The Sound of Silence: RNAi in Poly (ADP-Ribose) Research

Abstract
:1. The Life Cycle of Poly (ADP-Ribose)
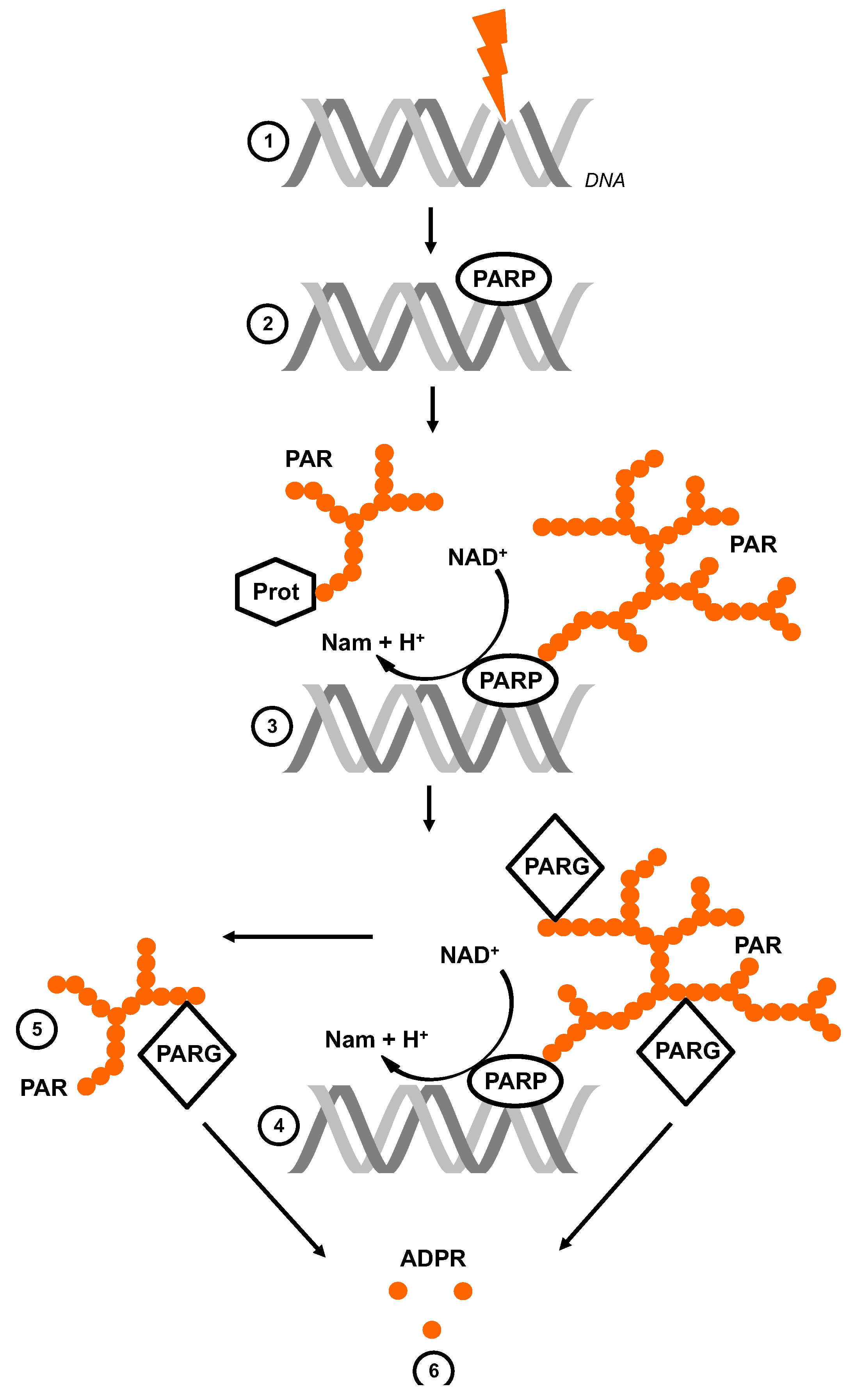
2. Experimental Tools to Investigate PAR and ADPR
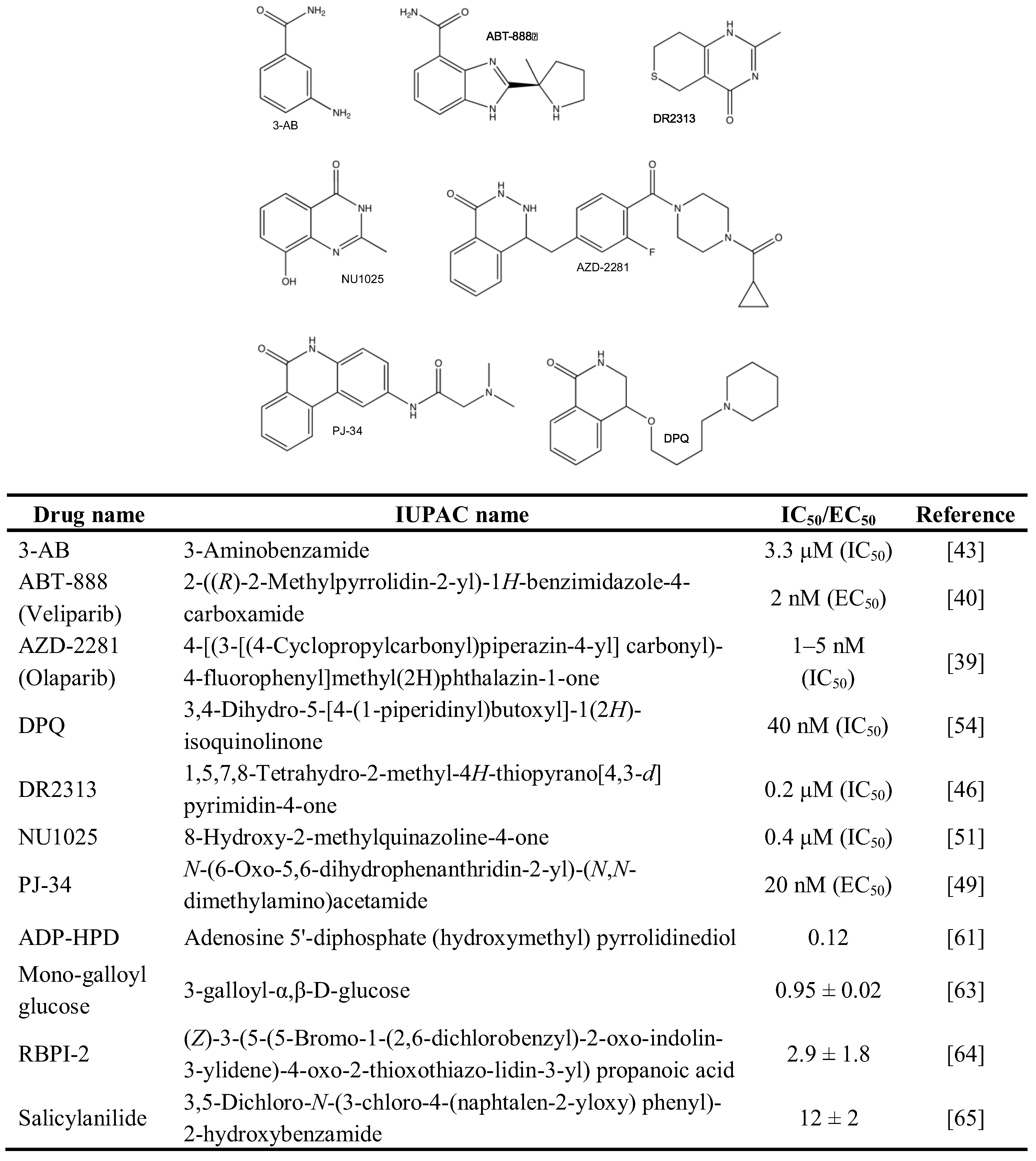
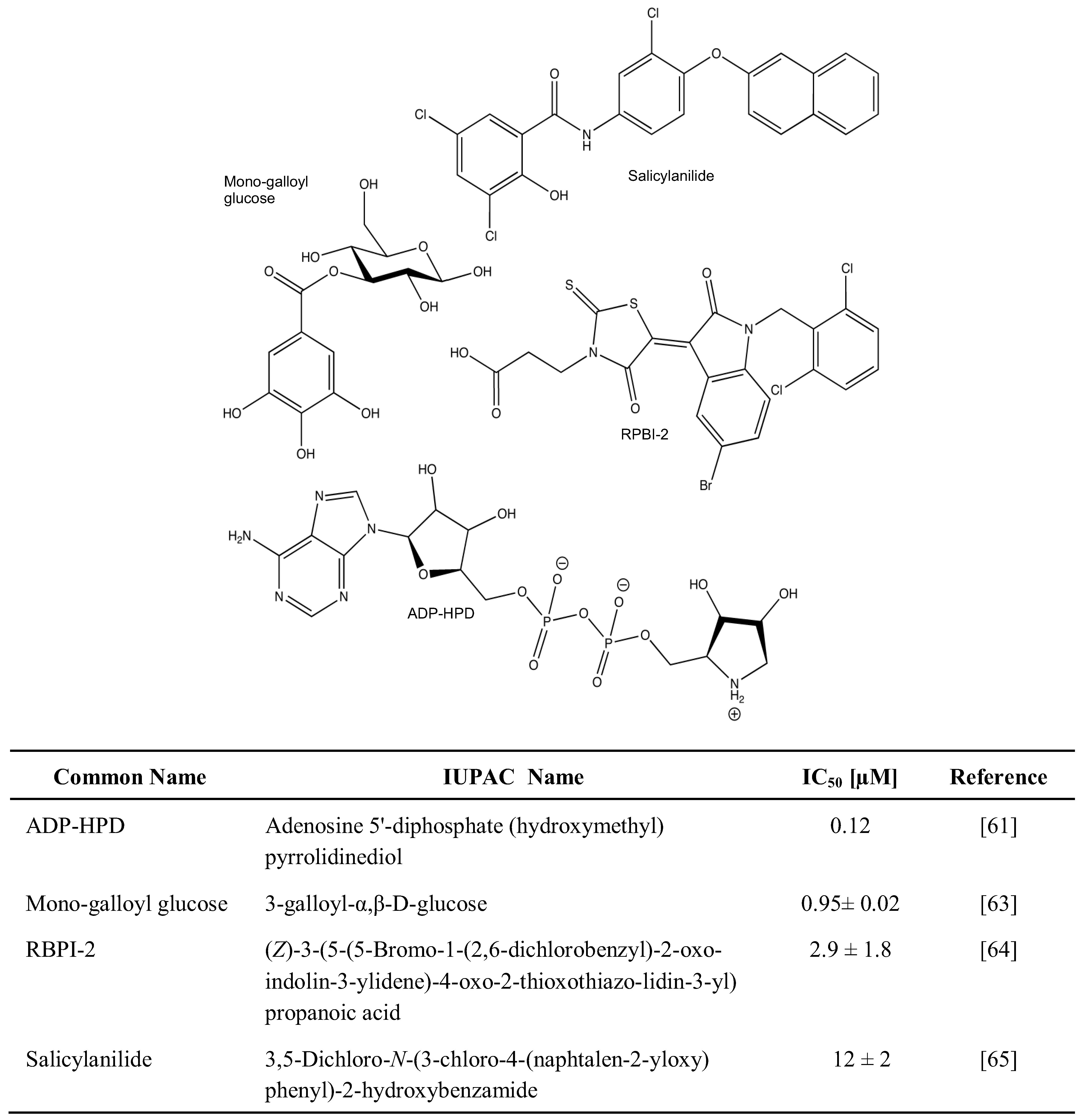
2.1. Chemical Inhibition of PAR Metabolizing Enzymes
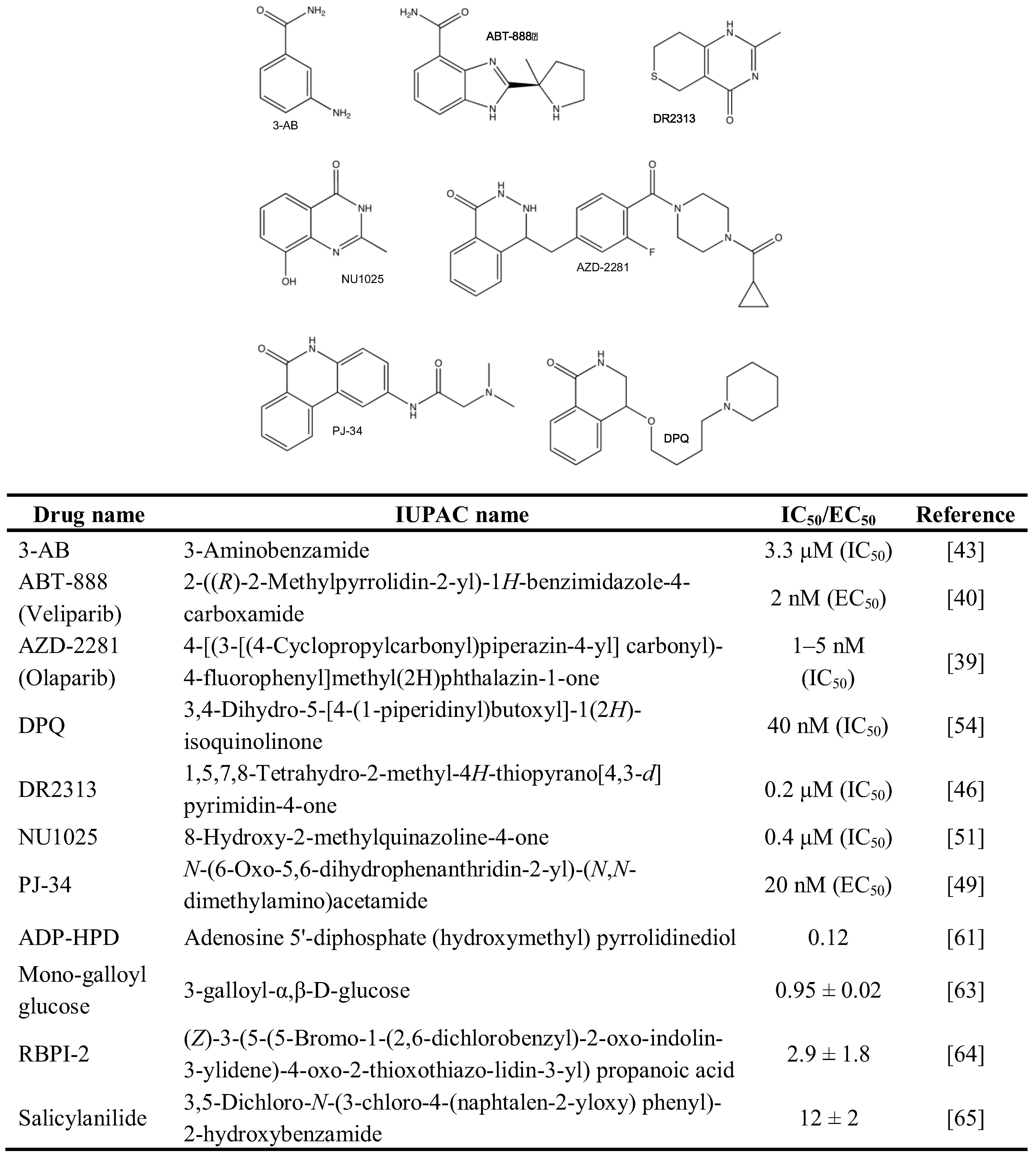
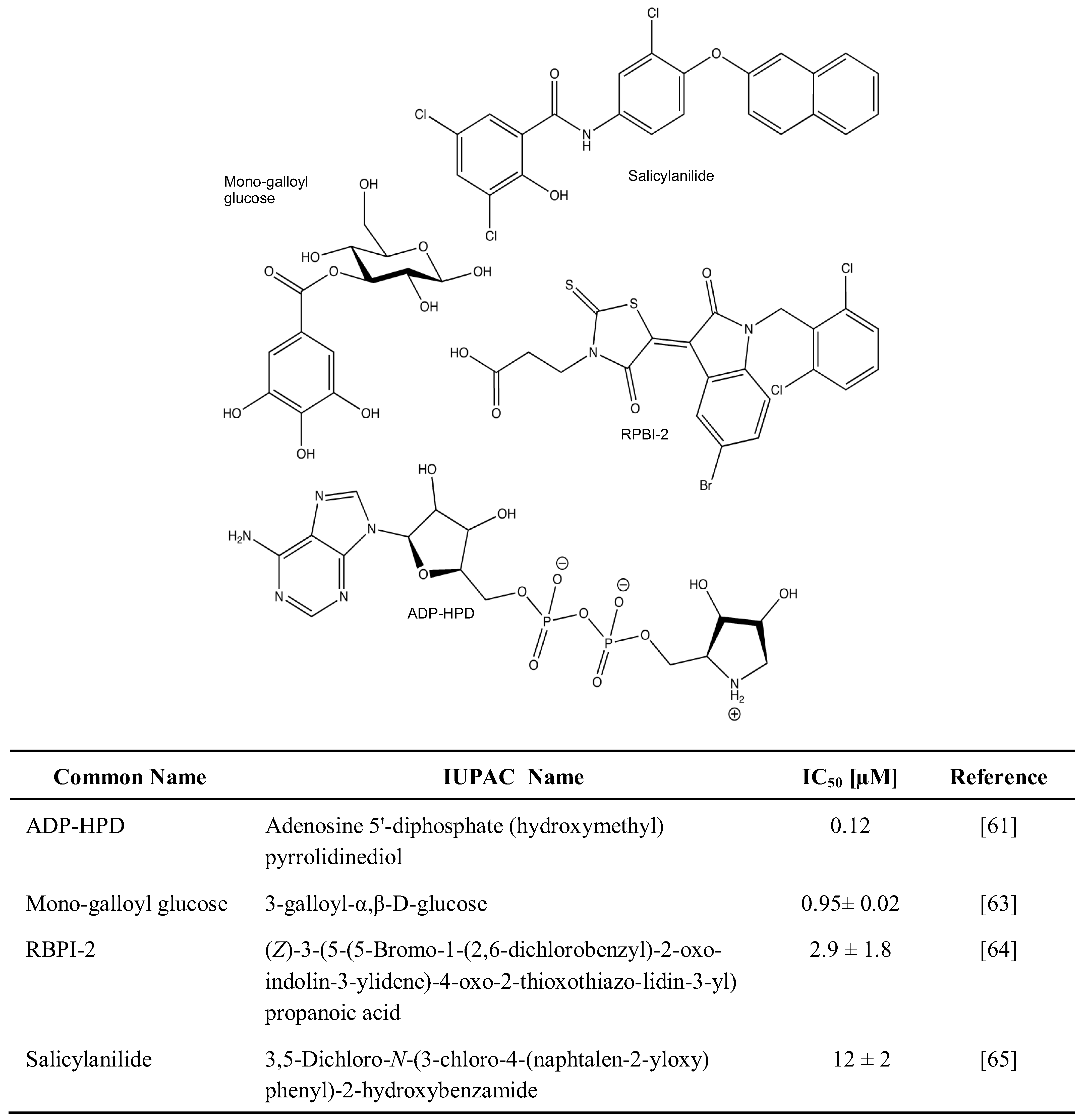
2.2. Genetic Disruption of PARPs and PARG
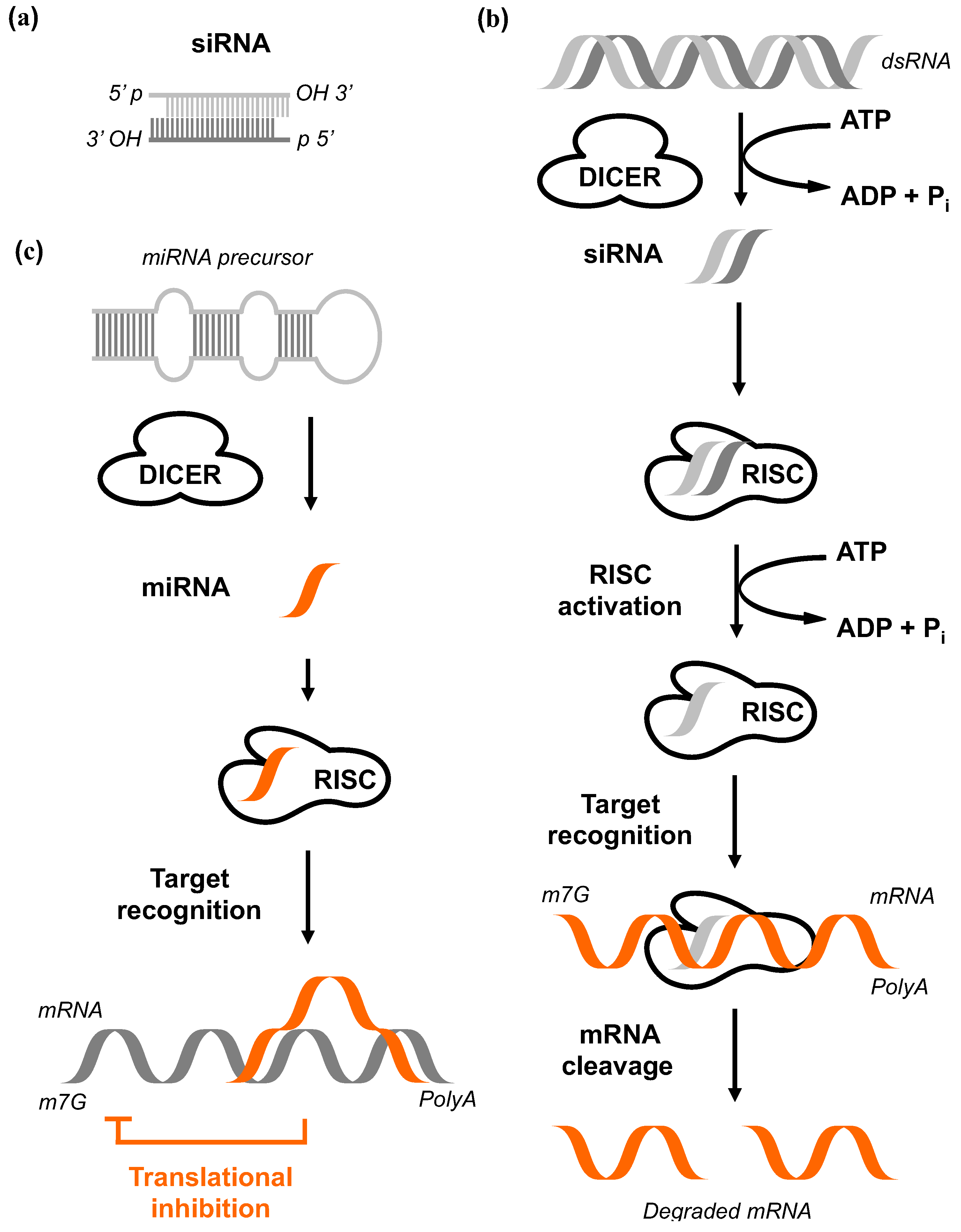
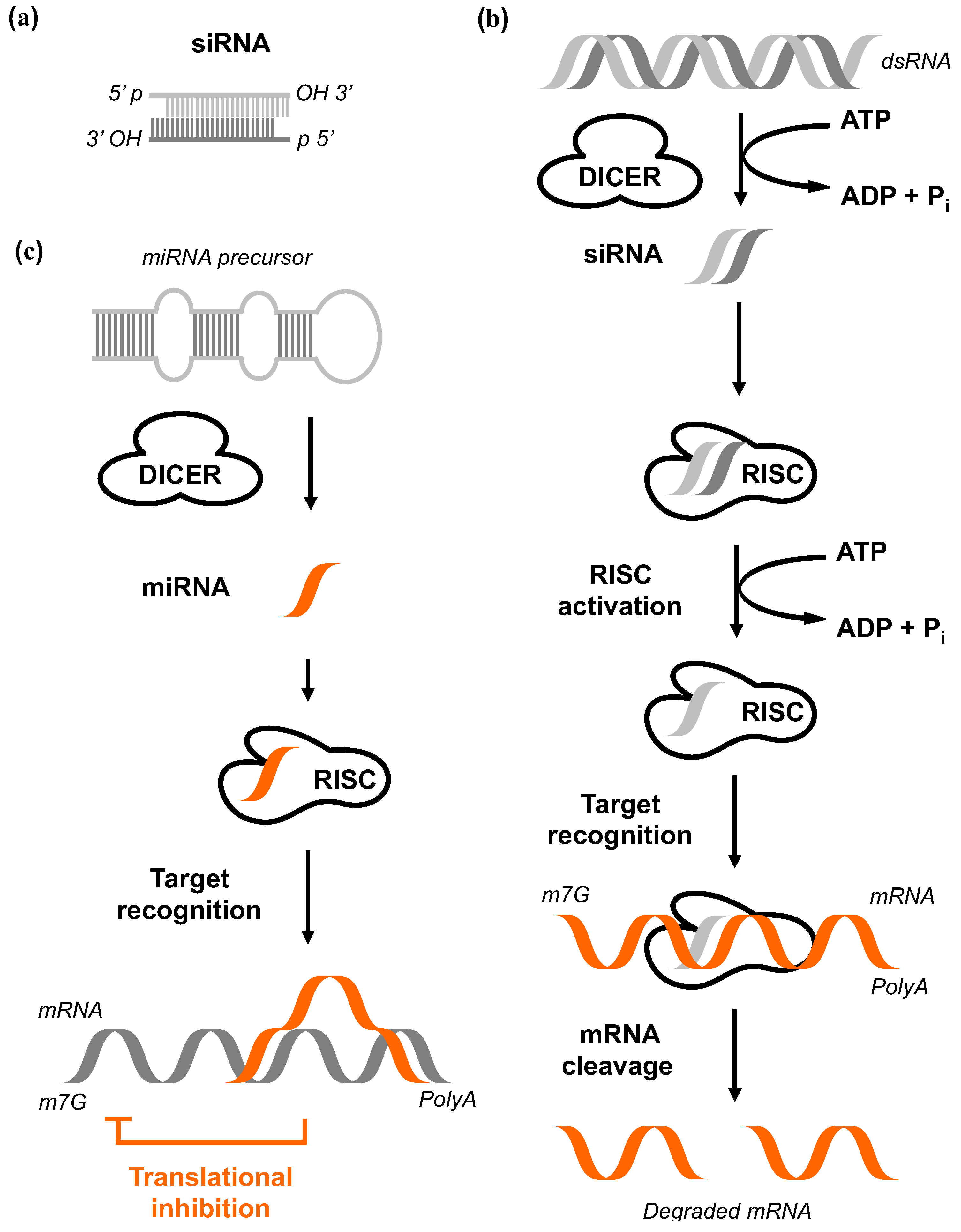
2.3. RNA Interference

{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Deletion | Phenotype | Ref. | |
|---|---|---|---|
| Parp1 | ◆ | Accumulation of DNA strand breaks and impaired DNA repair | [66,67,68] |
| ◆ | High genomic instability | ||
| ◆ | Hypersensitivity to γ-irradiation and alkylating agents | ||
| ◆ | No defects in viability, fertility, development or tissue differentiation | ||
| Parp2 | ◆ | High genomic instability | [70,73] |
| ◆ | Hypersensitivity to γ-irradiation and alkylating agents | ||
| ◆ | Impaired adipogenesis, thymopoiesis, and spermatogenesis | ||
| ◆ | No defects in viability, fertility, development or tissue differentiation | ||
| Parp1, Parp2 | ◆ | Embryonic lethality at onset of gastrulation | [70,75] |
| Parg | ◆ | Peri-implantation lethality | [76] |
| PargΔ2−3 | ◆ | Increased responses to genotoxic treatment and septic shock | [77,78] |
| ◆ | Phenotypically normal and viable | ||
(I) Short interfering RNA (siRNA)
(II) Short hairpin RNA (shRNA)
(III) MicroRNA (miRNA)
(IV) Bi-functional shRNA (bi-shRNA)
2.4. The Use of RNA Interference against PARP Enzymes
| Cell line | Cell type | Species | References |
|---|---|---|---|
| 293 | Embryonic kidney cells | Human | [105]*, [106,107] |
| 3T3-L1 | Preadipocytes | Mouse | [108]* |
| A20 | B-cell lymphoma | Mouse | [109] |
| A549 | Lung adenocarcinoma epithelial | Human | [110,111]*, [107,112,113,114] |
| AGYNB010 | Neuroblastoma | Mouse | [101]* |
| bEnd.3 | Cerebral vascular endothelial | Mouse | [114] |
| CHO | Ovarian cells | Chinese hamster | [103]* |
| EW7 | Ewing sarcoma | Human | [115]* |
| GM00637 | SV-40 transformed skin fibroblasts | Human | [116]* |
| H9 hESCs | Embryonic stem cells | Human | [117]* |
| HaCaT | Keratinocytes | Human | [118,119] |
| HCT-116 | Colon adenocarcinoma | Human | [120,121] |
| HeLa | Cervix carcinoma | Human | [102,122,123,124,125,126,127,128]*, [129,130] |
| HepG2 | Hepatocytes | Human | [131] |
| HUVEC | Endothelial cells | Human | [132] |
| J111 | Acute monocytic leukemia | Human | [102]* |
| Jurkat | T-cell lymphoma (Type II) | Human | [133]* |
| MCF-7 | Breast cancer | Human | [134]* |
| MEF | Fibroblasts | Mouse | [25,103,135,136]* |
| NB4 | Acute promyelocytic leukemia | Human | [115]* |
| NIT-1 | Insulinoma cells | Mouse | [137]* |
| PC12 | Prostate cancer | Human | [138] |
| Primary | Fibroblasts | Human | [139]* |
| Primary | Cerebral cortex neurons | Rat | [140] |
| Primary | Rheumatoid arthritis synovial cells | Human | [141,142] |
| Primary | Vascular smooth muscle | Rat | [143] |
| Ramos | Burkitt’s lymphoma | Human | [109] |
| SHSY5Y | Neuroblastoma | Human | [144] |
| SW480 | Colorectal adenocarcinoma | Human | [145] |
| WRL-68 | Liver cells | Human | [146] |
| Cell line | Cell type | Species | References |
|---|---|---|---|
| 293 | Embryonic kidney cells | Human | [72]*, [152] |
| A549 | Lung adenocarcinoma epithelial | Human | [111] |
| BHK | Baby hamster kidney fibroblast | Chinese hamster | [152] |
| C2C12 | Myoblasts | Mouse | [153]* |
| HeLa | Cervix carcinoma | Human | [122,126,127]* |
| MEF | Fibroblasts | Mouse | [135]* |
| MOVAS | Aortic smooth muscle | Mouse | [154]* |
| SW480 | Colorectal adenocarcinoma | Human | [145] |
2.5. The Use of RNA Interference against PARG
| Cell line | Cell type | Species | References |
|---|---|---|---|
| 16HBE | Bronchial epithelial | Human | [172]* |
| 293 | Embryonic kidney cells | Human | [107] |
| A549 | Lung adenocarcinoma epithelial | Human | [111]*, [107] |
| HeLa | Cervix carcinoma | Human | [122,163,165,166,173]* |
| LoVo | Colon carcinoma | Human | [174,175]* |
| MCF-7 | Breast cancer | Human | [134,163,164]*, [176] |
| MEF | Fibroblasts | Mouse | [25,38,163]* |
| Primary | Rheumatoid arthritis synovial cells | Human | [142] |
| Primary | Glioblastoma | Human | [177]* |
| RAW 264.7 | Macrophages | Mouse | [178]* |
3. Conclusions
Acknowledgments
References and Notes
- Ame, J.C.; Spenlehauer, C.; de Murcia, G. The parp superfamily. Bioessays 2004, 26, 882–893. [Google Scholar] [CrossRef]
- Schreiber, V.; Dantzer, F.; Ame, J.C.; de Murcia, G. Poly(adp-ribose): Novel functions for an old molecule. Nat. Rev. Mol. Cell. Biol. 2006, 7, 517–528. [Google Scholar] [CrossRef]
- Heeres, J.T.; Hergenrother, P.J. Poly(adp-ribose) makes a date with death. Current Opin. Chem. Biol. 2007, 11, 644–653. [Google Scholar] [CrossRef]
- Hottiger, M.O.; Hassa, P.O.; Luscher, B.; Schuler, H.; Koch-Nolte, F. Toward a unified nomenclature for mammalian adp-ribosyltransferases. Trends Biochem. Sci. 2010, 35, 208–219. [Google Scholar] [CrossRef]
- Gibson, B.A.; Kraus, W.L. New insights into the molecular and cellular functions of poly(adp-ribose) and parps. Nat. Rev. Mol. Cell Biol. 2012, 13, 411–424. [Google Scholar] [CrossRef]
- Langelier, M.F.; Servent, K.M.; Rogers, E.E.; Pascal, J.M. A third zinc-binding domain of human poly(adp-ribose) polymerase-1 coordinates DNA-dependent enzyme activation. J. Biol. Chem. 2008, 283, 4105–4114. [Google Scholar]
- Tao, Z.; Gao, P.; Hoffman, D.W.; Liu, H.W. Domain c of human poly(adp-ribose) polymerase-1 is important for enzyme activity and contains a novel zinc-ribbon motif. Biochemistry 2008, 47, 5804–5813. [Google Scholar] [CrossRef]
- Hassa, P.O.; Haenni, S.S.; Elser, M.; Hottiger, M.O. Nuclear adp-ribosylation reactions in mammalian cells: Where are we today and where are we going? Microbiol. Mol. Biol. Rev. MMBR 2006, 70, 789–829. [Google Scholar] [CrossRef]
- Sallmann, F.R.; Vodenicharov, M.D.; Wang, Z.Q.; Poirier, G.G. Characterization of sparp-1. An alternative product of parp-1 gene with poly(adp-ribose) polymerase activity independent of DNA strand breaks. J. Biol. Chem 2000, 275, 15504–15511. [Google Scholar]
- Gagne, J.P.; Isabelle, M.; Lo, K.S.; Bourassa, S.; Hendzel, M.J.; Dawson, V.L.; Dawson, T.M.; Poirier, G.G. Proteome-wide identification of poly(adp-ribose) binding proteins and poly(adp-ribose)-associated protein complexes. Nucleic Acids Res. 2008, 36, 6959–6976. [Google Scholar] [CrossRef] [Green Version]
- Timinszky, G.; Till, S.; Hassa, P.O.; Hothorn, M.; Kustatscher, G.; Nijmeijer, B.; Colombelli, J.; Altmeyer, M.; Stelzer, E.H.; Scheffzek, K.; et al. A macrodomain-containing histone rearranges chromatin upon sensing parp1 activation. Nat. Struct. Mol. Biol. 2009, 16, 923–929. [Google Scholar]
- Gottschalk, A.J.; Timinszky, G.; Kong, S.E.; Jin, J.; Cai, Y.; Swanson, S.K.; Washburn, M.P.; Florens, L.; Ladurner, A.G.; Conaway, J.W.; et al. Poly(adp-ribosyl)ation directs recruitment and activation of an atp-dependent chromatin remodeler. Proc. Natl. Acad. Sci. USA 2009, 106, 13770–13774. [Google Scholar]
- Ahel, D.; Horejsi, Z.; Wiechens, N.; Polo, S.E.; Garcia-Wilson, E.; Ahel, I.; Flynn, H.; Skehel, M.; West, S.C.; Jackson, S.P.; et al. Poly(adp-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme alc1. Science 2009, 325, 1240–1243. [Google Scholar]
- Realini, C.A.; Althaus, F.R. Histone shuttling by poly(adp-ribosylation). J. Biol. Chem. 1992, 267, 18858–18865. [Google Scholar]
- Malanga, M.; Pleschke, J.M.; Kleczkowska, H.E.; Althaus, F.R. Poly(adp-ribose) binds to specific domains of p53 and alters its DNA binding functions. J. Biol. Chem. 1998, 273, 11839–11843. [Google Scholar]
- Pleschke, J.M.; Kleczkowska, H.E.; Strohm, M.; Althaus, F.R. Poly(adp-ribose) binds to specific domains in DNA damage checkpoint proteins. J. Biol. Chem. 2000, 275, 40974–40980. [Google Scholar]
- Ogata, N.; Ueda, K.; Kagamiyama, H.; Hayaishi, O. Adp-ribosylation of histone h1. Identification of glutamic acid residues 2, 14, and the cooh-terminal lysine residue as modification sites. J. Biol. Chem. 1980, 255, 7616–7620. [Google Scholar]
- Tulin, A.; Spradling, A. Chromatin loosening by poly(adp)-ribose polymerase (parp) at drosophila puff loci. Science 2003, 299, 560–562. [Google Scholar] [CrossRef]
- Beneke, S. Regulation of chromatin structure by poly(adp-ribosyl)ation. Front. Genet. 2012, 3, 169. [Google Scholar]
- Ame, J.C.; Rolli, V.; Schreiber, V.; Niedergang, C.; Apiou, F.; Decker, P.; Muller, S.; Hoger, T.; Menissier-de Murcia, J.; de Murcia, G. Parp-2, a novel mammalian DNA damage-dependent poly(adp-ribose) polymerase. J. Biol. Chem. 1999, 274, 17860–17868. [Google Scholar]
- Yelamos, J.; Schreiber, V.; Dantzer, F. Toward specific functions of poly(adp-ribose) polymerase-2. Trends Mol. Med. 2008, 14, 169–178. [Google Scholar] [CrossRef]
- Pion, E.; Ullmann, G.M.; Ame, J.C.; Gerard, D.; de Murcia, G.; Bombarda, E. DNA-induced dimerization of poly(adp-ribose) polymerase-1 triggers its activation. Biochemistry 2005, 44, 14670–14681. [Google Scholar] [CrossRef]
- Schreiber, V. Parp-2, structure-function relationship. In Poly(adp-ribosyl)ation; Burkle, A., Ed.; Landes Bioscience: Austin, TX, USA, 2004; pp. 13–31. [Google Scholar]
- Schreiber, V.; Ame, J.C.; Dolle, P.; Schultz, I.; Rinaldi, B.; Fraulob, V.; Menissier-de Murcia, J.; de Murcia, G. Poly(adp-ribose) polymerase-2 (parp-2) is required for efficient base excision DNA repair in association with parp-1 and xrcc1. J. Biol. Chem. 2002, 277, 23028–23036. [Google Scholar]
- Blenn, C.; Wyrsch, P.; Bader, J.; Bollhalder, M.; Althaus, F.R. Poly(adp-ribose)glycohydrolase is an upstream regulator of ca2+ fluxes in oxidative cell death. Cell. Mol. Life Sci. CMLS 2011, 68, 1455–1466. [Google Scholar] [CrossRef] [Green Version]
- Wielckens, K.; Bredehorst, R.; Adamietz, P.; Hilz, H. Mono adp-ribosylation and poly adp-ribosylation of proteins in normal and malignant tissues. Adv. Enzyme Regul. 1982, 20, 23–37. [Google Scholar] [CrossRef]
- Wielckens, K.; Schmidt, A.; George, E.; Bredehorst, R.; Hilz, H. DNA fragmentation and nad depletion. Their relation to the turnover of endogenous mono(adp-ribosyl) and poly(adp-ribosyl) proteins. J. Biol. Chem. 1982, 257, 12872–12877. [Google Scholar]
- Alvarez-Gonzalez, R.; Althaus, F.R. Poly(adp-ribose) catabolism in mammalian cells exposed to DNA-damaging agents. Mutat. Res. 1989, 218, 67–74. [Google Scholar] [CrossRef]
- Brochu, G.; Shah, G.M.; Poirier, G.G. Purification of poly(adp-ribose) glycohydrolase and detection of its isoforms by a zymogram following one- or two-dimensional electrophoresis. Anal. Biochem. 1994, 218, 265–272. [Google Scholar]
- Davidovic, L.; Vodenicharov, M.; Affar, E.B.; Poirier, G.G. Importance of poly(adp-ribose) glycohydrolase in the control of poly(adp-ribose) metabolism. Exp. Cell. Res. 2001, 268, 7–13. [Google Scholar] [CrossRef]
- Braun, S.A.; Panzeter, P.L.; Collinge, M.A.; Althaus, F.R. Endoglycosidic cleavage of branched polymers by poly(adp-ribose) glycohydrolase. Eur. J. Biochem. 1994, 220, 369–375. [Google Scholar] [CrossRef]
- Alvarez-Gonzalez, R.; Jacobson, M.K. Characterization of polymers of adenosine diphosphate ribose generated in vitro and in vivo. Biochemistry 1987, 26, 3218–3224. [Google Scholar] [CrossRef]
- Ame, J.C.; Apiou, F.; Jacobson, E.L.; Jacobson, M.K. Assignment of the poly(adp-ribose) glycohydrolase gene (parg) to human chromosome 10q11.23 and mouse chromosome 14b by in situ hybridization. Cytogenet Cell. Genet. 1999, 85, 269–270. [Google Scholar] [CrossRef]
- Hatakeyama, K.; Nemoto, Y.; Ueda, K.; Hayaishi, O. Purification and characterization of poly(adp-ribose) glycohydrolase. Different modes of action on large and small poly(adp-ribose). J. Biol. Chem. 1986, 261, 14902–14911. [Google Scholar]
- Meyer, R.G.; Meyer-Ficca, M.L.; Whatcott, C.J.; Jacobson, E.L.; Jacobson, M.K. Two small enzyme isoforms mediate mammalian mitochondrial poly(adp-ribose) glycohydrolase (parg) activity. Exp. Cell. Res. 2007, 313, 2920–2936. [Google Scholar] [CrossRef]
- Oka, S.; Kato, J.; Moss, J. Identification and characterization of a mammalian 39-kda poly(adp-ribose) glycohydrolase. J. Biol. Chem. 2006, 281, 705–713. [Google Scholar]
- Niere, M.; Kernstock, S.; Koch-Nolte, F.; Ziegler, M. Functional localization of two poly(adp-ribose)-degrading enzymes to the mitochondrial matrix. Mol. Cell. Biol. 2008, 28, 814–824. [Google Scholar] [CrossRef]
- Niere, M.; Mashimo, M.; Agledal, L.; Dolle, C.; Kasamatsu, A.; Kato, J.; Moss, J.; Ziegler, M. Adp-ribosylhydrolase 3 (arh3), not poly(adp-ribose) glycohydrolase (parg) isoforms, is responsible for degradation of mitochondrial matrix-associated poly(adp-ribose. J. Biol. Chem. 2012, 287, 16088–16102. [Google Scholar]
- Menear, K.A.; Adcock, C.; Boulter, R.; Cockcroft, X.L.; Copsey, L.; Cranston, A.; Dillon, K.J.; Drzewiecki, J.; Garman, S.; Gomez, S.; et al. 4-[3-(4-cyclopropanecarbonylpiperazine-1-carbonyl)-4-fluorobenzyl]-2h-phthalazin- 1-one: A novel bioavailable inhibitor of poly(adp-ribose) polymerase-1. J. Med. Chem. 2008, 51, 6581–6591. [Google Scholar] [CrossRef]
- Penning, T.D.; Zhu, G.D.; Gandhi, V.B.; Gong, J.; Liu, X.; Shi, Y.; Klinghofer, V.; Johnson, E.F.; Donawho, C.K.; Frost, D.J.; et al. Discovery of the poly(adp-ribose) polymerase (parp) inhibitor 2-[(r)-2-methylpyrrolidin-2-yl]-1h-benzimidazole-4-carboxamide (abt-888) for the treatment of cancer. J. Med. Chem. 2009, 52, 514–523. [Google Scholar]
- Park, S.R.; Chen, A. Poly(adenosine diphosphate-ribose) polymerase inhibitors in cancer treatment. Hematol. Oncol. Clin. North. Am. 2012, 26, 649–670, ix. [Google Scholar] [CrossRef]
- Mangerich, A.; Burkle, A. How to kill tumor cells with inhibitors of poly(adp-ribosyl)ation. Int. J. Cancer 2011, 128, 251–265. [Google Scholar] [CrossRef]
- Rankin, P.W.; Jacobson, E.L.; Benjamin, R.C.; Moss, J.; Jacobson, M.K. Quantitative studies of inhibitors of adp-ribosylation in vitro and in vivo. J. Biol. Chem. 1989, 264, 4312–4317. [Google Scholar]
- Monti, D.; Cossarizza, A.; Salvioli, S.; Franceschi, C.; Rainaldi, G.; Straface, E.; Rivabene, R.; Malorni, W. Cell death protection by 3-aminobenzamide and other poly(adp-ribose)polymerase inhibitors: Different effects on human natural killer and lymphokine activated killer cell activities. Biochem. Biophys. Res. Commun. 1994, 199, 525–530. [Google Scholar] [CrossRef]
- Heller, B.; Wang, Z.Q.; Wagner, E.F.; Radons, J.; Burkle, A.; Fehsel, K.; Burkart, V.; Kolb, H. Inactivation of the poly(adp-ribose) polymerase gene affects oxygen radical and nitric oxide toxicity in islet cells. J. Biol. Chem. 1995, 270, 11176–11180. [Google Scholar]
- Nakajima, H.; Kakui, N.; Ohkuma, K.; Ishikawa, M.; Hasegawa, T. A newly synthesized poly(adp-ribose) polymerase inhibitor, dr2313 [2-methyl-3,5,7,8-tetrahydrothiopyrano[4,3-d]-pyrimidine-4-one]: Pharmacological profiles, neuroprotective effects, and therapeutic time window in cerebral ischemia in rats. J. Pharmacol. Exp. Ther. 2005, 312, 472–481. [Google Scholar]
- Abdelkarim, G.E.; Gertz, K.; Harms, C.; Katchanov, J.; Dirnagl, U.; Szabo, C.; Endres, M. Protective effects of pj34, a novel, potent inhibitor of poly(adp-ribose) polymerase (parp) in in vitro and in vivo models of stroke. Int. J. Mol. Med. 2001, 7, 255–260. [Google Scholar]
- Mabley, J.G.; Jagtap, P.; Perretti, M.; Getting, S.J.; Salzman, A.L.; Virag, L.; Szabo, E.; Soriano, F.G.; Liaudet, L.; Abdelkarim, G.E.; et al. Anti-inflammatory effects of a novel, potent inhibitor of poly (adp-ribose) polymerase. Inflamm. Res. 2001, 50, 561–569. [Google Scholar] [CrossRef]
- Garcia Soriano, F.; Virag, L.; Jagtap, P.; Szabo, E.; Mabley, J.G.; Liaudet, L.; Marton, A.; Hoyt, D.G.; Murthy, K.G.; Salzman, A.L.; et al. Diabetic endothelial dysfunction: The role of poly(adp-ribose) polymerase activation. Nat. Med. 2001, 7, 108–113. [Google Scholar] [CrossRef]
- Faro, R.; Toyoda, Y.; McCully, J.D.; Jagtap, P.; Szabo, E.; Virag, L.; Bianchi, C.; Levitsky, S.; Szabo, C.; Sellke, F.W. Myocardial protection by pj34, a novel potent poly (adp-ribose) synthetase inhibitor. Ann. Thorac. Surg. 2002, 73, 575–581. [Google Scholar] [CrossRef]
- Boulton, S.; Pemberton, L.C.; Porteous, J.K.; Curtin, N.J.; Griffin, R.J.; Golding, B.T.; Durkacz, B.W. Potentiation of temozolomide-induced cytotoxicity: A comparative study of the Biological effects of poly(adp-ribose) polymerase inhibitors. Br. J. Cancer 1995, 72, 849–856. [Google Scholar] [CrossRef]
- Griffin, R.J.; Srinivasan, S.; Bowman, K.; Calvert, A.H.; Curtin, N.J.; Newell, D.R.; Pemberton, L.C.; Golding, B.T. Resistance-modifying agents. 5. Synthesis and Biological properties of quinazolinone inhibitors of the DNA repair enzyme poly(adp-ribose) polymerase (parp). J. Med. Chem. 1998, 41, 5247–5256. [Google Scholar]
- Delaney, C.A.; Wang, L.Z.; Kyle, S.; White, A.W.; Calvert, A.H.; Curtin, N.J.; Durkacz, B.W.; Hostomsky, Z.; Newell, D.R. Potentiation of temozolomide and topotecan growth inhibition and cytotoxicity by novel poly(adenosine diphosphoribose) polymerase inhibitors in a panel of human tumor cell lines. Clin. Cancer Res. 2000, 6, 2860–2867. [Google Scholar]
- Suto, M.J.; Turner, W.R.; Arundel-Suto, C.M.; Werbel, L.M.; Sebolt-Leopold, J.S. Dihydroisoquinolinones: The design and synthesis of a new series of potent inhibitors of poly(adp-ribose) polymerase. Anticancer Drug Des. 1991, 6, 107–117. [Google Scholar]
- Moroni, F.; Meli, E.; Peruginelli, F.; Chiarugi, A.; Cozzi, A.; Picca, R.; Romagnoli, P.; Pellicciari, R.; Pellegrini-Giampietro, D.E. Poly(adp-ribose) polymerase inhibitors attenuate necrotic but not apoptotic neuronal death in experimental models of cerebral ischemia. Cell Death Differ. 2001, 8, 921–932. [Google Scholar] [CrossRef]
- Eliasson, M.J.; Sampei, K.; Mandir, A.S.; Hurn, P.D.; Traystman, R.J.; Bao, J.; Pieper, A.; Wang, Z.Q.; Dawson, T.M.; Snyder, S.H.; et al. Poly(adp-ribose) polymerase gene disruption renders mice resistant to cerebral ischemia. Nat. Med. 1997, 3, 1089–1095. [Google Scholar]
- Espinoza, L.A.; Smulson, M.E.; Chen, Z. Prolonged poly(adp-ribose) polymerase-1 activity regulates jp-8-induced sustained cytokine expression in alveolar macrophages. Free Radical Biol. Med. 2007, 42, 1430–1440. [Google Scholar] [CrossRef]
- Moroni, F.; Formentini, L.; Gerace, E.; Camaioni, E.; Pellegrini-Giampietro, D.E.; Chiarugi, A.; Pellicciari, R. Selective parp-2 inhibitors increase apoptosis in hippocampal slices but protect cortical cells in models of post-ischaemic brain damage. Br. J. Pharmacol 2009, 157, 854–862. [Google Scholar] [CrossRef]
- Banasik, M.; Stedeford, T.; Strosznajder, R.P. Natural inhibitors of poly(adp-ribose) polymerase-1. Mol. Neurobiol. 2012. Submitted. [Google Scholar]
- Blenn, C.; Wyrsch, P.; Althaus, F.R. The ups and downs of tannins as inhibitors of poly(adp-ribose)glycohydrolase. Molecules 2011, 16, 1854–1877. [Google Scholar]
- Slama, J.T.; Aboul-Ela, N.; Goli, D.M.; Cheesman, B.V.; Simmons, A.M.; Jacobson, M.K. Specific inhibition of poly(adp-ribose) glycohydrolase by adenosine diphosphate (hydroxymethyl)pyrrolidinediol. J. Med. Chem. 1995, 38, 389–393. [Google Scholar] [CrossRef]
- Slama, J.T.; Aboul-Ela, N.; Jacobson, M.K. Mechanism of inhibition of poly(adp-ribose) glycohydrolase by adenosine diphosphate (hydroxymethyl)pyrrolidinediol. J. Med. Chem. 1995, 38, 4332–4336. [Google Scholar] [CrossRef]
- Formentini, L.; Arapistas, P.; Pittelli, M.; Jacomelli, M.; Pitozzi, V.; Menichetti, S.; Romani, A.; Giovannelli, L.; Moroni, F.; Chiarugi, A. Mono-galloyl glucose derivatives are potent poly(adp-ribose) glycohydrolase (parg) inhibitors and partially reduce parp-1-dependent cell death. Br. J. Pharmacol. 2008, 155, 1235–1249. [Google Scholar] [CrossRef]
- Finch, K.E.; Knezevic, C.E.; Nottbohm, A.C.; Partlow, K.C.; Hergenrother, P.J. Selective small molecule inhibition of poly(adp-ribose) glycohydrolase (parg). ACS Chem Biol. 2012, 7, 563–570. [Google Scholar] [CrossRef]
- Steffen, J.D.; Coyle, D.L.; Damodaran, K.; Beroza, P.; Jacobson, M.K. Discovery and structure-activity relationships of modified salicylanilides as cell permeable inhibitors of poly(adp-ribose) glycohydrolase (parg). J. Med. Chem. 2011, 54, 5403–5413. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Auer, B.; Stingl, L.; Berghammer, H.; Haidacher, D.; Schweiger, M.; Wagner, E.F. Mice lacking adprt and poly(adp-ribosyl)ation develop normally but are susceptible to skin disease. Genes Dev. 1995, 9, 509–520. [Google Scholar] [CrossRef]
- de Murcia, J.M.; Niedergang, C.; Trucco, C.; Ricoul, M.; Dutrillaux, B.; Mark, M.; Oliver, F.J.; Masson, M.; Dierich, A.; LeMeur, M.; et al. Requirement of poly(adp-ribose) polymerase in recovery from DNA damage in mice and in cells. Proc. Natl. Acad. Sci. USA 1997, 94, 7303–7307. [Google Scholar]
- Le Rhun, Y.; Kirkland, J.B.; Shah, G.M. Cellular responses to DNA damage in the absence of poly(adp-ribose) polymerase. Biochem. Biophys. Res. Commun. 1998, 245, 1–10. [Google Scholar] [CrossRef]
- Wang, Z.Q.; Stingl, L.; Morrison, C.; Jantsch, M.; Los, M.; Schulze-Osthoff, K.; Wagner, E.F. Parp is important for genomic stability but dispensable in apoptosis. Genes Dev. 1997, 11, 2347–2358. [Google Scholar] [CrossRef]
- Menissier de Murcia, J.; Ricoul, M.; Tartier, L.; Niedergang, C.; Huber, A.; Dantzer, F.; Schreiber, V.; Ame, J.C.; Dierich, A.; LeMeur, M.; et al. Functional interaction between parp-1 and parp-2 in chromosome stability and embryonic development in mouse. EMBO J. 2003, 22, 2255–2263. [Google Scholar] [CrossRef]
- Dantzer, F.; Mark, M.; Quenet, D.; Scherthan, H.; Huber, A.; Liebe, B.; Monaco, L.; Chicheportiche, A.; Sassone-Corsi, P.; de Murcia, G.; et al. Poly(adp-ribose) polymerase-2 contributes to the fidelity of male meiosis i and spermiogenesis. Proc. Natl. Acad. Sci. USA 2006, 103, 14854–14859. [Google Scholar]
- Bai, P.; Houten, S.M.; Huber, A.; Schreiber, V.; Watanabe, M.; Kiss, B.; de Murcia, G.; Auwerx, J.; Menissier-de Murcia, J. Poly(adp-ribose) polymerase-2 [corrected] controls adipocyte differentiation and adipose tissue function through the regulation of the activity of the retinoid x receptor/peroxisome proliferator-activated receptor-gamma [corrected] heterodimer. J. Biol. Chem. 2007, 282, 37738–37746. [Google Scholar]
- Yelamos, J.; Monreal, Y.; Saenz, L.; Aguado, E.; Schreiber, V.; Mota, R.; Fuente, T.; Minguela, A.; Parrilla, P.; de Murcia, G.; et al. Parp-2 deficiency affects the survival of cd4+cd8+ double-positive thymocytes. EMBO J. 2006, 25, 4350–4360. [Google Scholar] [CrossRef]
- Xi, H.; Kersh, G.J. Sustained early growth response gene 3 expression inhibits the survival of cd4/cd8 double-positive thymocytes. J. Immunol. 2004, 173, 340–348. [Google Scholar]
- Oei, S.L.; Keil, C.; Ziegler, M. Poly(adp-ribosylation) and genomic stability. Biochem. Cell Biol. 2005, 83, 263–269. [Google Scholar] [CrossRef]
- Koh, D.W.; Lawler, A.M.; Poitras, M.F.; Sasaki, M.; Wattler, S.; Nehls, M.C.; Stoger, T.; Poirier, G.G.; Dawson, V.L.; Dawson, T.M. Failure to degrade poly(adp-ribose) causes increased sensitivity to cytotoxicity and early embryonic lethality. Proc. Natl. Acad. Sci. USA 2004, 101, 17699–17704. [Google Scholar]
- Cortes, U.; Tong, W.M.; Coyle, D.L.; Meyer-Ficca, M.L.; Meyer, R.G.; Petrilli, V.; Herceg, Z.; Jacobson, E.L.; Jacobson, M.K.; Wang, Z.Q. Depletion of the 110-kilodalton isoform of poly(adp-ribose) glycohydrolase increases sensitivity to genotoxic and endotoxic stress in mice. Mol. Cell. Biol. 2004, 24, 7163–7178. [Google Scholar] [CrossRef]
- Meyer-Ficca, M.L.; Lonchar, J.; Credidio, C.; Ihara, M.; Li, Y.; Wang, Z.Q.; Meyer, R.G. Disruption of poly(adp-ribose) homeostasis affects spermiogenesis and sperm chromatin integrity in mice. Biol. Reprod. 2009, 81, 46–55. [Google Scholar] [CrossRef]
- Montgomery, M.K. Rna interference: Historical overview and significance. Meth. Mol. Biol. 2004, 265, 3–21. [Google Scholar]
- Guo, S.; Kemphues, K.J. Par-1, a gene required for establishing polarity in c. Elegans embryos, encodes a putative ser/thr kinase that is asymmetrically distributed. Cell 1995, 81, 611–620. [Google Scholar] [CrossRef]
- Rocheleau, C.E.; Downs, W.D.; Lin, R.; Wittmann, C.; Bei, Y.; Cha, Y.H.; Ali, M.; Priess, J.R.; Mello, C.C. Wnt signaling and an apc-related gene specify endoderm in early c. Elegans embryos. Cell 1997, 90, 707–716. [Google Scholar] [CrossRef]
- Fire, A.; Xu, S.; Montgomery, M.K.; Kostas, S.A.; Driver, S.E.; Mello, C.C. Potent and specific genetic interference by double-stranded rna in caenorhabditis elegans. Nature 1998, 391, 806–811. [Google Scholar]
- Napoli, C.; Lemieux, C.; Jorgensen, R. Introduction of a chimeric chalcone synthase gene into petunia results in reversible co-suppression of homologous genes in trans. Plant. Cell. 1990, 2, 279–289. [Google Scholar]
- Romano, N.; Macino, G. Quelling: Transient inactivation of gene expression in neurospora crassa by transformation with homologous sequences. Mol. Microbiol. 1992, 6, 3343–3353. [Google Scholar] [CrossRef]
- Hutvagner, G.; Zamore, P.D. Rnai: Nature abhors a double-strand. Curr. Opin. Genet. Dev. 2002, 12, 225–232. [Google Scholar] [CrossRef]
- Bernstein, E.; Caudy, A.A.; Hammond, S.M.; Hannon, G.J. Role for a bidentate ribonuclease in the initiation step of rna interference. Nature 2001, 409, 363–366. [Google Scholar]
- Hammond, S.M.; Bernstein, E.; Beach, D.; Hannon, G.J. An rna-directed nuclease mediates post-transcriptional gene silencing in drosophila cells. Nature 2000, 404, 293–296. [Google Scholar] [CrossRef]
- Zamore, P.D.; Tuschl, T.; Sharp, P.A.; Bartel, D.P. Rnai: Double-stranded rna directs the atp-dependent cleavage of mrna at 21 to 23 nucleotide intervals. Cell 2000, 101, 25–33. [Google Scholar] [CrossRef]
- Elbashir, S.M.; Harborth, J.; Lendeckel, W.; Yalcin, A.; Weber, K.; Tuschl, T. Duplexes of 21-nucleotide rnas mediate rna interference in cultured mammalian cells. Nature 2001, 411, 494–498. [Google Scholar]
- Robb, G.B.; Brown, K.M.; Khurana, J.; Rana, T.M. Specific and potent rnai in the nucleus of human cells. Nat. Struct. Mol. Biol. 2005, 12, 133–137. [Google Scholar]
- Yu, J.Y.; DeRuiter, S.L.; Turner, D.L. Rna interference by expression of short-interfering rnas and hairpin rnas in mammalian cells. Proc. Natl. Acad. Sci. USA 2002, 99, 6047–6052. [Google Scholar]
- Brummelkamp, T.R.; Bernards, R.; Agami, R. A system for stable expression of short interfering rnas in mammalian cells. Science 2002, 296, 550–553. [Google Scholar] [CrossRef]
- Rao, D.D.; Vorhies, J.S.; Senzer, N.; Nemunaitis, J. Shrna: Similarities and differences. Adv. Drug Deliv. Rev. 2009, 61, 746–759. [Google Scholar] [CrossRef]
- Bartel, D.P. Micrornas: Genomics, biogenesis, mechanism, and function. Cell 2004, 116, 281–297. [Google Scholar] [CrossRef]
- Liu, J.; Rivas, F.V.; Wohlschlegel, J.; Yates, J.R., 3rd; Parker, R.; Hannon, G.J. A role for the p-body component gw182 in microrna function. Nat. Cell. Biol. 2005, 7, 1261–1266. [Google Scholar]
- John, B.; Enright, A.J.; Aravin, A.; Tuschl, T.; Sander, C.; Marks, D.S. Human microrna targets. PLoS Biol. 2004, 2, e363. [Google Scholar] [CrossRef] [Green Version]
- Chang, T.C.; Yu, D.; Lee, Y.S.; Wentzel, E.A.; Arking, D.E.; West, K.M.; Dang, C.V.; Thomas-Tikhonenko, A.; Mendell, J.T. Widespread microrna repression by myc contributes to tumorigenesis. Nat. Genet. 2008, 40, 43–50. [Google Scholar]
- Wang, Z.; Rao, D.D.; Senzer, N.; Nemunaitis, J. Rna interference and cancer therapy. Pharm. Res. 2011, 28, 2983–2995. [Google Scholar] [CrossRef]
- Seyhan, A.A. Rnai: A potential new class of therapeutic for human genetic disease. Hum. Genet. 2011, 130, 583–605. [Google Scholar] [CrossRef]
- Bora, R.S.; Gupta, D.; Mukkur, T.K.; Saini, K.S. Rna interference therapeutics for cancer: Challenges and opportunities (review). Mol. Med. Report. 2012, 6, 9–15. [Google Scholar]
- Gan, L.; Anton, K.E.; Masterson, B.A.; Vincent, V.A.; Ye, S.; Gonzalez-Zulueta, M. Specific interference with gene expression and gene function mediated by long dsrna in neural cells. J. Neurosci. Meth. 2002, 121, 151–157. [Google Scholar] [CrossRef]
- Kameoka, M.; Nukuzuma, S.; Itaya, A.; Tanaka, Y.; Ota, K.; Ikuta, K.; Yoshihara, K. Rna interference directed against poly(adp-ribose) polymerase 1 efficiently suppresses human immunodeficiency virus type 1 replication in human cells. J. Virol. 2004, 78, 8931–8934. [Google Scholar] [CrossRef]
- Shah, R.G.; Ghodgaonkar, M.M.; Affar el, B.; Shah, G.M. DNA vector-based rnai approach for stable depletion of poly(adp-ribose) polymerase-1. Biochem. Biophys. Res. Commun. 2005, 331, 167–174. [Google Scholar] [CrossRef]
- Dykxhoorn, D.M.; Novina, C.D.; Sharp, P.A. Killing the messenger: Short rnas that silence gene expression. Nature reviews. Mol. Cell Biol. 2003, 4, 457–467. [Google Scholar]
- Bai, P.; Canto, C.; Oudart, H.; Brunyanszki, A.; Cen, Y.; Thomas, C.; Yamamoto, H.; Huber, A.; Kiss, B.; Houtkooper, R.H.; et al. Parp-1 inhibition increases mitochondrial metabolism through sirt1 activation. Cell. Metab. 2011, 13, 461–468. [Google Scholar] [CrossRef]
- Tempera, I.; Deng, Z.; Atanasiu, C.; Chen, C.J.; D'Erme, M.; Lieberman, P.M. Regulation of epstein-barr virus orip replication by poly(adp-ribose) polymerase 1. J. Virol. 2010, 84, 4988–4997. [Google Scholar] [CrossRef]
- Erdelyi, K.; Bai, P.; Kovacs, I.; Szabo, E.; Mocsar, G.; Kakuk, A.; Szabo, C.; Gergely, P.; Virag, L. Dual role of poly(adp-ribose) glycohydrolase in the regulation of cell death in oxidatively stressed a549 cells. Faseb J. 2009.
- Erener, S.; Hesse, M.; Kostadinova, R.; Hottiger, M.O. Poly(adp-ribose)polymerase-1 (parp1) controls adipogenic gene expression and adipocyte function. Mol. Endocrinol. 2012, 26, 79–86. [Google Scholar] [CrossRef] [Green Version]
- Ambrose, H.E.; Papadopoulou, V.; Beswick, R.W.; Wagner, S.D. Poly-(adp-ribose) polymerase-1 (parp-1) binds in a sequence-specific manner at the bcl-6 locus and contributes to the regulation of bcl-6 transcription. Oncogene 2007, 26, 6244–6252. [Google Scholar] [CrossRef]
- Huang, X.; Dong, Y.; Bey, E.A.; Kilgore, J.A.; Bair, J.S.; Li, L.S.; Patel, M.; Parkinson, E.I.; Wang, Y.; Williams, N.S.; et al. An nqo1 substrate with potent antitumor activity that selectively kills by parp1-induced programmed necrosis. Cancer Res. 2012, 72, 3038–3047. [Google Scholar]
- Fisher, A.E.; Hochegger, H.; Takeda, S.; Caldecott, K.W. Poly(adp-ribose) polymerase 1 accelerates single-strand break repair in concert with poly(adp-ribose) glycohydrolase. Mol. Cell. Biol. 2007, 27, 5597–5605. [Google Scholar] [CrossRef]
- Strom, C.E.; Johansson, F.; Uhlen, M.; Szigyarto, C.A.; Erixon, K.; Helleday, T. Poly (adp-ribose) polymerase (parp) is not involved in base excision repair but parp inhibition traps a single-strand intermediate. Nucleic Acids Res. 2011, 39, 3166–3175. [Google Scholar] [CrossRef]
- Hegan, D.C.; Lu, Y.; Stachelek, G.C.; Crosby, M.E.; Bindra, R.S.; Glazer, P.M. Inhibition of poly(adp-ribose) polymerase down-regulates brca1 and rad51 in a pathway mediated by e2f4 and p130. Proc. Natl. Acad. Sci. USA 2010, 107, 2201–2206. [Google Scholar]
- Modis, K.; Gero, D.; Erdelyi, K.; Szoleczky, P.; DeWitt, D.; Szabo, C. Cellular bioenergetics is regulated by parp1 under resting conditions and during oxidative stress. Biochem. Pharmacol. 2012, 83, 633–643. [Google Scholar]
- Mathieu, J.; Flexor, M.; Lanotte, M.; Besancon, F. A parp-1/jnk1 cascade participates in the synergistic apoptotic effect of tnfalpha and all-trans retinoic acid in apl cells. Oncogene 2008, 27, 3361–3370. [Google Scholar] [CrossRef]
- Kandan-Kulangara, F.; Shah, R.G.; Affar el, B.; Shah, G.M. Persistence of different forms of transient rnai during apoptosis in mammalian cells. PloS one 2010, 5, e12263. [Google Scholar] [CrossRef]
- Cimadamore, F.; Curchoe, C.L.; Alderson, N.; Scott, F.; Salvesen, G.; Terskikh, A.V. Nicotinamide rescues human embryonic stem cell-derived neuroectoderm from parthanatic cell death. Stem Cells 2009, 27, 1772–1781. [Google Scholar]
- Ding, W.; Liu, W.; Cooper, K.L.; Qin, X.J.; de Souza Bergo, P.L.; Hudson, L.G.; Liu, K.J. Inhibition of poly(adp-ribose) polymerase-1 by arsenite interferes with repair of oxidative DNA damage. J. Biol. Chem. 2009, 284, 6809–6817. [Google Scholar]
- Qin, X.J.; Hudson, L.G.; Liu, W.; Timmins, G.S.; Liu, K.J. Low concentration of arsenite exacerbates uvr-induced DNA strand breaks by inhibiting parp-1 activity. Toxicol. Appl. Pharmacol 2008, 232, 41–50. [Google Scholar] [CrossRef]
- Zhang, N.; Chen, Y.; Jiang, R.; Li, E.; Chen, X.; Xi, Z.; Guo, Y.; Liu, X.; Zhou, Y.; Che, Y.; et al. Parp and rip 1 are required for autophagy induced by 11'-deoxyverticillin a, which precedes caspase-dependent apoptosis. Autophagy 2011, 7, 598–612. [Google Scholar] [CrossRef]
- Vernole, P.; Muzi, A.; Volpi, A.; Terrinoni, A.; Dorio, A.S.; Tentori, L.; Shah, G.M.; Graziani, G. Common fragile sites in colon cancer cell lines: Role of mismatch repair, rad51 and poly(adp-ribose) polymerase-1. Mutation Res. 2011, 712, 40–48. [Google Scholar] [CrossRef]
- Cohausz, O.; Blenn, C.; Malanga, M.; Althaus, F.R. The roles of poly(adp-ribose)-metabolizing enzymes in alkylation-induced cell death. Cell. Mol. Life Sci. 2008, 65, 644–655. [Google Scholar] [CrossRef]
- Ouararhni, K.; Hadj-Slimane, R.; Ait-Si-Ali, S.; Robin, P.; Mietton, F.; Harel-Bellan, A.; Dimitrov, S.; Hamiche, A. The histone variant mh2a1.1 interferes with transcription by down-regulating parp-1 enzymatic activity. Genes Dev. 2006, 20, 3324–3336. [Google Scholar] [CrossRef]
- Ghosh, U.; Das, N.; Bhattacharyya, N.P. Inhibition of telomerase activity by reduction of poly(adp-ribosyl)ation of tert and tep1/tp1 expression in hela cells with knocked down poly(adp-ribose) polymerase-1 (parp-1) gene. Mutation Res. 2007, 615, 66–74. [Google Scholar] [CrossRef]
- Muthumani, K.; Choo, A.Y.; Zong, W.X.; Madesh, M.; Hwang, D.S.; Premkumar, A.; Thieu, K.P.; Emmanuel, J.; Kumar, S.; Thompson, C.B.; et al. The hiv-1 vpr and glucocorticoid receptor complex is a gain-of-function interaction that prevents the nuclear localization of parp-1. Nat. Cell. Biol. 2006, 8, 170–179. [Google Scholar]
- Chang, P.; Coughlin, M.; Mitchison, T.J. Tankyrase-1 polymerization of poly(adp-ribose) is required for spindle structure and function. Nat. Cell. Biol. 2005, 7, 1133–1139. [Google Scholar]
- Cohausz, O.; Althaus, F.R. Role of parp-1 and parp-2 in the expression of apoptosis-regulating genes in hela cells. Cell. Biol. Toxicol. 2009, 25, 379–391. [Google Scholar] [CrossRef]
- Beneke, S.; Cohausz, O.; Malanga, M.; Boukamp, P.; Althaus, F.; Burkle, A. Rapid regulation of telomere length is mediated by poly(adp-ribose) polymerase-1. Nucleic Acids Res. 2008, 36, 6309–6317. [Google Scholar] [CrossRef]
- Szanto, A.; Hellebrand, E.E.; Bognar, Z.; Tucsek, Z.; Szabo, A.; Gallyas, F., Jr.; Sumegi, B.; Varbiro, G. Parp-1 inhibition-induced activation of pi-3-kinase-akt pathway promotes resistance to taxol. Biochem. Pharmacol. 2009, 77, 1348–1357. [Google Scholar]
- Byun, J.Y.; Kim, M.J.; Eum, D.Y.; Yoon, C.H.; Seo, W.D.; Park, K.H.; Hyun, J.W.; Lee, Y.S.; Lee, J.S.; Yoon, M.Y.; et al. Reactive oxygen species-dependent activation of bax and poly(adp-ribose) polymerase-1 is required for mitochondrial cell death induced by triterpenoid pristimerin in human cervical cancer cells. Mol. Pharmacol 2009, 76, 734–744. [Google Scholar] [CrossRef]
- Pang, J.; Gong, H.; Xi, C.; Fan, W.; Dai, Y.; Zhang, T.M. Poly(adp-ribose) polymerase 1 is involved in glucose toxicity through sirt1 modulation in hepg2 hepatocytes. J. Cell. Biochem 2011, 112, 299–306. [Google Scholar] [CrossRef]
- Mathews, M.T.; Berk, B.C. Parp-1 inhibition prevents oxidative and nitrosative stress-induced endothelial cell death via transactivation of the vegf receptor 2. Arterioscler Thromb Vasc Biol. 2008, 28, 711–717. [Google Scholar] [CrossRef]
- Park, M.T.; Kim, M.J.; Kang, Y.H.; Choi, S.Y.; Lee, J.H.; Choi, J.A.; Kang, C.M.; Cho, C.K.; Kang, S.; Bae, S.; et al. Phytosphingosine in combination with ionizing radiation enhances apoptotic cell death in radiation-resistant cancer cells through ros-dependent and -independent aif release. Blood 2005, 105, 1724–1733. [Google Scholar]
- Frizzell, K.M.; Gamble, M.J.; Berrocal, J.G.; Zhang, T.; Krishnakumar, R.; Cen, Y.; Sauve, A.A.; Kraus, W.L. Global analysis of transcriptional regulation by poly(adp-ribose) polymerase-1 and poly(adp-ribose) glycohydrolase in mcf-7 human breast cancer cells. J. Biol. Chem. 2009, 284, 33926–33938. [Google Scholar]
- Wyrsch, P.; Blenn, C.; Bader, J.; Althaus, F.R. Cell death and autophagy under oxidative stress: Roles of poly(adp-ribose)polymerases and ca2+. Mol. Cell. Biol. 2012.
- Ariumi, Y.; Turelli, P.; Masutani, M.; Trono, D. DNA damage sensors atm, atr, DNA-pkcs, and parp-1 are dispensable for human immunodeficiency virus type 1 integration. J. Virol. 2005, 79, 2973–2978. [Google Scholar] [CrossRef]
- Lin, Y.; Tang, X.; Zhu, Y.; Shu, T.; Han, X. Identification of parp-1 as one of the transcription factors binding to the repressor element in the promoter region of cox-2. Arch. Biochem. Biophys. 2011, 505, 123–129. [Google Scholar] [CrossRef]
- Kondo, K.; Obitsu, S.; Ohta, S.; Matsunami, K.; Otsuka, H.; Teshima, R. Poly(adp-ribose) polymerase (parp)-1-independent apoptosis-inducing factor (aif) release and cell death are induced by eleostearic acid and blocked by alpha-tocopherol and mek inhibition. J. Biol. Chem. 2010, 285, 13079–13091. [Google Scholar]
- Woodhouse, B.C.; Dianova, I.I.; Parsons, J.L.; Dianov, G.L. Poly(adp-ribose) polymerase-1 modulates DNA repair capacity and prevents formation of DNA double strand breaks. DNA Repair 2008, 7, 932–940. [Google Scholar] [CrossRef]
- Diaz-Hernandez, J.I.; Moncada, S.; Bolanos, J.P.; Almeida, A. Poly(adp-ribose) polymerase-1 protects neurons against apoptosis induced by oxidative stress. Cell. Death Differ. 2007, 14, 1211–1221. [Google Scholar] [CrossRef]
- Kitamura, T.; Sekimata, M.; Kikuchi, S.; Homma, Y. Involvement of poly(adp-ribose) polymerase 1 in erbb2 expression in rheumatoid synovial cells. Am. J. Physiol. Cell. Physiol. 2005, 289, C82–88. [Google Scholar] [CrossRef]
- Garcia, S.; Bodano, A.; Pablos, J.L.; Gomez-Reino, J.J.; Conde, C. Poly(adp-ribose) polymerase inhibition reduces tumor necrosis factor-induced inflammatory response in rheumatoid synovial fibroblasts. Ann. Rheum. Dis. 2008, 67, 631–637. [Google Scholar]
- Huang, D.; Wang, Y.; Wang, L.; Zhang, F.; Deng, S.; Wang, R.; Zhang, Y.; Huang, K. Poly(adp-ribose) polymerase 1 is indispensable for transforming growth factor-beta induced smad3 activation in vascular smooth muscle cell. PloS One 2011, 6, e27123. [Google Scholar]
- Lapucci, A.; Pittelli, M.; Rapizzi, E.; Felici, R.; Moroni, F.; Chiarugi, A. Poly(adp-ribose) polymerase-1 is a nuclear epigenetic regulator of mitochondrial DNA repair and transcription. Mol. Pharmacol. 2011, 79, 932–940. [Google Scholar] [CrossRef]
- Bryant, H.E.; Schultz, N.; Thomas, H.D.; Parker, K.M.; Flower, D.; Lopez, E.; Kyle, S.; Meuth, M.; Curtin, N.J.; Helleday, T. Specific killing of brca2-deficient tumours with inhibitors of poly(adp-ribose) polymerase. Nature 2005, 434, 913–917. [Google Scholar]
- Racz, B.; Hanto, K.; Tapodi, A.; Solti, I.; Kalman, N.; Jakus, P.; Kovacs, K.; Debreceni, B.; Gallyas, F., Jr.; Sumegi, B. Regulation of mkp-1 expression and mapk activation by parp-1 in oxidative stress: A new mechanism for the cytoplasmic effect of parp-1 activation. Free Radical Biol. Med. 2010, 49, 1978–1988. [Google Scholar]
- Boehler, C.; Gauthier, L.R.; Mortusewicz, O.; Biard, D.S.; Saliou, J.M.; Bresson, A.; Sanglier-Cianferani, S.; Smith, S.; Schreiber, V.; Boussin, F.; et al. Poly(adp-ribose) polymerase 3 (parp3), a newcomer in cellular response to DNA damage and mitotic progression. Proc. Natl. Acad. Sci. USA 2011, 108, 2783–2788. [Google Scholar]
- Loseva, O.; Jemth, A.S.; Bryant, H.E.; Schuler, H.; Lehtio, L.; Karlberg, T.; Helleday, T. Parp-3 is a mono-adp-ribosylase that activates parp-1 in the absence of DNA. J. Biol. Chem. 2010, 285, 8054–8060. [Google Scholar]
- Chang, W.; Dynek, J.N.; Smith, S. Numa is a major acceptor of poly(adp-ribosyl)ation by tankyrase 1 in mitosis. Biochem. J 2005, 391, 177–184. [Google Scholar] [CrossRef]
- Diani-Moore, S.; Ram, P.; Li, X.; Mondal, P.; Youn, D.Y.; Sauve, A.A.; Rifkind, A.B. Identification of the aryl hydrocarbon receptor target gene tiparp as a mediator of suppression of hepatic gluconeogenesis by 2,3,7,8-tetrachlorodibenzo-p-dioxin and of nicotinamide as a corrective agent for this effect. J. Biol. Chem. 2010, 285, 38801–38810. [Google Scholar]
- Di Paola, S.; Micaroni, M.; Di Tullio, G.; Buccione, R.; Di Girolamo, M. Parp16/artd15 is a novel endoplasmic-reticulum-associated mono-adp-ribosyltransferase that interacts with, and modifies karyopherin-ss1. PloS one 2012, 7, e37352. [Google Scholar]
- Hung, C.F.; Cheng, T.L.; Wu, R.H.; Teng, C.F.; Chang, W.T. A novel bidirectional expression system for simultaneous expression of both the protein-coding genes and short hairpin rnas in mammalian cells. Biochem. Biophys. Res. Commun. 2006, 339, 1035–1042. [Google Scholar] [CrossRef]
- Bai, P.; Canto, C.; Brunyanszki, A.; Huber, A.; Szanto, M.; Cen, Y.; Yamamoto, H.; Houten, S.M.; Kiss, B.; Oudart, H.; et al. Parp-2 regulates sirt1 expression and whole-body energy expenditure. Cell Metab. 2011, 13, 450–460. [Google Scholar] [CrossRef]
- Szanto, M.; Rutkai, I.; Hegedus, C.; Czikora, A.; Rozsahegyi, M.; Kiss, B.; Virag, L.; Gergely, P.; Toth, A.; Bai, P. Poly(adp-ribose) polymerase-2 depletion reduces doxorubicin-induced damage through sirt1 induction. Cardiovasc. Res. 2011, 92, 430–438. [Google Scholar] [CrossRef]
- Gravel, C.; Stergiou, L.; Gagnon, S.N.; Desnoyers, S. The c. Elegans gene pme-5: Molecular cloning and role in the DNA-damage response of a tankyrase orthologue. DNA Repair 2004, 3, 171–182. [Google Scholar] [CrossRef]
- St-Laurent, J.F.; Gagnon, S.N.; Dequen, F.; Hardy, I.; Desnoyers, S. Altered DNA damage response in caenorhabditis elegans with impaired poly(adp-ribose) glycohydrolases genes expression. DNA Repair 2007, 6, 329–343. [Google Scholar] [CrossRef]
- Boamah, E.K.; Kotova, E.; Garabedian, M.; Jarnik, M.; Tulin, A.V. Poly(adp-ribose) polymerase 1 (parp-1) regulates ribosomal biogenesis in drosophila nucleoli. PLoS Genet. 2012, 8, e1002442. [Google Scholar]
- Tulin, A.; Naumova, N.M.; Menon, A.K.; Spradling, A.C. Drosophila poly(adp-ribose) glycohydrolase mediates chromatin structure and sir2-dependent silencing. Genetics 2006, 172, 363–371. [Google Scholar]
- Popoff, I.; Jijon, H.; Monia, B.; Tavernini, M.; Ma, M.; McKay, R.; Madsen, K. Antisense oligonucleotides to poly(adp-ribose) polymerase-2 ameliorate colitis in interleukin-10-deficient mice. J. Pharmacol. Exp. Ther. 2002, 303, 1145–1154. [Google Scholar] [CrossRef]
- Goldberg, M.S.; Xing, D.; Ren, Y.; Orsulic, S.; Bhatia, S.N.; Sharp, P.A. Nanoparticle-mediated delivery of sirna targeting parp1 extends survival of mice bearing tumors derived from brca1-deficient ovarian cancer cells. Proc. Natl. Acad. Sci. USA 2011, 108, 745–750. [Google Scholar]
- De Block, M.; Verduyn, C.; De Brouwer, D.; Cornelissen, M. Poly(adp-ribose) polymerase in plants affects energy homeostasis, cell death and stress tolerance. Plant. J Cell Mol. Biol. 2005, 41, 95–106. [Google Scholar]
- Vanderauwera, S.; De Block, M.; Van de Steene, N.; van de Cotte, B.; Metzlaff, M.; Van Breusegem, F. Silencing of poly(adp-ribose) polymerase in plants alters abiotic stress signal transduction. Proc. Natl. Acad. Sci. USA 2007, 104, 15150–15155. [Google Scholar]
- Blenn, C.; Althaus, F.R.; Malanga, M. Poly(adp-ribose) glycohydrolase silencing protects against h2o2-induced cell death. Biochem. J 2006, 396, 419–429. [Google Scholar] [CrossRef]
- Feng, X.; Zhou, Y.; Proctor, A.M.; Hopkins, M.M.; Liu, M.; Koh, D.W. Silencing of apoptosis-inducing factor and poly(adp-ribose) glycohydrolase reveals novel roles in breast cancer cell death after chemotherapy. Mol. Cancer 2012, 11, 48. [Google Scholar] [CrossRef]
- Andrabi, S.A.; Kim, N.S.; Yu, S.W.; Wang, H.; Koh, D.W.; Sasaki, M.; Klaus, J.A.; Otsuka, T.; Zhang, Z.; Koehler, R.C.; et al. Poly(adp-ribose) (par) polymer is a death signal. Proc. Natl Acad Sci. USA 2006, 103, 18308–18313. [Google Scholar]
- Ame, J.C.; Fouquerel, E.; Gauthier, L.R.; Biard, D.; Boussin, F.D.; Dantzer, F.; de Murcia, G.; Schreiber, V. Radiation-induced mitotic catastrophe in parg-deficient cells. J. Cell Sci. 2009, 122, 1990–2002. [Google Scholar] [CrossRef]
- Norberg, E.; Gogvadze, V.; Ott, M.; Horn, M.; Uhlen, P.; Orrenius, S.; Zhivotovsky, B. An increase in intracellular ca2+ is required for the activation of mitochondrial calpain to release aif during cell death. Cell. Death Differ. 2008, 15, 1857–1864. [Google Scholar] [CrossRef]
- Naziroglu, M. Trpm2 cation channels, oxidative stress and neurological diseases: Where are we now? Neurochemical. Res. 2011, 36, 355–366. [Google Scholar] [CrossRef]
- Perraud, A.L.; Fleig, A.; Dunn, C.A.; Bagley, L.A.; Launay, P.; Schmitz, C.; Stokes, A.J.; Zhu, Q.; Bessman, M.J.; Penner, R.; et al. Adp-ribose gating of the calcium-permeable ltrpc2 channel revealed by nudix motif homology. Nature 2001, 411, 595–599. [Google Scholar]
- Toth, B.; Csanady, L. Identification of direct and indirect effectors of the transient receptor potential melastatin 2 (trpm2) cation channel. J. Biol. Chem. 2010, 285, 30091–30102. [Google Scholar] [CrossRef]
- Buelow, B.; Song, Y.; Scharenberg, A.M. The poly(adp-ribose) polymerase parp-1 is required for oxidative stress-induced trpm2 activation in lymphocytes. J. Biol. Chem. 2008, 283, 24571–24583. [Google Scholar]
- Huang, H.Y.; Cai, J.F.; Liu, Q.C.; Hu, G.H.; Xia, B.; Mao, J.Y.; Wu, D.S.; Liu, J.J.; Zhuang, Z.X. Role of poly(adp-ribose) glycohydrolase in the regulation of cell fate in response to benzo(a)pyrene. Exp. cell Res. 2012, 318, 682–690. [Google Scholar]
- Biard, D.S. Untangling the relationships between DNA repair pathways by silencing more than 20 DNA repair genes in human stable clones. Nucleic Acids Res. 2007, 35, 3535–3550. [Google Scholar] [CrossRef]
- Li, Q.; Li, M.; Wang, Y.L.; Fauzee, N.J.; Yang, Y.; Pan, J.; Yang, L.; Lazar, A. Rna interference of parg could inhibit the metastatic potency of colon carcinoma cells via pi3-kinase/akt pathway. Cell. Physiol. Biochem. 2012, 29, 361–372. [Google Scholar] [CrossRef]
- Pan, J.; Fauzee, N.J.; Wang, Y.L.; Sheng, Y.T.; Tang, Y.; Wang, J.Q.; Wu, W.Q.; Yan, J.X.; Xu, J. Effect of silencing parg in human colon carcinoma lovo cells on the ability of huvec migration and proliferation. Cancer Gene Ther. 2012. [Google Scholar]
- Fathers, C.; Drayton, R.M.; Solovieva, S.; Bryant, H.E. Inhibition of poly(adp-ribose) glycohydrolase (parg) specifically kills brca2-deficient tumor cells. Cell. Cycle 2012, 11, 990–997. [Google Scholar] [CrossRef]
- Tang, J.B.; Svilar, D.; Trivedi, R.N.; Wang, X.H.; Goellner, E.M.; Moore, B.; Hamilton, R.L.; Banze, L.A.; Brown, A.R.; Sobol, R.W. N-methylpurine DNA glycosylase and DNA polymerase beta modulate ber inhibitor potentiation of glioma cells to temozolomide. Neuro Oncol. 2011, 13, 471–486. [Google Scholar] [CrossRef]
- Rapizzi, E.; Fossati, S.; Moroni, F.; Chiarugi, A. Inhibition of poly(adp-ribose) glycohydrolase by gallotannin selectively up-regulates expression of proinflammatory genes. Mol. Pharmacol. 2004, 66, 890–898. [Google Scholar] [CrossRef]
© 2012 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Blenn, C.; Wyrsch, P.; Althaus, F.R. The Sound of Silence: RNAi in Poly (ADP-Ribose) Research. Genes 2012, 3, 779-805. https://doi.org/10.3390/genes3040779
Blenn C, Wyrsch P, Althaus FR. The Sound of Silence: RNAi in Poly (ADP-Ribose) Research. Genes. 2012; 3(4):779-805. https://doi.org/10.3390/genes3040779
Chicago/Turabian StyleBlenn, Christian, Philippe Wyrsch, and Felix R. Althaus. 2012. "The Sound of Silence: RNAi in Poly (ADP-Ribose) Research" Genes 3, no. 4: 779-805. https://doi.org/10.3390/genes3040779