Sustained Delivery of Chondroitinase ABC from Hydrogel System




 ,
,
Abstract
:
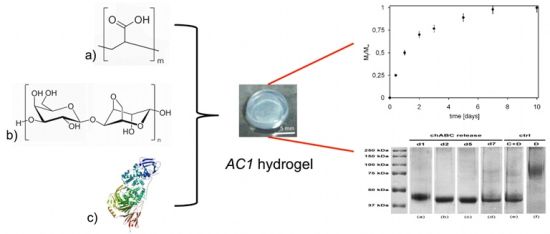{kind=link}
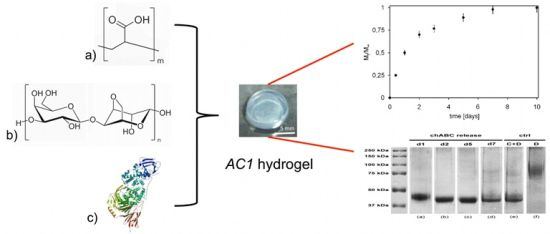{kind=link}
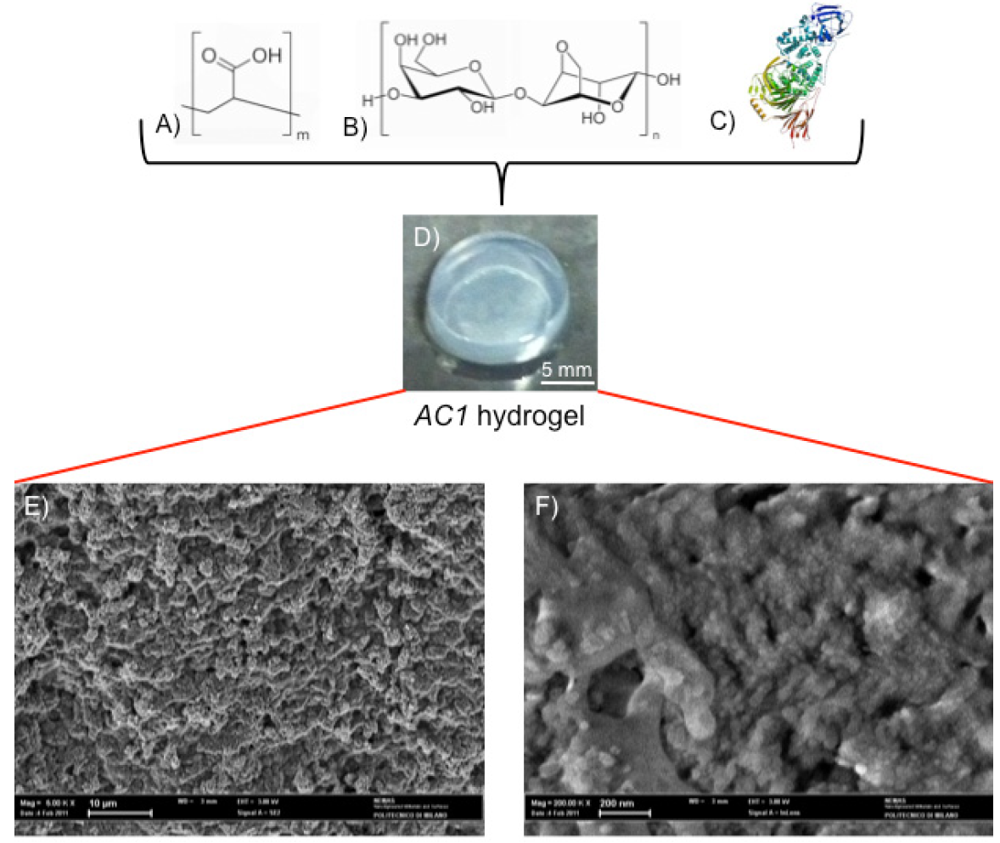{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Experimental Section
2.1. Hydrogel Synthesis
2.2. Morphological Studies: Environmental Scanning Electron Microscopy (E/SEM) Analysis
2.3. Rheology
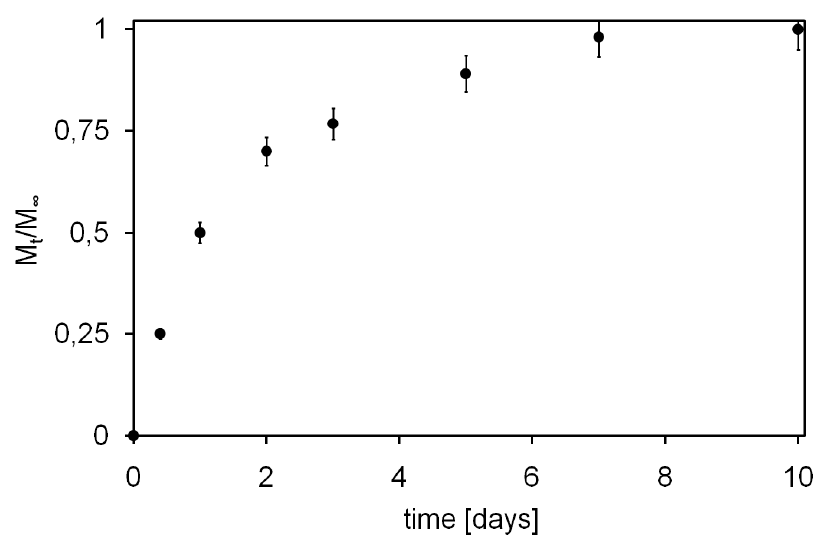
2.4. Tx Loading and Release Studies
2.5 Delivery and Enzymatic Activity Evaluation
2.6. Statistical Analysis
3. Results and Discussion

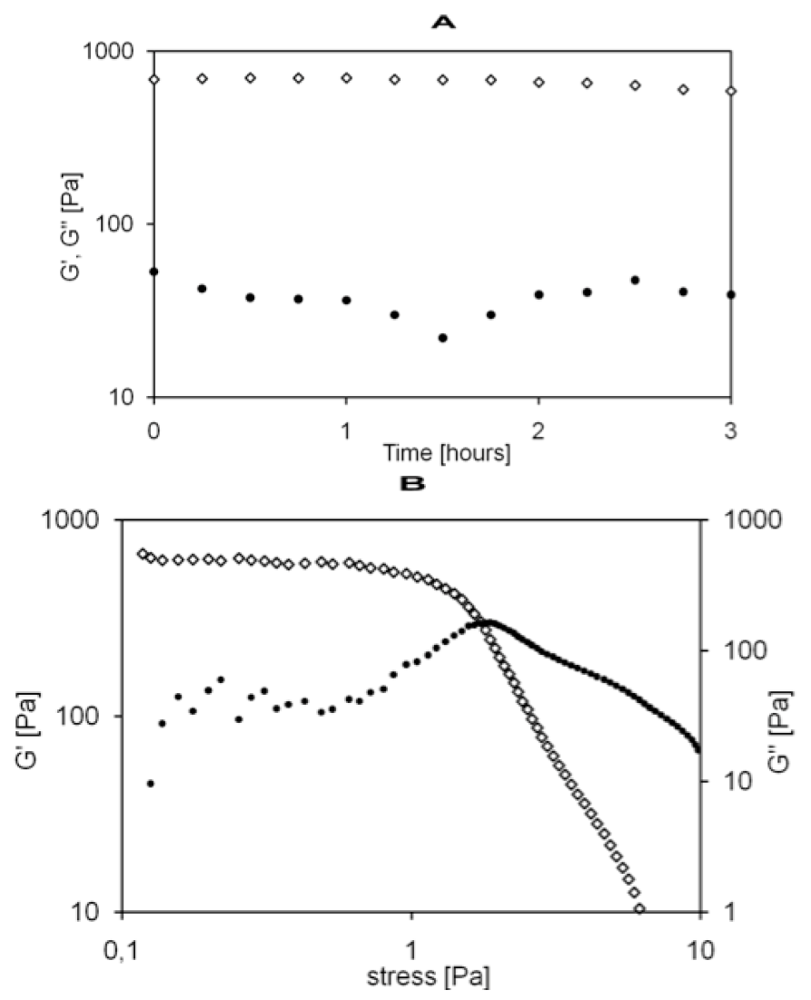
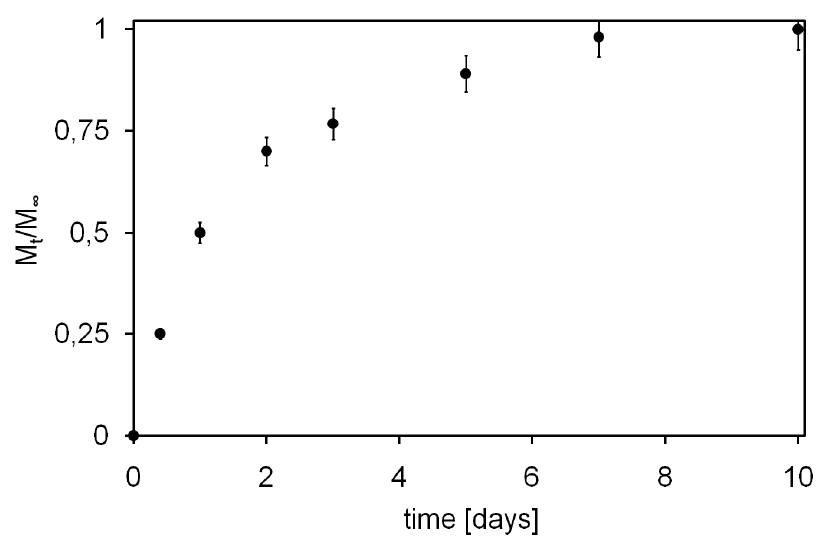
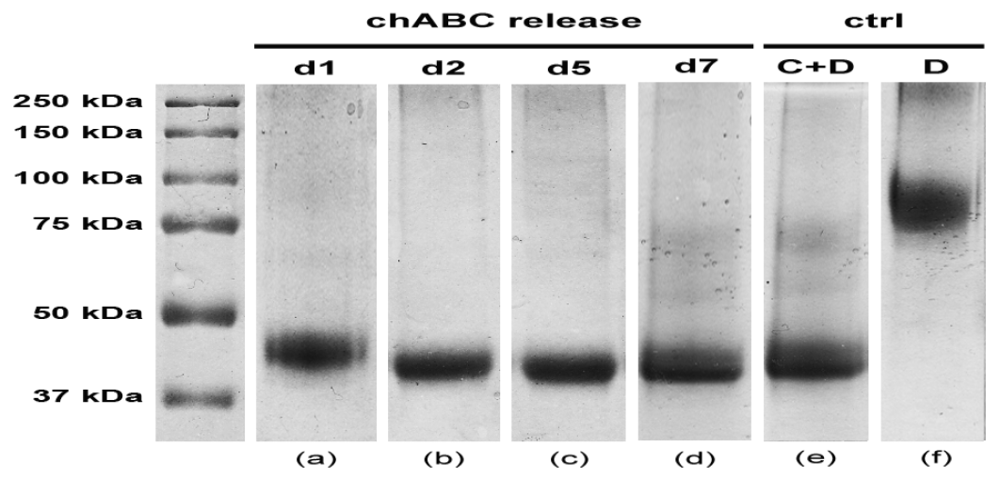
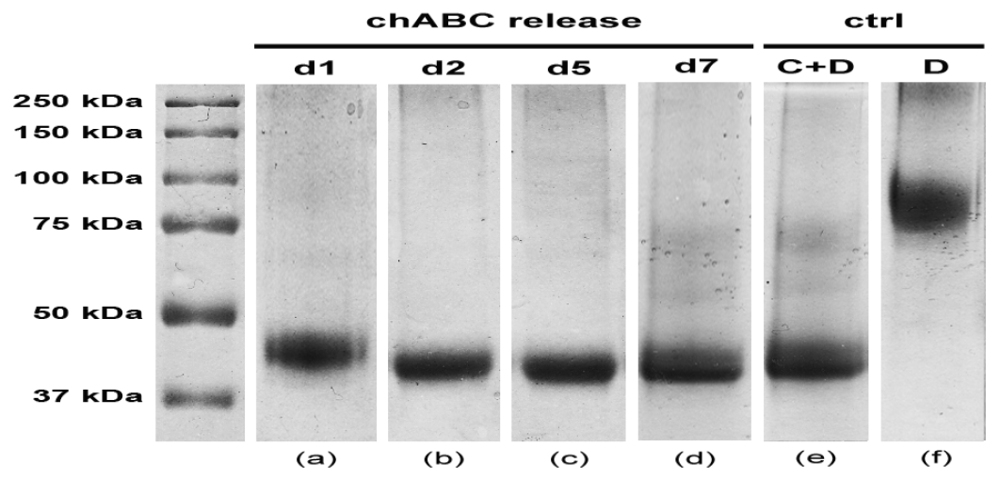
4. Conclusions
Acknowledgments
References
- van den Berg, M.E.L.; Castellote, J.M.; Mahillo-Fernandez, I.; de Pedro-Cuesta, J. Incidence of spinal cord injury worldwide: A systematic review. Neuroepidemiology 2010, 34(3), 184–192. [Google Scholar]
- Ditunno, J.F. Outcome measures: Evolution in clinical trials of neurological/functional recovery in spinal cord injury. Spinal Cord. 2010, 48(9), 674–684. [Google Scholar] [CrossRef]
- van Leeuwen, C.M.; Post, M.W.; Hoekstra, T.; van der Woude, L.H.; de Groot, S.; Snoek, G.J.; Mulder, D.G.; Lindeman, E. Trajectories in the course of life satisfaction after spinal cord injury: Identification and predictors. Arch. Phys. Med. Rehabil. 2011, 92(2), 207–213. [Google Scholar]
- Kubinova, S.; Sykova, E. Nanotechnology for treatment of stroke and spinal cord injury. Nanomedicine 2010, 5(1), 99–108. [Google Scholar] [CrossRef]
- Bradbury, E.J.; Carter, L.M. Manipulating the glial scar: Chondroitinase ABC as a therapy for spinal cord injury. Brain Res. Bull. 2011, 84(4–5), 306–316. [Google Scholar] [CrossRef]
- Bradbury, E.J.; McMahon, S.B. Opinion—Spinal cord repair strategies: Why do they work? Nat. Rev. Neurosci. 2006, 7(8), 644–653. [Google Scholar] [CrossRef]
- Lee, H.; McKeon, R.J.; Bellamkonda, R.V. Sustained delivery of thermostabilized chABC enhances axonal sprouting and functional recovery after spinal cord injury. Proc. Natl. Acad. Sci. USA 2010, 107(8), 3340–3345. [Google Scholar]
- Siebert, J.R.; Stelzner, D.J.; Osterhout, D.J. Chondroitinase treatment following spinal contusion injury increases migration of oligodendrocyte progenitor cells. Exp Neurol 2011, 123(1), 19–29. [Google Scholar]
- Hyatt, A.J.; Wang, D.; Kwok, J.C.; Fawcett, J.W.; Martin, K.R. Controlled release of chondroitinase ABC from fibrin gel reduces the level of inhibitory glycosaminoglycan chains in lesioned spinal cord. J. Control Release 2010, 147(1), 24–29. [Google Scholar]
- Langer, R. Perspectives and challenges in tissue engineering and regenerative medicine. Adv. Mater. 2009, 21(32–33), 3235–3236. [Google Scholar] [CrossRef]
- Baumann, M.D.; Kang, C.E.; Tator, C.H.; Shoichet, M.S. Intrathecal delivery of a polymeric nanocomposite hydrogel after spinal cord injury. Biomaterials 2010, 31(30), 7631–7639. [Google Scholar]
- Slaughter, B.V.; Khurshid, S.S.; Fisher, O.Z.; Khademhosseini, A.; Peppas, N.A. Hydrogels in regenerative medicine. Adv. Mater. 2009, 21(32–33), 3307–3329. [Google Scholar]
- Perale, G.; Rossi, F.; Sundstrom, E.; Bacchiega, S.; Masi, M.; Forloni, G.; Veglianese, P. Hydrogels in spinal cord injury repair strategies. ACS Chem. Neurosci. 2011, 2(7), 345–366. [Google Scholar]
- Perale, G.; Veglianese, P.; Rossi, F.; Peviani, M.; Santoro, M.; Llupi, D.; Micotti, E.; Forloni, G.; Masi, M. In situ agar-carbomer polycondensation: A chemical approach to regenerative medicine. Mater. Lett. 2011, 65(11), 1688–1692. [Google Scholar]
- Rossi, F.; Santoro, M.; Casalini, T.; Veglianese, P.; Masi, M.; Perale, G. Characterization and degradation behavior of agar—Carbomer based hydrogels for drug delivery applications: Solute effect. Int. J. Mol. Sci. 2011, 12(6), 3394–3408. [Google Scholar]
- Santoro, M.; Marchetti, P.; Rossi, F.; Perale, G.; Castiglione, F.; Mele, A.; Masi, M. Smart approach to evaluate drug diffusivity in injectable agar-carbomer hydrogels for drug delivery. J. Phys. Chem. B 2011, 115(11), 2503–2510. [Google Scholar]
- Perale, G.; Rossi, F.; Santoro, M.; Peviani, M.; Papa, S.; Llupi, D.; Torriani, P.; Micotti, E.; Previdi, S.; Cervo, L.; Sundstrom, E.; Boccaccini, A.R.; Masi, M.; Forloni, G.; Veglianese, P. Multiple drug delivery hydrogel system for spinal cord injury repair strategies. J. Control Release 2012, in press. [Google Scholar]
- Thorne, R.G.; Nicholson, C. In vivo diffusion analysis with quantum dots and dextrans predicts the width of brain extracellular space. Proc. Natl. Acad. Sci. USA 2006, 103(14), 5567–5572. [Google Scholar] [CrossRef]
- Huang, W.; Lunin, V.V.; Li, Y.; Suzuki, S.; Sugiura, N.; Miyazono, H.; Cygler, M. Crystal structure of proteus vulgaris chondroitin sulfate ABC lyase I at 1.9 A resolution. J. Mol. Biol. 2003, 328(3), 623–634. [Google Scholar]
- Rossi, F.; Perale, G.; Masi, M. Biological buffered saline solution as solvent in agar-carbomer hydrogel synthesis. Chem. Pap. 2010, 64(5), 573–578. [Google Scholar] [CrossRef]
- Yu, L.; Ding, J.D. Injectable hydrogels as unique biomedical materials. Chem. Soc. Rev. 2008, 37(8), 1473–1481. [Google Scholar]
- Zhao, Q.; Sun, J.Z.; Ling, Q.C.; Zhou, Q.Y. Synthesis of macroporous thermosensitive hydrogels: A novel method of controlling pore size. Langmuir 2009, 25(5), 3249–3254. [Google Scholar]
- Tan, H.; Ramirez, C.M.; Miljkovic, N.; Li, H.; Rubin, J.P.; Marra, K.G. Thermosensitive injectable hyaluronic acid hydrogel for adipose tissue engineering. Biomaterials 2009, 30(36), 6844–6853. [Google Scholar]
- Balgude, A.P.; Yu, X.; Szymanski, A.; Bellamkonda, R.V. Agarose gel stiffness determines rate of DRG neurite extension in 3D cultures. Biomaterials 2001, 22(10), 1077–1084. [Google Scholar]
- Jones, D.S.; Bruschi, M.L.; de Freitas, O.; Gremiao, M.P.D.; Lara, E.H.G.; Andrews, G.P. Rheological, mechanical and mucoadhesive properties of thermoresponsive, bioadhesive binary mixtures composed of poloxamer 407 and carbopol 974P designed as platforms for implantable drug delivery systems for use in the oral cavity. Int. J. Pharm. 2009, 372(1–2), 49–58. [Google Scholar]
- Barbucci, R.; Pasqui, D.; Favaloro, R.; Panariello, G. A thixotropic hydrogel from chemically cross-linked guar gum: Synthesis, characterization and rheological behaviour. Carbohydr. Res. 2008, 343(18), 3058–3065. [Google Scholar]
- Koutsopoulos, S.; Unsworth, L.D.; Nagaia, Y.; Zhang, S.G. Controlled release of functional proteins through designer self-assembling peptide nanofiber hydrogel scaffold. Proc. Natl. Acad. Sci. USA 2009, 106(12), 4623–4628. [Google Scholar] [CrossRef]
- Imai, K.; Hiramatsu, A.; Fukushima, D.; Pierschbacher, M.D.; Okada, Y. Degradation of decorin by matrix metalloproteinases: Identification of the cleavage sites, kinetic analyses and transforming growth factor-beta1 release. Biochem. J. 1997, 322 (Part 3), 809–814. [Google Scholar]
- Honda, E.; Munakata, H. Purification and characterization of decorin from the culture media of MRC-5 cells. Int. J. Biochem. Cell Biol. 2004, 36(8), 1635–1644. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Rossi, F.; Veglianese, P.; Santoro, M.; Papa, S.; Rogora, C.; Dell’Oro, V.; Forloni, G.; Masi, M.; Perale, G. Sustained Delivery of Chondroitinase ABC from Hydrogel System. J. Funct. Biomater. 2012, 3, 199-208. https://doi.org/10.3390/jfb3010199
Rossi F, Veglianese P, Santoro M, Papa S, Rogora C, Dell’Oro V, Forloni G, Masi M, Perale G. Sustained Delivery of Chondroitinase ABC from Hydrogel System. Journal of Functional Biomaterials. 2012; 3(1):199-208. https://doi.org/10.3390/jfb3010199
Chicago/Turabian StyleRossi, Filippo, Pietro Veglianese, Marco Santoro, Simonetta Papa, Cristina Rogora, Valentina Dell’Oro, Gianluigi Forloni, Maurizio Masi, and Giuseppe Perale. 2012. "Sustained Delivery of Chondroitinase ABC from Hydrogel System" Journal of Functional Biomaterials 3, no. 1: 199-208. https://doi.org/10.3390/jfb3010199
APA StyleRossi, F., Veglianese, P., Santoro, M., Papa, S., Rogora, C., Dell’Oro, V., Forloni, G., Masi, M., & Perale, G. (2012). Sustained Delivery of Chondroitinase ABC from Hydrogel System. Journal of Functional Biomaterials, 3(1), 199-208. https://doi.org/10.3390/jfb3010199