Notch1 Activation Up-Regulates Pancreatic and Duodenal Homeobox-1

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Results and Discussion
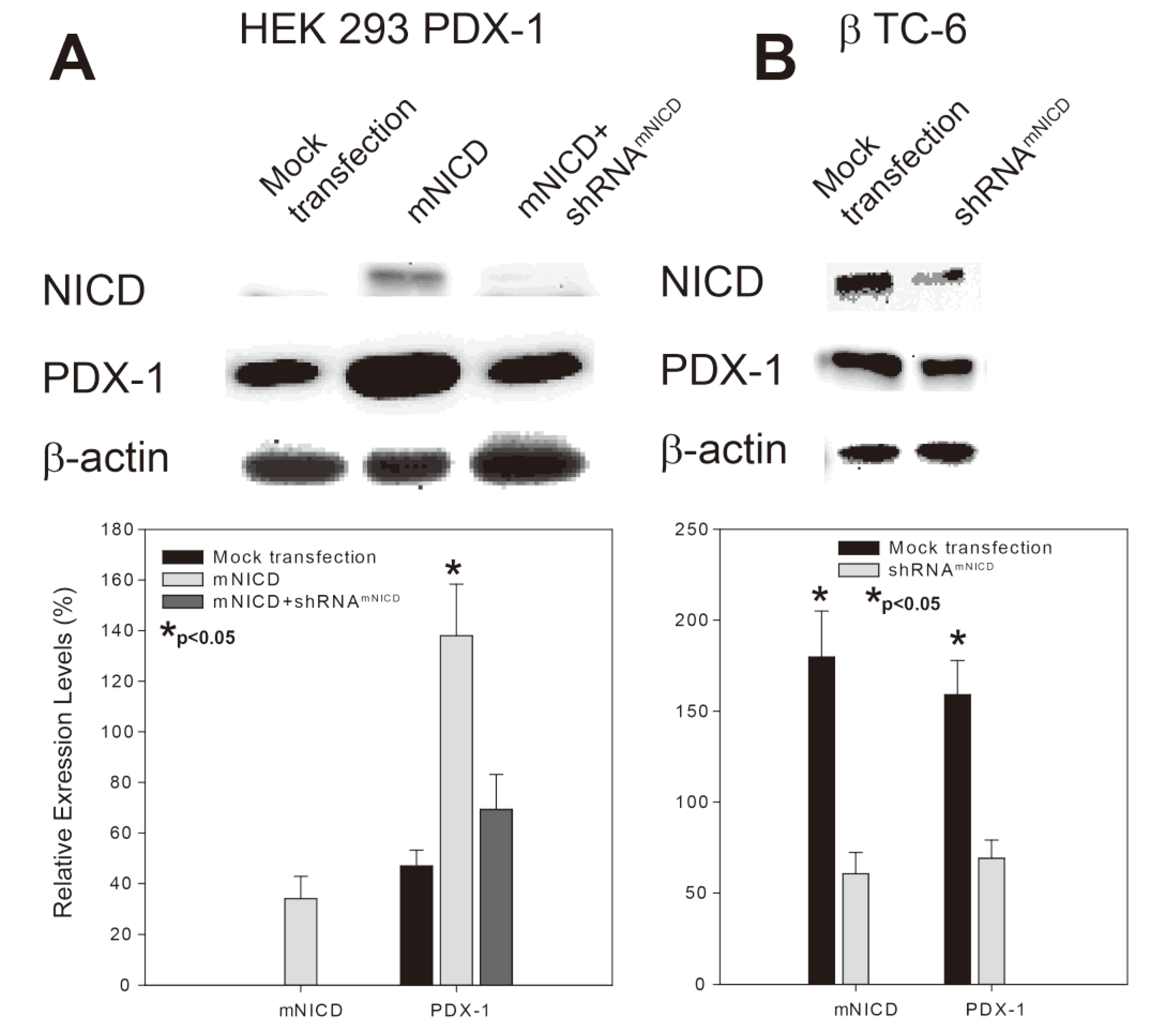
2.1. Notch1 Intracellular Domain (NICD) Enhances PDX-1 Expression in PDX-1 Stable HEK 293 Cells and β-TC-6 Cells
2.2. Notch1 Activation-Enhanced PDX-1 Expression Is Accompanied by Increased Insulin Expression/Secretion and Cell Proliferation in β-TC-6 Cells
2.3. Notch1 Activation Induces Hypoglycemia and Hyperinsulinemia in a Transgenic βNICD+/+ Mouse Model


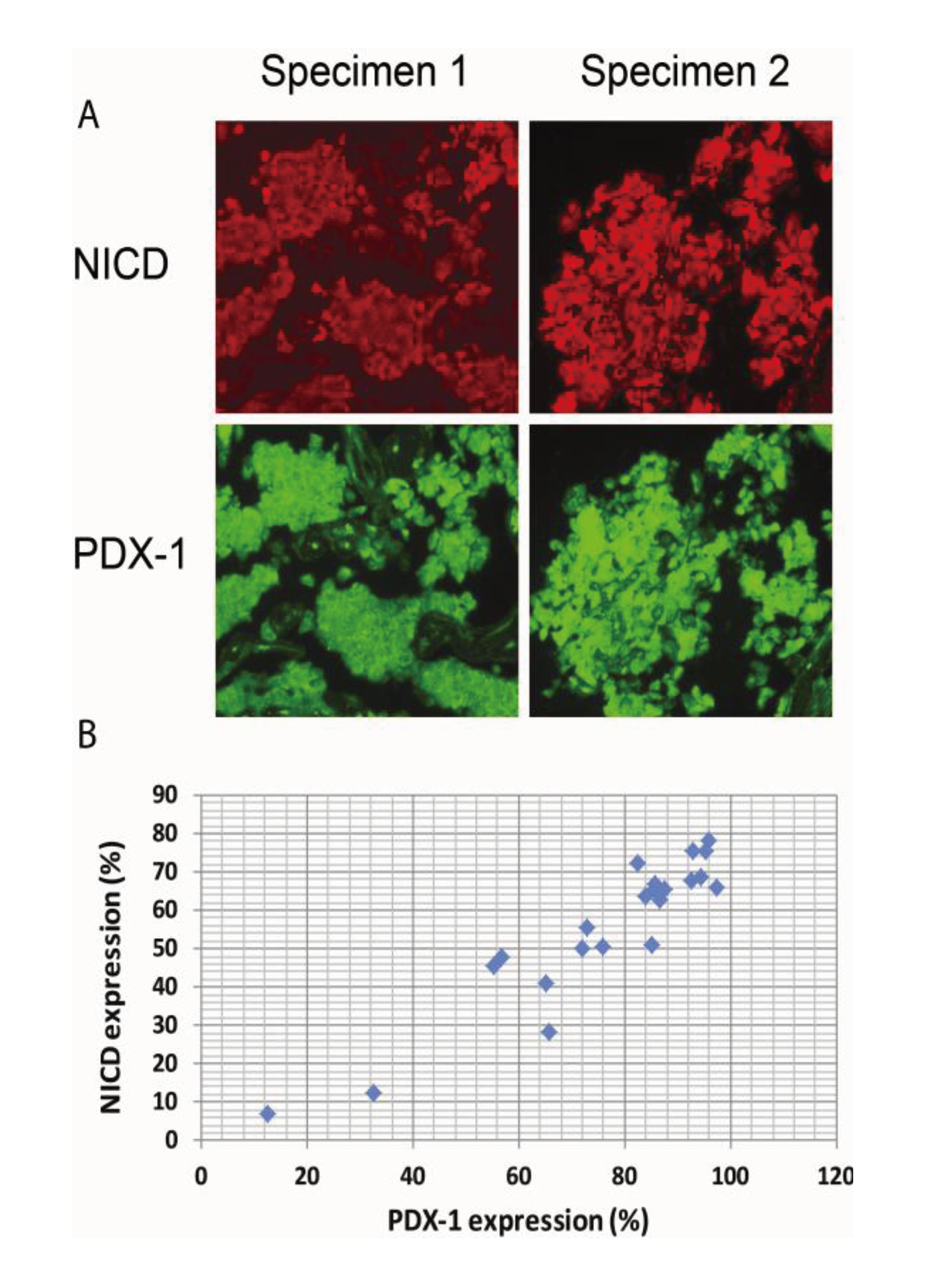
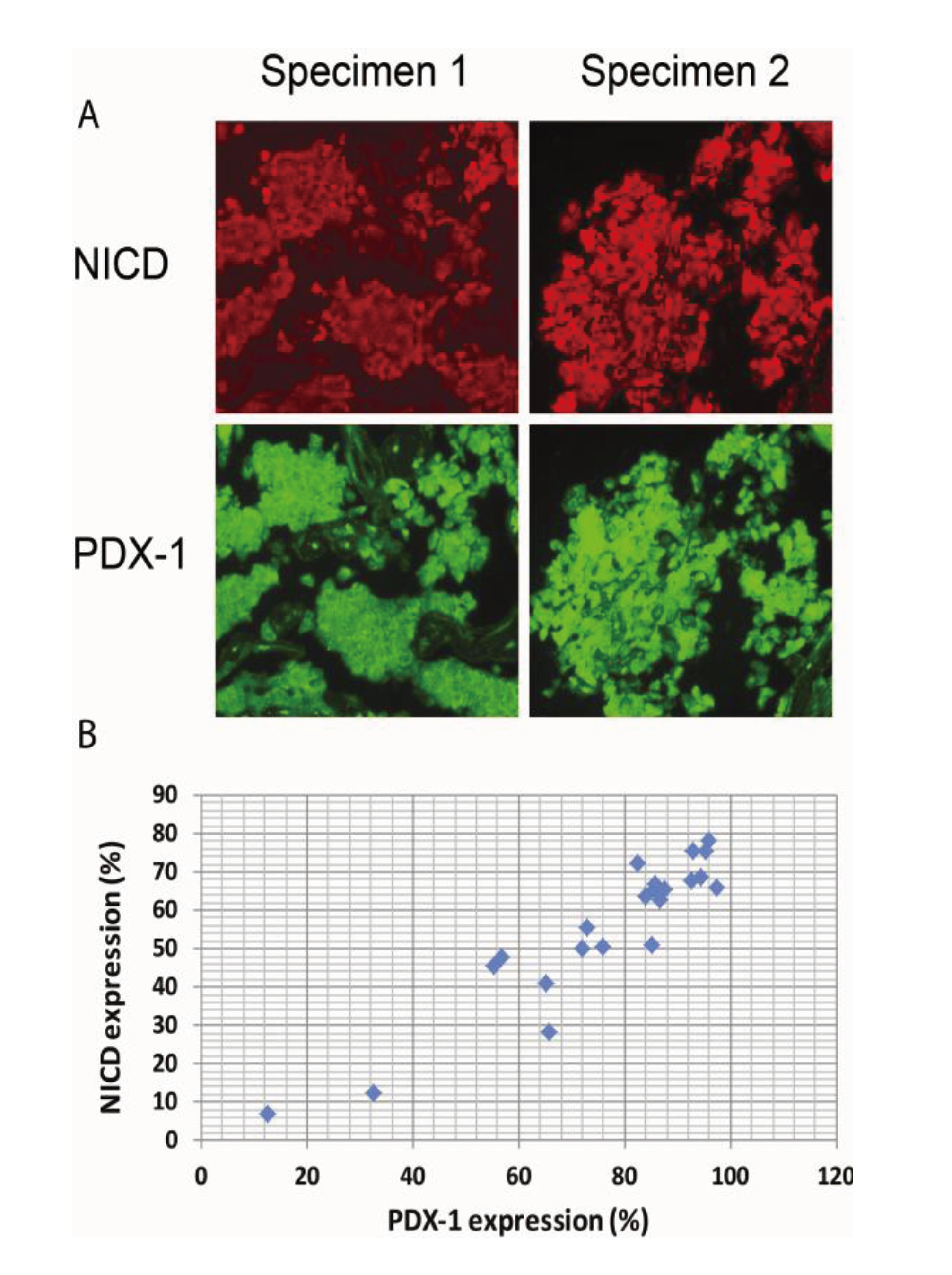
2.4. Activation of Notch1 Is Associated with PDX-1 Overexpression in Human Pancreatic Neuroendocrine Tumors (PNETs)
3. Experimental Section
3.1. Cell Lines, Vectors, Antibodies and PNET Specimens
3.2. Transient and Stable Transfection
3.3. Cell Proliferation Assays
3.4. Western Blotting
3.5. Generation of βNICD+/+ Mice
3.6. Insulin and Glucose Measurements
3.7. Intraperitoneal Glucose Tolerance Test (IPGTT)
3.8. Immunohistochemical Staining
3.9. Statistical Analysis
4. Conclusions
Acknowledgements
Conflict of Interest
References
- Jonsson, J.; Carlsson, L.; Edlund, T.; Edlund, H. Insulin-promoter-factor 1 is required for pancreas development in mice. Nature 1994, 371, 606–609. [Google Scholar] [CrossRef]
- Stoffers, D.A.; Zinkin, N.T.; Stanojevic, V.; Clarke, W.L.; Habener, J.F. Pancreatic agenesis attributable to a single nucleotide deletion in the human IPF1 gene coding sequence. Nat. Genet. 1997, 15, 106–110. [Google Scholar]
- Ahlgren, U.; Jonsson, J.; Jonsson, L.; Simu, K.; Edlund, H. beta-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the beta-cell phenotype and maturity onset diabetes. Genes Dev. 1998, 12, 1763–1768. [Google Scholar] [CrossRef]
- Brissova, M.; Shiota, M.; Nicholson, W.E.; Gannon, M.; Knobel, S.M.; Piston, D.W.; Wright, C.V.; Powers, A.C. Reduction in pancreatic transcription factor PDX-1 impairs glucose-stimulated insulin secretion. J. Biol. Chem. 2002, 277, 11225–11232. [Google Scholar] [CrossRef]
- Johnson, J.D.; Ahmed, N.T.; Luciani, D.S.; Han, Z.; Tran, H.; Fujita, J.; Misler, S.; Edlund, H.; Polonsky, K.S. Increased islet apoptosis in Pdx1+/− mice. J. Clin. Invest. 2003, 111, 1147–1160. [Google Scholar]
- Al-Quobaili, F.; Montenarh, M. Pancreatic duodenal homeobox factor-1 and diabetes mellitus type 2 (review). Int. J. Mol. Med. 2008, 21, 399–404. [Google Scholar]
- Ballian, N.; Liu, S.H.; Brunicardi, F.C. Transcription factor PDX-1 in human colorectal adenocarcinoma: A potential tumor marker? World J. Gastroenterol. 2008, 14, 5823–5826. [Google Scholar]
- Jonmarker, S.; Glaessgen, A.; Culp, W.D.; Pisa, P.; Lewensohn, R.; Ekman, P.; Valdman, A.; Egevad, L. Expression of PDX-1 in prostate cancer, prostatic intraepithelial neoplasia and benign prostatic tissue. APMIS 2008, 116, 491–498. [Google Scholar] [CrossRef]
- Koizumi, M.; Doi, R.; Toyoda, E.; Masui, T.; Tulachan, S.S.; Kawaguchi, Y.; Fujimoto, K.; Gittes, G.K.; Imamura, M. Increased PDX-1 expression is associated with outcome in patients with pancreatic cancer. Surgery 2003, 134, 260–266. [Google Scholar] [CrossRef]
- Leys, C.M.N.S.; Rudzinski, E.; Kaminishi, M.; Montgomery, E.; Washington, M.K.; Goldenring, J.R. Expression of PDX-1 in human gastric metaplasia and gastric adenocarcinoma. Hum. Pathol. 2006, 37, 1162–1168. [Google Scholar] [CrossRef]
- Liu, S.H.; Rao, D.D.; Nemunaitis, J.; Senzer, N.; Zhou, G.; Dawson, D.; Gingras, M.C.; Wang, Z.; Gibbs, R.; Norman, M.; et al. PDX-1 Is a Therapeutic Target for Pancreatic Cancer, Insulinoma and Islet Neoplasia Using a Novel RNA Interference Platform. PLoS One 2012, 7, e40452. [Google Scholar] [CrossRef]
- Sakai, H.E.Y.; Li, X.L.; Akiyama, Y.; Miyake, S.; Takizawa, T.; Konishi, N.; Tatematsu, M.; Koike, M.; Yuasa, Y. PDX-1 homeobox protein expression in pseudopyloric glands and gastric carcinomas. Gut 2004, 53, 323–330. [Google Scholar] [CrossRef]
- Wang, X.P.; Li, Z.J.; Magnusson, J.; Brunicardi, F.C. Tissue MicroArray analyses of pancreatic duodenal homeobox-1 in human cancers. World J. Surg. 2005, 29, 334–338. [Google Scholar] [CrossRef]
- Liu, T.; Gou, S.M.; Wang, C.Y.; Wu, H.S.; Xiong, J.X.; Zhou, F. Pancreas duodenal homeobox-1 expression and significance in pancreatic cancer. World J. Gastroenterol. 2007, 13, 2615–2618. [Google Scholar]
- Liu, S.H.; Patel, S.; Gingras, M.C.; Nemunaitis, J.; Zhou, G.; Chen, C.; Li, M.; Fisher, W.; Gibbs, R.; Brunicardi, F.C. PDX-1: Demonstration of oncogenic properties in pancreatic cancer. Cancer 2011, 117, 723–733. [Google Scholar] [CrossRef]
- Gidekel Friedlander, S.Y.; Chu, G.C.; Snyder, E.L.; Girnius, N.; Dibelius, G.; Crowley, D.; Vasile, E.; DePinho, R.A.; Jacks, T. Context-dependent transformation of adult pancreatic cells by oncogenic K-Ras. Cancer Cell 2009, 16, 379–389. [Google Scholar] [CrossRef]
- Liu, S.; Ballian, N.; Belaguli, N.S.; Patel, S.; Li, M.; Templeton, N.S.; Gingras, M.C.; Gibbs, R.; Fisher, W.; Brunicardi, F.C. PDX-1 acts as a potential molecular target for treatment of human pancreatic cancer. Pancreas 2008, 37, 210–220. [Google Scholar] [CrossRef]
- Guo, D.; Ye, J.; Dai, J.; Li, L.; Chen, F.; Ma, D.; Ji, C. Notch-1 regulates Akt signaling pathway and the expression of cell cycle regulatory proteins cyclin D1, CDK2 and p21 in T-ALL cell lines. Leuk. Res. 2009, 33, 678–685. [Google Scholar] [CrossRef]
- Cohen, B.; Shimizu, M.; Izrailit, J.; Ng, N.F.; Buchman, Y.; Pan, J.G.; Dering, J.; Reedijk, M. Cyclin D1 is a direct target of JAG1-mediated Notch signaling in breast cancer. Breast Cancer Res. Treat. 2010, 123, 113–124. [Google Scholar] [CrossRef]
- Palomero, T.; Lim, W.K.; Odom, D.T.; Sulis, M.L.; Real, P.J.; Margolin, A.; Barnes, K.C.; O'Neil, J.; Neuberg, D.; Weng, A.P.; et al. NOTCH1 directly regulates c-MYC and activates a feed-forward-loop transcriptional network promoting leukemic cell growth. Proc. Natl. Acad. Sci. USA 2006, 103, 18261–18266. [Google Scholar] [CrossRef]
- Jiao, X.; Wood, L.D.; Lindman, M.; Jones, S.; Buckhaults, P.; Polyak, K.; Sukumar, S.; Carter, H.; Kim, D.; Karchin, R.; et al. Somatic mutations in the Notch, NF-KB, PIK3CA, and Hedgehog pathways in human breast cancers. Genes Chromosomes Cancer 2012, 51, 480–489. [Google Scholar] [CrossRef]
- Lindsay, J.; Jiao, X.; Sakamaki, T.; Casimiro, M.C.; Shirley, L.A.; Tran, T.H.; Ju, X.; Liu, M.; Li, Z.; Wang, C.; et al. ErbB2 induces Notch1 activity and function in breast cancer cells. Clin. Transl. Sci. 2008, 1, 107–115. [Google Scholar] [CrossRef]
- Borggrefe, T.; Oswald, F. The Notch signaling pathway: Transcriptional regulation at Notch target genes. Cell. Mol. Life Sci. 2009, 66, 1631–1646. [Google Scholar] [CrossRef]
- Bray, S.J. Notch signalling: A simple pathway becomes complex. Nat. Rev. Mol. Cell. Biol. 2006, 7, 678–689. [Google Scholar] [CrossRef]
- D'Souza, B.; Miyamoto, A.; Weinmaster, G. The many facets of Notch ligands. Oncogene 2008, 27, 5148–5167. [Google Scholar] [CrossRef]
- Radtke, F.; Raj, K. The role of Notch in tumorigenesis: Oncogene or tumour suppressor? Nat. Rev. Cancer 2003, 3, 756–767. [Google Scholar] [CrossRef]
- Ranganathan, P.; Weaver, K.L.; Capobianco, A.J. Notch signalling in solid tumours: A little bit of everything but not all the time. Nat. Rev. Cancer 2011, 11, 338–351. [Google Scholar] [CrossRef]
- Pajvani, U.B.; Shawber, C.J.; Samuel, V.T.; Birkenfeld, A.L.; Shulman, G.I.; Kitajewski, J.; Accili, D. Inhibition of Notch signaling ameliorates insulin resistance in a FoxO1-dependent manner. Nat. Med. 2011, 17, 961–967. [Google Scholar] [CrossRef]
- Fowler, J.C.; Zecchini, V.R.; Jones, P.H. Intestinal activation of Notch signaling induces rapid onset hepatic steatosis and insulin resistance. PLoS One 2011, 6, e20767. [Google Scholar] [CrossRef]
- Horn, S.; Kobberup, S.; Jorgensen, M.C.; Kalisz, M.; Klein, T.; Kageyama, R.; Gegg, M.; Lickert, H.; Lindner, J.; Magnuson, M.A.; et al. Mind bomb 1 is required for pancreatic beta-cell formation. Proc. Natl. Acad. Sci. USA 2012, 109, 7356–7361. [Google Scholar] [CrossRef]
- Koo, B.K.; Lim, H.S.; Song, R.; Yoon, M.J.; Yoon, K.J.; Moon, J.S.; Kim, Y.W.; Kwon, M.C.; Yoo, K.W.; Kong, M.P.; et al. Mind bomb 1 is essential for generating functional Notch ligands to activate Notch. Development 2005, 132, 3459–3470. [Google Scholar] [CrossRef]
- Leal, M.C.; Surace, E.I.; Holgado, M.P.; Ferrari, C.C.; Tarelli, R.; Pitossi, F.; Wisniewski, T.; Castano, E.M.; Morelli, L. Notch signaling proteins HES-1 and Hey-1 bind to insulin degrading enzyme (IDE) proximal promoter and repress its transcription and activity: Implications for cellular Abeta metabolism. Biochim. Biophys. Acta 2012, 1823, 227–235. [Google Scholar]
- McKinnon, C.M.; Docherty, K. Pancreatic duodenal homeobox-1, PDX-1, a major regulator of beta cell identity and function. Diabetologia 2001, 44, 1203–1214. [Google Scholar] [CrossRef]
- Buteau, J.; Roduit, R.; Susini, S.; Prentki, M. Glucagon-like peptide-1 promotes DNA synthesis, activates phosphatidylinositol 3-kinase and increases transcription factor pancreatic and duodenal homeobox gene 1 (PDX-1) DNA binding activity in beta (INS-1)-cells. Diabetologia 1999, 42, 856–864. [Google Scholar] [CrossRef]
- Wang, X.; Cahill, C.M.; Pineyro, M.A.; Zhou, J.; Doyle, M.E.; Egan, J.M. Glucagon-like peptide-1 regulates the beta cell transcription factor, PDX-1, in insulinoma cells. Endocrinology 1999, 140, 4904–4907. [Google Scholar] [CrossRef]
- Arantes, V.C.; Reis, M.A.; Latorraca, M.Q.; Ferreira, F.; Stoppiglia, L.F.; Carneiro, E.M.; Boschero, A.C. Palmitic acid increase levels of pancreatic duodenal homeobox-1 and p38/stress-activated protein kinase in islets from rats maintained on a low protein diet. Br. J. Nutr. 2006, 96, 1006–1012. [Google Scholar] [CrossRef]
- Watanabe, H.; Saito, H.; Ueda, J.; Evers, B.M. Regulation of pancreatic duct cell differentiation by phosphatidylinositol-3 kinase. Biochem. Biophys. Res. Commun. 2008, 370, 33–37. [Google Scholar] [CrossRef]
- Lebrun, P.; Montminy, M.R.; van Obberghen, E. Regulation of the pancreatic duodenal homeobox-1 protein by DNA-dependent protein kinase. J. Biol. Chem. 2005, 280, 38203–38210. [Google Scholar] [CrossRef]
- Boucher, M.J.; Selander, L.; Carlsson, L.; Edlund, H. Phosphorylation marks IPF1/PDX1 protein for degradation by glycogen synthase kinase 3-dependent mechanisms. J. Biol. Chem. 2006, 281, 6395–6403. [Google Scholar] [CrossRef]
- Zhou, G.; Gingras, M.C.; Liu, S.H.; Li, D.; Li, Z.; Catania, R.L.; Stehling, K.M.; Li, M.; Paganelli, G.; Gibbs, R.A.; et al. The hypofunctional effect of P335L single nucleotide polymorphism on SSTR5 function. World J. Surg. 2011, 35, 1715–1724. [Google Scholar] [CrossRef]
- Zhou, G.; Liu, S.H.; Shahi, K.M.; Wang, H.; Duan, X.; Lin, X.; Feng, X.H.; Li, M.; Fisher, W.E.; Demayo, F.J.; et al. Negative regulation of pancreatic and duodenal homeobox-1 by somatostatin receptor subtype 5. Mol. Endocrinol. 2012, 26, 1225–1234. [Google Scholar] [CrossRef]
- Fujitani, Y.; Fujitani, S.; Boyer, D.F.; Gannon, M.; Kawaguchi, Y.; Ray, M.; Shiota, M.; Stein, R.W.; Magnuson, M.A.; Wright, C.V. Targeted deletion of a cis-regulatory region reveals differential gene dosage requirements for Pdx1 in foregut organ differentiation and pancreas formation. Genes Dev. 2006, 20, 253–266. [Google Scholar] [CrossRef]
- Wu, K.L.; Gannon, M.; Peshavaria, M.; Offield, M.F.; Henderson, E.; Ray, M.; Marks, A.; Gamer, L.W.; Wright, C.V.; Stein, R. Hepatocyte nuclear factor 3beta is involved in pancreatic beta-cell-specific transcription of the pdx-1 gene. Mol. Cell. Biol. 1997, 17, 6002–6013. [Google Scholar]
- Gao, N.; LeLay, J.; Vatamaniuk, M.Z.; Rieck, S.; Friedman, J.R.; Kaestner, K.H. Dynamic regulation of Pdx1 enhancers by Foxa1 and Foxa2 is essential for pancreas development. Genes Dev. 2008, 22, 3435–3448. [Google Scholar] [CrossRef]
- Humphrey, R.K.; Yu, S.M.; Flores, L.E.; Jhala, U.S. Glucose regulates steady-state levels of PDX1 via the reciprocal actions of GSK3 and AKT kinases. J. Biol. Chem. 2010, 285, 3406–3416. [Google Scholar] [CrossRef]
- Kishi, A.; Nakamura, T.; Nishio, Y.; Maegawa, H.; Kashiwagi, A. Sumoylation of Pdx1 is associated with its nuclear localization and insulin gene activation. Am. J. Physiol. Endocrinol. Metab. 2003, 284, E830–E840. [Google Scholar]
- Kim, Y.C.; Kim, S.Y.; Mellado-Gil, J.M.; Yadav, H.; Neidermyer, W.; Kamaraju, A.K.; Rane, S.G. RB regulates pancreas development by stabilizing Pdx1. EMBO J. 2011, 30, 1563–1576. [Google Scholar] [CrossRef]
- Rafiq, I.; da Silva Xavier, G.; Hooper, S.; Rutter, G.A. Glucose-stimulated preproinsulin gene expression and nuclear trans-location of pancreatic duodenum homeobox-1 require activation of phosphatidylinositol 3-kinase but not p38 MAPK/SAPK2. J. Biol. Chem. 2000, 275, 15977–15984. [Google Scholar]
- Furukawa, N.; Shirotani, T.; Araki, E.; Kaneko, K.; Todaka, M.; Matsumoto, K.; Tsuruzoe, K.; Motoshima, H.; Yoshizato, K.; Kishikawa, H.; et al. Possible involvement of atypical protein kinase C (PKC) in glucose-sensitive expression of the human insulin gene: DNA-binding activity and transcriptional activity of pancreatic and duodenal homeobox gene-1 (PDX-1) are enhanced via calphostin C-sensitive but phorbol 12-myristate 13-acetate (PMA) and Go 6976-insensitive pathway. Endocr. J. 1999, 46, 43–58. [Google Scholar] [CrossRef]
- Kawamori, D.; Kaneto, H.; Nakatani, Y.; Matsuoka, T.A.; Matsuhisa, M.; Hori, M.; Yamasaki, Y. The forkhead transcription factor Foxo1 bridges the JNK pathway and the transcription factor PDX-1 through its intracellular translocation. J. Biol. Chem. 2006, 281, 1091–1098. [Google Scholar]
- Zhou, G.; Wang, H.; Liu, S.-H.; Shahi, K.M.; Lin, X.; Wu, J.; Feng, X.-H.; Qin, J.; Tan, T.-H.; Brunicardi, F.C. p38 MAP Kinase Interacts with and Stabilizes Pancreatic and Duodenal Homeobox-1. Curr. Mol. Med. 2013, 13, 377–386. [Google Scholar]
- Oswald, F.; Tauber, B.; Dobner, T.; Bourteele, S.; Kostezka, U.; Adler, G.; Liptay, S.; Schmid, R.M. p300 acts as a transcriptional coactivator for mammalian Notch-1. Mol. Cell. Biol. 2001, 21, 7761–7774. [Google Scholar] [CrossRef]
- Tang, Z.; Kadesch, T. Identification of a novel activation domain in the Notch-responsive transcription factor CSL. Nucleic Acids Res. 2001, 29, 2284–2291. [Google Scholar] [CrossRef]
- Feanny, M.A.; Fagan, S.P.; Ballian, N.; Liu, S.H.; Li, Z.; Wang, X.; Fisher, W.; Brunicardi, F.C.; Belaguli, N.S. PDX-1 expression is associated with islet proliferation in vitro and in vivo. J. Surg. Res. 2008, 144, 8–16. [Google Scholar] [CrossRef]
- Murtaugh, L.C.; Stanger, B.Z.; Kwan, K.M.; Melton, D.A. Notch signaling controls multiple steps of pancreatic differentiation. Proc. Natl. Acad. Sci. USA 2003, 100, 14920–14925. [Google Scholar]
- Dohda, T.; Maljukova, A.; Liu, L.; Heyman, M.; Grander, D.; Brodin, D.; Sangfelt, O.; Lendahl, U. Notch signaling induces SKP2 expression and promotes reduction of p27Kip1 in T-cell acute lymphoblastic leukemia cell lines. Exp. Cell Res. 2007, 313, 3141–3152. [Google Scholar] [CrossRef]
- Wang, H.; Chen, Y.; Fernandez-Del, C.; Yilmaz, O.; Deshpande, V. Heterogeneity in signaling pathways of gastroenteropancreatic neuroendocrine tumors: A critical look at notch signaling pathway. Mod. Pathol. 2013, 26, 139–147. [Google Scholar] [CrossRef]
- Zhou, G.; Gingras, M.C.; Liu, S.H.; Sanchez, R.; Edwards, D.; Dawson, D.; Christensen, K.; Paganelli, G.; Gibbs, R.; Fisher, W.; et al. SSTR5 P335L monoclonal antibody differentiates pancreatic neuroendocrine neuroplasms with different SSTR5 genotypes. Surgery 2011, 150, 1136–1142. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Liu, S.-H.; Zhou, G.; Yu, J.; Wu, J.; Nemunaitis, J.; Senzer, N.; Dawson, D.; Li, M.; Fisher, W.E.; Brunicardi, F.C. Notch1 Activation Up-Regulates Pancreatic and Duodenal Homeobox-1. Genes 2013, 4, 358-374. https://doi.org/10.3390/genes4030358
Liu S-H, Zhou G, Yu J, Wu J, Nemunaitis J, Senzer N, Dawson D, Li M, Fisher WE, Brunicardi FC. Notch1 Activation Up-Regulates Pancreatic and Duodenal Homeobox-1. Genes. 2013; 4(3):358-374. https://doi.org/10.3390/genes4030358
Chicago/Turabian StyleLiu, Shi-He, Guisheng Zhou, Juehua Yu, James Wu, John Nemunaitis, Neil Senzer, David Dawson, Min Li, William E. Fisher, and F. Charles Brunicardi. 2013. "Notch1 Activation Up-Regulates Pancreatic and Duodenal Homeobox-1" Genes 4, no. 3: 358-374. https://doi.org/10.3390/genes4030358