottom: An invitation to enter a new field of physics. J. Microelectromec. Syst. 1992, 1, 60–66. [Google Scholar] [CrossRef]
- Silva, G.A. Introduction to nanotechnology and its applications to medicine. Surg. Neurol. 2004, 61, 216–220. [Google Scholar] [CrossRef]
- Sandhiya, S.; Dkhar, S.A.; Surendiran, A. Emerging trends of nanomedicine—An overview. Fundam. Clin. Pharmacol. 2009, 23, 263–269. [Google Scholar] [CrossRef]
- Silva, G.A. Neuroscience nanotechnology: Progress, opportunities and challenges. Nat. Rev. Neurosci. 2006, 7, 65–74. [Google Scholar] [CrossRef]
- Lee, N.; Hyeon, T. Designed synthesis of uniformly sized iron oxide nanoparticles for efficient magnetic resonance imaging contrast agents. Chem. Soc. Rev. 2012, 41, 2575–2589. [Google Scholar] [CrossRef]
- Lu, C.; Stewart, D.J.; Lee, J.J.; Ji, L.; Ramesh, R.; Jayachandran, G.; Nunez, M.I.; Wistuba, I.I.; Erasmus, J.J.; Hicks, M.E.; Grimm, E.A.; Reuben, J.M.; Baladandayuthapani, V.; Templeton, N.S.; McMannis, J.D.; Roth, J.A. Phase I clinical trial of systemically administered TUSC2 (FUS1)-nanoparticles mediating functional gene transfer in humans. PLoS One 2012, 7, e34833. [Google Scholar]
- Rupp, R.; Rosenthal, S.L.; Stanberry, L.R. VivaGelTM (SPL7013 Gel): A candidate dendrimer-microbicide for the prevention of HIV and HSV infection. Int. J. Nanomed. 2007, 2, 561–566. [Google Scholar]
- Morachis, J.M.; Mahmoud, E.A.; Almutairi, A. Physical and chemical strategies for therapeutic delivery by using polymeric nanoparticles. Pharmacol. Rev. 2012, 64, 505–519. [Google Scholar] [CrossRef]
- Barandeh, F.; Nguyen, P.-L.; Kumar, R.; Iacobucci, G.J.; Kuznicki, M.L.; Kosterman, A.; Bergey, E.J.; Prasad, P.N.; Gunawardena, S. Organically modified silica nanoparticles are biocompatible and can be targeted to neurons in vivo. PLoS One 2012, 2, 1–15. [Google Scholar]
- Deamer, D.W. From “Banghasomes” to liposomes: A memoir of Alec Bangham, 1921-2010. FASEB J. 2010, 24, 1308–1310. [Google Scholar] [CrossRef]
- Torchilin, V.P. Recent advances with liposomes as pharmaceutical carriers. Nat. Rev. Drug Discov. 2005, 4, 145–160. [Google Scholar] [CrossRef]
- Schnyder, A.; Huwyler, J. Drug transport to brain with targeted liposomes. NeuroRx 2005, 2, 99–107. [Google Scholar] [CrossRef]
- Martins, S.; Sarmento, B.; Ferreira, D.C.; Souto, E.B. Lipid-based colloidal carriers for peptide and protein delivery—Liposomes versus lipid nanoparaticles. Int. J. Nanomed. 2007, 2, 595–607. [Google Scholar]
- Micheli, M.R.; Bova, R.; Magini, A.; Polidoro, M.; Emiliani, C. Lipid-based nanocarriers for CNS-targeted drug delivery. Recent Pat. CNS Drug Discov. 2012, 1, 71–86. [Google Scholar]
- Juliano, R.I.; Alam, R.; Dixit, V.; Kang, H.M. Cell-targeting and cell-penetrating peptides for delivery of therapeutic and imaging agents. Wiley Interdiscip. Rev. Nanomed. Nanobiotechnol. 2009, 1, 324–335. [Google Scholar] [CrossRef]
- Zhang, Y.; Zhang, Y.F.; Bryant, J.; Charles, A.; Boado, R.J.; Pardridge, W.M. Intravenous RNA interference gene therapy targeting the human epidermal growth factor receptor prolongs survival in intracranial brain cancer. Clin. Cancer Res. 2004, 1, 3667–3677. [Google Scholar]
- Gunawan, R.C.; Auguste, D.T. Immunoliposomes that target endothelium in vitro are dependent on lipid raft formation. Mol. Pharm. 2010, 7, 1569–1575. [Google Scholar] [CrossRef]
- Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C. Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 10, 454–477. [Google Scholar]
- Uner, M.; Yener, G. Importance of solid lipid nanoparticles (SLN) in various administration routes and future perspectives. Int. J. Nanomed. 2007, 2, 289–300. [Google Scholar]
- Kaur, I.P.; Bhandari, R.; Bhandari, S.; Kakkar, V. Potential of solid lipid nanoparticles in brain targeting. J. Control. Release 2008, 127, 97–109. [Google Scholar] [CrossRef]
- Sinha, V.R.; Srivastava, S.; Goel, H.; Jindal, V. Solid lipid nanoparticles (SLN’S)—Trends and implications in drug targeting. Int. J. Adv. Pharm. Sci. 2010, 1, 212–238. [Google Scholar]
- Jawahar, N.; Gowthamarajan, K.; Meyyanathan, S.N.; Sood, S. Brain delivery by solid lipid nanoparticles for CNS drugs. Int. J. Pharm. Res. Dev. 2011, 3, 206–216. [Google Scholar]
- Eldem, T.; Speiser, P.; Hincal, A. Optimization of spray-dried and –congealed lipid micropellets and characterization of their surface morphology by scanning electron microscopy. Pharm. Res. 1991, 8, 47–54. [Google Scholar] [CrossRef]
- Puri, A.; Loomis, K.; Smith, B.; Lee, J.H.; Yavlovich, A.; Heldman, E.; Blumenthal, R. Lipid-based nanoparticles as pharmaceutical drug carriers: From concepts to clinic. Crit. Rev. Ther. Drug Carrier Syst. 2009, 26, 523–580. [Google Scholar] [CrossRef]
- Jain, A.; Agarwal, A.; Majumder, S.; Lariya, N.; Khaya, A.; Agrawal, H.; Majumdar, S.; Agrawal, G.P. Mannosylated solid lipid nanoparticles as vectors for site-specific delivery of an anti-cancer drug. J. Control. Release 2010, 20, 359–367. [Google Scholar]
- Blasi, P.; Giovagnoli, S.; Schoubben, A.; Ricci, M.; Rossi, C. Solid lipid nanoparticles for targeted brain drug delivery. Adv. Drug Deliv. Rev. 2007, 10, 454–477. [Google Scholar]
- Kreuter, J.; Petrov, V.E.; Kharkevich, D.A.; Alyautdin, R.N. Influence of the type of surfactant on the analgesic effects induced by the peptide dalagin after its delivery across the blood-brain barrier using surfactant-coated nanoparticles. J. Control. Release 1997, 49, 81–87. [Google Scholar] [CrossRef]
- Alyautdin, R.N.; Petrov, V.E.; Langer, K.; Berthold, A.; Kharkevich, D.A.; Kreuter, J. Delivery of loperamide across the blood-brain barrier with polysorbate-80 coated polybutylcyanoacrylate nanoparticles. Pharm. Res. 1997, 14, 325–328. [Google Scholar] [CrossRef]
- Friese, A.; Seiller, E.; Quack, G.; Lorenz, B.; Kreuter, J. Increase of the duration of the anticonvulsive activity of a novel NMDA receptor antagonist using poly(butylcyanoacrylate) nanoparticles as a parenteral controlled release system. Eur. J. Pharm. Biopharm. 2000, 49, 103–109. [Google Scholar] [CrossRef]
- Gulyaev, A.E.; Gelperina, S.E.; Skidan, I.N.; Antropov, A.S.; Kivman, G.Y.; Kreuter, J. Significant transport of doxorubicin into the brain with polysorbate 80-coated nanoparticles. Pharm. Res. 1999, 16, 1564–1569. [Google Scholar] [CrossRef]
- Wilson, B.; Samanta, M.K.; Santhi, K.; Perumal, K.; Kumar, S.; Paramakrishnan, N.; Suresh, B. Targeted delivery of tacrine into the brain with polysorbate 80-coated poly(butylcyanoacrylate) nanoparticles. Eur. J. Pharm. Biopharm. 2008, 70, 75–84. [Google Scholar] [CrossRef]
- Toffano, G.; Mazzari, S.; Zanotti, A.; Bruni, A. Synergistic effect of phosphatidylserine with gamma-aminobutyric acid in antagonizing the isoniazid-induced convulsions in mice. Neurochem. Res. 1984, 9, 1065–1073. [Google Scholar] [CrossRef]
- Loeb, C.; Benassi, E.; Besio, G.; Maffini, M.; Tanganelli, P. Liposome-entrapped GABA modifies behavioral and electrographic changes of penicillin-induced epileptic activity. Neurology 1982, 32, 1234–1238. [Google Scholar]
- Snodgrass, S.R. GABA and epilepsy: Their complex relationship and the evolution of our understanding. J. Child Neurol. 1992, 7, 77–86. [Google Scholar] [CrossRef]
- Bennewitz, M.F.; Saltzman, M.W. Nanotechnology for the delivery of drugs to the brain for epilepsy. Neurotherapeutics 2009, 6, 323–336. [Google Scholar] [CrossRef]
- Ogden, K.K.; Traynelis, S.F. New advances in NMDA receptor pharmacology. Trends Pharmacol. Sci. 2011, 32, 726–733. [Google Scholar] [CrossRef]
- Klassen, T.; Davis, C.; Goldman, A.; Burgess, D.; Chen, T.; Wheeler, D.; McPherson, J.; Bourquin, T.; Lewis, L.; Villasana, D.; Morgan, M.; Muzny, D.; Gibbs, R.; Noebels, J. Exome sequencing of ion channel genes reveals complex profiles confounding personal risk assessment in epilepsy. Cell 2011, 145, 1036–1048. [Google Scholar] [CrossRef]
- During, M.J.; Freese, A.; Deutsch, A.Y.; Kibat, P.G.; Sabel, B.A.; Langer, R.; Roth, R.H. Biochemical and behavioral recovery in a rodent model of Parkinson’s disease following stereotactic implantation of dopamine-containing liposomes. Exp. Neurol. 1992, 115, 193–199. [Google Scholar] [CrossRef]
- Jain, N.K.; Rana, N.C.; Jain, S.K. Brain drug delivery system bearing dopamine hydrochloride for effective management of parkinsonism. Drug Dev. Ind. Pharm. 1998, 24, 671–675. [Google Scholar] [CrossRef]
- Khare, P.; Jain, A.; Jain, N.K.; Soni, V.; Jain, S.K. Glutamate-conjugated liposomes of dopamine hydrochloride for effective management of Parkinsonism’s. PDA J. Pharm. Sci.Technol. 2009, 63, 372–379. [Google Scholar]
- Zeevalk, G.D.; Razmpour, R.; Bernard, L.P. Glutathione and Parkinson’s disease: Is this the elephant in the room? Biomed. Pharmacother. 2008, 62, 236–249. [Google Scholar] [CrossRef]
- Martin, H.L.; Teismann, P. Glutathione—A review on its role and significance in Parkinson’s disease. FASEB J. 2009, 23, 3263–3272. [Google Scholar] [CrossRef]
- Lecker, S.H.; Goldberg, A.L.; Mitch, W.E. Protein degradation by the ubiquitin-proteasome pathway in normal and disease states. J. Am. Soc. Nephrol. 2006, 17, 1807–1819. [Google Scholar] [CrossRef]
- Zeevalk, G.D.; Manzino, L.; Sonsalla, P.K.; Bernard, L.P. Characterization of intracellular elevation of glutathione (GSH) with glutathione monoethyl ester and GSH in brain and neuronal cultures: Relevance to Parkinson’s disease. Exp. Neurol. 2007, 203, 512–520. [Google Scholar] [CrossRef]
- Wade, L.A.; Brady, H.M. Cysteine and cystine transport at the blood-brain barrier. J. Neurochem. 1981, 37, 730–734. [Google Scholar]
- Zeevalk, G.D.; Bernard, L.P.; Guilford, F.T. Liposomal-glutathione provides maintenance of intracellular glutathione and neuroprotection in mesencephalic neuronal cells. Neurochem. Res. 2010, 35, 1575–1587. [Google Scholar] [CrossRef]
- Gardoni, F.; Ghiglieri, V.; Luca, M.; Calabresi, P. Assemblies of glutamate receptor subunits with post-synaptic density proteins and their alterations in Parkinson’s disease. Prog. Brain Res. 2010, 183, 169–182. [Google Scholar] [CrossRef]
- Suzuki, T.; Zhang, J.; Miyazawa, S.; Liu, Q.; Farzan, M.R.; Yao, W.D. Association of membrane rafts and postsynaptic density: Proteomics, biochemical, and ultrastructural analyses. J. Neurochem. 2011, 119, 64–77. [Google Scholar] [CrossRef]
- Puthenveedu, M.A.; Yudowski, G.A.; von Zastrow, M. Endocytosis of neurotransmitter receptors: Location matters. Cell 2007, 130, 988–989. [Google Scholar] [CrossRef]
- El-Agnaf, O.; Paleologol, K.E.; Greer, B.; Abogrein, A.M.; King, J.E.; Salem, S.A.; Fullwood, N.G.; Benson, F.E.; Hewitt, R.; Ford, K.J.; Martin, F.L.; Harriott, P.; Cookson, M.R.; Allsop, D. A strategy for designing inhibitors of α-synuclein aggregation and toxicity as a novel treatment for Parkinson’s disease and related disorders. FASEB J. 2004, 18, 1315–1317. [Google Scholar]
- Ghosh, A.K.; Gemma, S.; Tang, J. β-Secretase as a therapeutic target for Alzheimer’s disease. Neurotherapeutics 2008, 5, 399–408. [Google Scholar] [CrossRef]
- Meredith, J.E., Jr.; Thompson, L.A.; Toyn, J.H.; Marcin, L.; Barten, D.M.; Marcinkeviciene, J.; Kopcho, L.; Kim, Y.; Lin, A.; Guss, V.; Burton, C.; Iben, L.; Polson, C.; Cantone, J.; Ford, M.; Drexler, D.; Fiedler, T.; Lentz, K.A.; Grace, J.E., Jr.; Kolb, J.; Corsa, J.; Pierdominico, M.; Jones, K.; Olsen, R.E.; Macor, J.E.; Albright, C.F. P-glycoprotein efflux and other factors limit brain amyloid beta reduction by beta-site amyloid precursor protein-cleaving enzyme 1 inhibitors in mice. J. Pharmacol. Exp. Ther. 2008, 326, 502–513. [Google Scholar]
- Mutlu, N.B.; Değim, Z.; Yilmaz, S.; Eşsiz, D.; Nacar, A. New perspective for the treatment of Alzheimer diseases: Liposomal rivastigmine formulations. Drug Dev. Ind. Pharm. 2011, 37, 775–789. [Google Scholar] [CrossRef]
- Phaconpai, W.; Wattanathorn, J.; Muchimapura, S.; Tong-un, T.; Preechagoon, D. Neuroprotective effect of quercetin encapsulated liposomes: A novel therapeutic strategy against Alzheimer’s disease. Am. J. Appl. Sci. 2010, 7, 480–485. [Google Scholar] [CrossRef]
- Mourtas, S.; Canovi, M.; Zona, C.; Aurilia, D.; Niarakis, A.; LaFerla, B.; Salmona, M.; Nicotra, F.; Gobbi, M.; Antimisiaris, S.G. Curcumin-decorated nanoliposomes with very high affinity for amyloid-β1-42 peptide. Biomaterials 2011, 32, 1635–1645. [Google Scholar]
- Picone, P.; Bondi, M.L.; Montana, G.; Bruno, A.; Pitarresi, G.; Giammona, G.; Di Carlo, M. Ferulic acid inhibits oxidative stress and cell death induced by Ab oligomers: Improved delivery by solid lipid nanoparticles. Free Radic. Res. 2009, 43, 1133–1145. [Google Scholar] [CrossRef]
- DeMattos, R.B.; Bales, K.R.; Cummins, D.J.; Dodart, J.C.; Paul, S.M.; Holtzman, D.M. Peripheral anti-A beta antibody alters CNS and plasma A beta clearance and decreases brain A beta burden in a mouse model of Alzheimer’s disease. Proc. Natl. Acad. Sci. USA 2001, 98, 8850–8855. [Google Scholar]
- Gobbi, M.; Re, F.; Canovi, M.; Beeg, M.; Gregori, M.; Sesana, S.; Sonnino, S.; Brogioli, D.; Musicanti, C.; Gasco, P.; Salmona, M.; Masserini, M.E. Lipid-based nanoparticles with high binding affinity for amyloid-beta1-42 peptide. Biomaterials 2010, 31, 6519–6529. [Google Scholar]
- Muhs, A.; Hickman, D.T.; Pihlgren, M.; Chuard, N.; Giriens, V.; Meerschman, C.; van der Auwera, I.; van Leuven, F.; Sugawara, M.; Weingertner, M.C.; Bechinger, B.; Greferath, R.; Kolonko, N.; Nagel-Steger, L.; Riesner, D.; Brady, R.O.; Pfeifer, A.; Nicolau, C. Liposomal vaccines with conformation-specific amyloid peptide antigens define immune response and efficacy in APP transgenic mice. Proc. Natl. Acad. Sci. USA 2007, 104, 9810–9815. [Google Scholar]
- Skovronsky, D.M.; Lee, V.M.; Trojanowski, J.Q. Neurodegenerative diseases: New concepts of pathogenesis and their therapeutic implications. Annu. Rev. Pathol. 2006, 1, 151–170. [Google Scholar] [CrossRef]
- Fasman, G.D.; Perczel, A.; Moore, C.D. Solubilization of beta-amyloid-(1-42)-peptide: Reversing the beta-sheet conformation induced by aluminum with silicates. Proc. Natl. Acad. Sci. USA 1995, 92, 369–371. [Google Scholar] [CrossRef]
- Eckert, G.P.; Chang, S.; Eckmann, J.; Copanaki, E.; Hagi, S.; Hener, U.; Müller, W.E.; Kögel, D. Liposome-incorporated DHA increases neuronal survival by enhancing non-amyloidogenic APP processing. Biochim. Biophys. Acta 2011, 1808, 236–243. [Google Scholar] [CrossRef]
- Kojro, E.; Gimpi, G.; Lammich, S.; Marz, W.; Fahrenholz, F. Low cholesterol stimulates the nonamyloidogenic pathway by its effect on the alpha-secretase ADAM 10. Proc. Natl. Acad. Sci. USA 2001, 98, 5815–5820. [Google Scholar]
- Cossec, J.C.; Simon, A.; Marquer, C.; Moldrich, R.X.; Leterrier, C.; Rossier, J.; Duyckaerts, C.; Lenkei, Z.; Poitier, M.C. Clathrin-dependent APP endocytosis and Abeta secretion are highly sensitive to the level of plasma membrane cholesterol. Biochim. Biophys. Acta 2010, 1801, 846–852. [Google Scholar] [CrossRef]
- Rajendran, L.; Schneider, A.; Schlechtingen, G.; Weidlich, S.; Ries, J.; Braxmeier, T.; Schwille, P.; Schulz, J.B.; Schroeder, C.; Simons, M.; Jennings, G.; Knölker, H.J.; Simons, K. Efficient inhibition of the Alzheimer’s disease beta-secretase by membrane targeting. Science 2008, 320, 520–523. [Google Scholar]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Gulati, V.; Wallace, R. Rafts, Nanoparticles and Neural Disease. Nanomaterials 2012, 2, 217-250. https://doi.org/10.3390/nano2030217
Gulati V, Wallace R. Rafts, Nanoparticles and Neural Disease. Nanomaterials. 2012; 2(3):217-250. https://doi.org/10.3390/nano2030217
Chicago/Turabian StyleGulati, Vishal, and Ron Wallace. 2012. "Rafts, Nanoparticles and Neural Disease" Nanomaterials 2, no. 3: 217-250. https://doi.org/10.3390/nano2030217
APA StyleGulati, V., & Wallace, R. (2012). Rafts, Nanoparticles and Neural Disease. Nanomaterials, 2(3), 217-250. https://doi.org/10.3390/nano2030217