The Role of Epigenetics in Resistance to Cisplatin Chemotherapy in Lung Cancer



Abstract
: Non-small cell lung cancer (NSCLC) is the most common cause of cancer related death in the world. Cisplatin and carboplatin are the most commonly used cytotoxic chemotherapeutic agents to treat the disease. These agents, usually combined with drugs such as gemcitabine or pemetrexed, induce objective tumor responses in only 20–30% of patients. Aberrant epigenetic regulation of gene expression is a frequent event in NSCLC. In this article we review the emerging evidence that epigenetics and the cellular machinery involved with this type of regulation may be key elements in the development of cisplatin resistance in NSCLC.1. Introduction
Lung cancer is the cancer with the highest mortality accounting for 28% of all cancer deaths, estimated at 1.3 million deaths worldwide every year [1]. In the USA the incidence and mortality for cancers of the lung and bronchus are expected to be 219,440 and 159,390, respectively, in 2009 [2]. Lung cancer itself is subdivided into two broad categories, non-small-cell lung cancer (NSCLC) and small cell lung cancer (SCLC). NSCLC can then be further divided into three major types, squamous cell carcinoma (SCC), adenocarcinoma and large cell carcinoma. Mortality in lung cancer is high due in part to (a) difficulties in detecting it at an early stage and (b) associated resistance to currently available chemotherapy and radiotherapy regimes [3]. While lung cancer is often considered to be preventable as most cases can be attributed to smoking, approximately 25% of all lung cancers worldwide are not caused by smoking. If considered as a separate entity, lung cancer in never smokers would still rank as the seventh most common cause of cancer death worldwide [3].
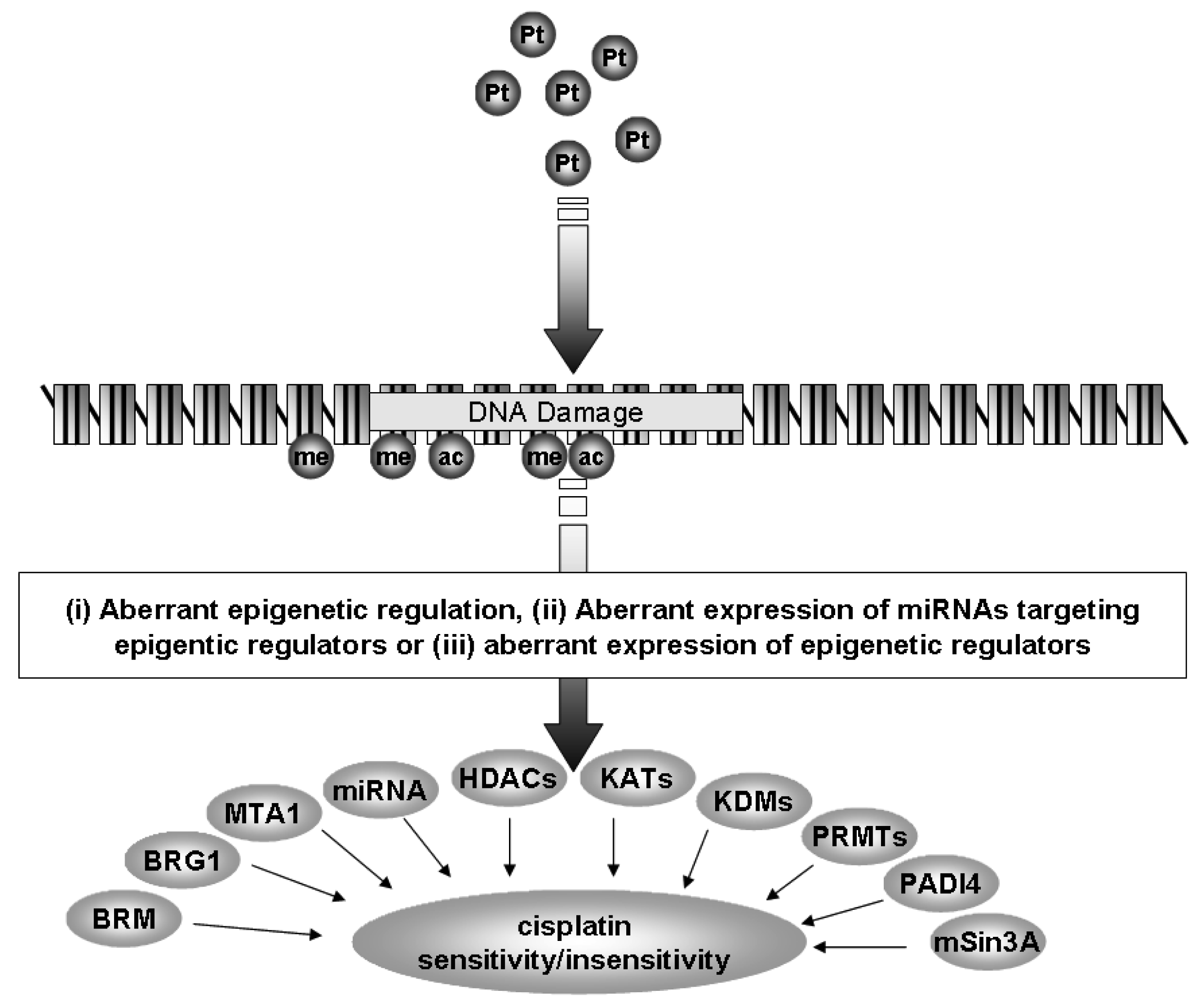
Currently, the standard of care for NSCLC includes treatment with a platinum-based chemotherapy regimen [4]. However, many patients do not benefit from this treatment and tumors often develop resistance to platinum based therapy. In the following review we shall discuss how epigenetics, a specialized form of gene regulation, and the cellular machinery involved with this regulation may be of critical importance in the development of resistance to cisplatin in NSCLC (Figure 1).
2. Epigenetics
A modern definition of epigenetics is considered to be stable and heritable changes in gene expression which are not due to changes in the primary DNA sequence. Current known epigenetic mechanisms involve the following: DNA CpG methylation, histone post-translational modifications (PTMs), gene imprinting and non-coding RNA (ncRNA).
2.1. miRNAs
miRNAs are specialized forms of ncRNA. They consist of small, approximately 22 nucleotide ncRNAs that regulate gene expression through posttranscriptional silencing of target genes, by binding to complementary sequences on target messenger RNA transcripts (mRNAs), resulting in either mRNA degradation or translational repression and gene silencing. Their primary roles are to regulate the self-renewal, differentiation, and division of cells and their levels are frequently altered in cancer [5]. This differential expression has proven useful to distinguish between small cell lung cancer (SCLC) and non-small cell lung cancer (NSCLC) [6], histological subtypes of NSCLC (squamous versus adenocarcinomatous) [7-9], as blood based (plasma or serum) biomarkers for the identification of NSCLC [10,11], prognosis [8,12,13] and for the identification of those miRNAs associated with NSCLC tumorigenesis [14]. It has also been shown that miRNAs can be epigenetically regulated [15,16], and that a specific set of miRNAs can directly regulate the epigenetic machinery (leading to the term epi-miRNAs) [15,16]. In subsequent sections we shall discuss how miRNAs can affect cisplatin resistance in lung cancer either as miRNAs or through epi-miRNA effects (Figure 1).
2.2. DNA CpG Methylation and Lung Cancer
DNA can be methylated on cytosine residues. In many cases this methylation takes place on cytosine residues adjacent to guanine residues, also known as CpGs. Methylation of CpG sites within gene promoters can lead to transcriptional repression, a feature found for important genes such as tumor suppressors in a number of human cancers. The importance of DNA methylation in the development of lung cancer was recently demonstrated when it was shown that the transformation efficiency for immortalization of normal bronchial epithelial cells could be enhanced by low dose exposure to carcinogens. The mechanism underpinning this involved hypermethylation of 5–10 genes due to elevated expression of DNA methyltransferase 1 (DNMT1). Ablation of DNMT1 was shown to reverse this process. Moreover, stable “knock-down” of DNMT1 prior to carcinogen exposure was sufficient to prevent cellular transformation [17], and it is well established that aberrant DNA CpG methylation is a well frequent event in lung cancer leading to the inactivation/dysregulation of critical genes [18].
A full discussion of the role of aberrant DNA methylation in cancer is beyond the scope of this review and the reader is directed to the following overviews of this topic [19-21]
2.3. miRNAs, DNA Methyltransferases and Lung Cancer
Studies have shown that expression of the enzymes responsible for DNA CpG methylation (DNA methyltransferases or DNMTs), are both upregulated and associated with prognosis in lung cancer [22-24]. One particular miRNA family, the miR-29 family (comprising mIR-29a, -29b, and -29c) has been shown to directly target DNMT3A and -3B and indirectly DNMT1 [25-27], In this regard, the mIR-29 family was found to be downregulated in NSCLC and enforced expression of miR-29s in lung cancer cell lines restored normal patterns of DNA methylation, induced reexpression of methylation-silenced tumor suppressor genes, and inhibited tumorigenicity in vitro and in vivo [25] (Table 1). Other miRNAs identified in other cancer types also target DNMTs include mIR-148a and mIR-152 (DNMT1) [28], and miR-143 (DNMT3a) [29]. Of these mIR-143 has been shown to be both downregulated in NSCLC and associated with smoking status [30] (Table 1), while in effusions taken from lung cancer patients lower levels of cell-free miR-152 were present in effusions taken from patients who were docetaxol resistant compared to effusions taken from patients who were docetaxol sensitive [31] (Table 1/Figure 1)).
2.4. Methylated Genes Associated with Sensitivity to Cisplatin Based Therapy
Significant evidence is emerging linking loss of gene expression in NSCLC by DNA CpG methylation with cisplatin resistance. Indeed, pulsed exposure to cisplatin has been shown to result in drug-induced DNA hypermethylation both in vitro and in vivo [32,33]. Checkpoint kinase 2 (CHK2) was one of the first examples of one such gene whose downregulation by DNA CpG methylation in NSCLC was associated with resistance to standard chemotherapies including cisplatin [34]. Another gene Empty Spiracles, Drosophila, 2, HOMOLOG OF; (EMX2) has also been shown to be dramatically downregulated in lung cancer tissue samples by methylation of its promoter, and restoration of EMX2 gene expression sensitized lung cancer cells to cisplatin [35]. The serine protease HtrA3 has also been shown to be reduced or completely lost in over 50% of lung cancer cell lines and primary lung tumors from heavy smokers. The loss of HtrA3 expression is due to DNA methylation and results in resistance to both resistance to etoposide and cisplatin [36]. Expression of transglutaminase 2 (TGM2) has been linked to cisplatin resistance in ovarian cancer [37]. Hong and colleagues have now shown that the TGM2 gene is silenced by promoter CpG methylation in approximately one-third of all NSCLC cell lines examined. Cell lines which had loss of TGM2 were more sensitive to cisplatin, and targeting TGM2 using siRNA also resulted in increased sensitivity to this drug [38]. Using microarray technology to compare isogenic parent/resistant cell lines Perona and colleagues identified loss of expression of insulin-like binding protein -3 (IGFBP3) by DNA CpG methylation in cisplatin resistant cells and found a strong correlation between IGFBP3 methylation status and cisplatin response in patients, where methylated promoters were mostly found in patients with cisplatin resistant tumors [39].
The potential for the use of DNA CpG methylation as a biomarker for response to chemotherapy in NSCLC came from a study of circulating serum DNA, where methylation-dependent transcriptional silencing of 14-3-3sigma, a major G2-M checkpoint control gene, was found to be a predictor for longer survival in cisplatin-plus-gemcitabine-treated NSCLC patients [40]. The results discussed above for IGFBP-3, TGM2, CHK2, HtrA3 and EMX2 indicate that a pretreatment analysis of these genes in patients prior to chemotherapy may have translational benefit. More recently Rosell and colleagues have shown that in serum DNA taken from patients enrolled in a multicenter, randomized study of customized cisplatin-based chemotherapy in stage IV NSCLC (clinicaltrials.gov.identifier: NCT00174629) who subsequently underwent second-line chemotherapy or treatment with EGFR tyrosine kinase inhibitors (TKIs) that if the gene for checkpoint with forkhead-associated [41] and ring finger (RF) (CHFR) gene was hypo- or unmethylated in patients receiving second-line EGFR (TKIs), this was associated with longer survival [42].
2.5. miRNAs and Sensitivity to Cisplatin Based Therapy
As previously discussed, miRNAs have been shown to have altered expression in lung cancer. But miRNAs themselves have also been linked to cisplatin resistance (Figure 1). In the lung cancer cell line A549 the miRNAs miR-181a, miR-181b and miR-630 have been shown to be involved with cellular responses to cisplatin (Table 1) [43,44]. miR-181a was found to enhance cisplatin triggered cell death by inducing apoptosis through Bax oligomerization, mitochondrial transmembrane potential dissipation, and proteolytic maturation of caspase-9 and caspase-3 [44]. miR-181b was found to be downregulated in an isogenic cisplatin resistant A549 cell line (A549/CDDP), and overexpression of this microRNA decreased levels of BCL2 with resultant enhanced sensitivity to cisplatin induced cell death [43]. It is interesting to note that miR-181a has been found to be both significantly downregulated and associated with poor survival in primary NSCLC tissues [30].
mIR-630 was found to block the early manifestations of the DNA damage response (phosphorylation of ATM, histone H2AX and p53), with concomitant induction of p27(Kip1), reductions in rates of cell proliferation and arrest at the G0-G1 phase of the cell cycle as opposed to the late S-G2-M cell cycle arrest normally mediated by cisplatin [44]
The microRNA-200 family plays important roles in regulating epithelial-to-mesenchymal transition [45]. In NSCLC one of these, miR-200c has been shown to be downregulated in NSCLC as a consequence of DNA CpG methylation (Table 1), and restoration of its expression was shown to restore the sensitivity of a resistant cell line to cisplatin and cetuximab [46]. As such it is becoming very clear that miRNAs may play important roles in tumor cell responses to cisplatin.
2.6. Predictive and Prognostic Value of HISTONE Post-Translational Modifications in Cancer
Post-translational modifications of histones or the “histone code” have emerged as a major mechanism by which cells regulate gene expression and cellular function. Aberrant histone post-translational modifications (PTMs) have now been shown to have both predictive and prognostic value in many cancers including adult acute lymphoblastic leukemia (ALL) [47], acute myeloid leukemia AML [48], breast cancer [49,50], colorectal cancer [51], gastric cancer [52], glioma [53], hepatocellular carcinoma [54], lymphoma [51], pancreatic cancer [55], prostate cancer [50,56,57], esophageal cancer [58-60], ovarian cancer [61], and renal cell carcinoma [62-65].
Histone PTMs have also been shown to have both predictive and prognostic value in NSCLC [66,67]. Deregulation of some of the enzymes involved with regulating these modifications in a bronchial epithelial cell transformation model suggest that they play important roles in the transformation process [68]. In addition, strong evidence links aberrant expression of epigenetic regulators, in particular histone deacetylases (HDACs) to chronic obstructive pulmonary disease (COPD), a condition with an increased risk of developing NSCLC [69,70].
2.7. Aberrant Levels of Histone Modifying Enzymes in NSCLC
Histone PTMs are carried out by several diverse families of proteins. The best studied of these families are the lysine acetyltransferases (KATs), histone deacetylases (HDACs), K-methyltransferases (KMTs) and K-demethylases (KDMs). The expression of many of these enzymes has now been shown to be altered in NSCLC.
2.7.1. HDACs
The family of HDACs is separated into several classes (Classes I-IV) based on their homology to yeast proteins [71,72]. The Class I members comprise HDACs 1-3 and HDAC8, and in NSCLC, elevated levels of HDAC1 mRNA are found in higher stage (Stage III or IV) cancers [69,70,73], while other members of the class I HDACs have also been observed to have altered expression (Table 2, Figure 1) [69,70,73]. Elevated levels of HDAC3 protein are found in 92% of the SCC subtype (Table 2) [69,70,73], and more recently high expression of HDAC3 has been shown to correlate with poor prognosis in the adenocarcinoma subtype of NSCLC (Figure 1) [74].
The Class II family members comprise HDACs 4, 5, 6, 7, 9 and 10. An analysis of this subclass in NSCLC revealed that reduced mRNA expression for each family member occurred in NSCLC (Figure 1) and was associated with poor prognosis and could act as an independent predictor of poor prognosis with HDAC10 having the strongest predictive capacity (Table 2) [75].
Of the Class III family members Sirtuins (Sirts1-7), Sirt1 has been shown to have altered expression in lung cancer, with 46.4% (45/97) of tumors showing an absence or low expression of SIRT1 protein (Table 2), which was linked to poor prognosis [76].
HDACs form large multi-protein complexes to regulate gene expression [77]. mSin3A, a critical component serving as a scaffold on which the multi-component HDAC co-repressor complex assembles, has also been observed to have decreased expression in NSCLC (Table 2) [78].
ATP-dependent SWI/SNF chromatin remodeling complexes members have also been shown to be altered in the lung. In NSCLC cell lines, the SWI/SNF complex has been found to form a larger complex containing neuron-restrictive silencer factor (NRSF) and its co-repressors, mSin3A and CoREST and it has been suggested that deregulation of NRSF-regulated genes in NSCLC could in fact contribute to enhanced tumorigenicity [69,70,73]. Indeed, expression of the SWI/SNF ATPase subunits, BRG1 and BRM (BRG1/BRM), have been shown to be either mutated or lost in approximately 30% of human non-small lung cancer cell lines (Table 2, Figure 1) [69]. In primary NSCLC tumors, 10% had loss of both BRG1 and BRM, correlating with the poorest prognosis [69]. Using multiple tissue arrays 12 core proteins involved with chromatin remodeling machinery were examined in 300 NSCLC samples (150 adenocarcinomas and 150 squamous cell carcinomas). Two distinct clusters emerged: one containing BRM, Ini-1, retinoblastoma, mSin3A, HDAC1, and HAT1, the other BRG1, BAF155, HDAC2, BAF170, and RbAP48 [69]. Positive nuclear BRM (N-BRM) staining correlated with a favorable prognosis in patients with a five year-survival of 53.5% compared with 32.3% for those patients with tumors that were negative for N-BRM (P = 0.015). Copositivity for both N-BRM and nuclear BRG1 had an increased five year-survival of 72% compared with 33.6% (P = 0.013) in patients whose tumors were positive for either, or negative for both markers. In contrast, membranous BRM (M-BRM) staining correlated with a poorer prognosis in adenocarcinoma patients with a five year-survival of 16.7% compared with those without M-BRM staining (38.1%; P = 0.016) [69].
The expression of Metastasis-associated protein 1 (MTA-1) has been shown to be significantly elevated in NSCLC and was found to be associated with both tumor invasiveness and metastasis (Table 2) [79]. Both MTA-1 and MTA-2 have been shown to functionally associate with histone deacetylases [80], suggesting that the overexpression of MTA's may cause aberrant HDAC activity which may be involved with invasiveness and metastasis of NSCLC.
The E2F transcription factor 1 (E2F1) positively regulates cell cycle progression and also functions as a potent inducer of apoptosis, especially when activated by DNA damage. Studies have now identified miR-449a and mIR449b as microRNAs regulated by this transcription factor [81,82]. mIR-449a has now been shown to target both SIRT1 [82] and HDAC1 [83] (Table 1). Furthermore, levels of miR-449a have been shown to be reduced in lung cancers compared to normal lung tissue (Table 1) [84], and this has functionally been associated with an aberrant epigenetic chromatin configuration through histone H3 Lys27 trimethylation [81]. As a consequence of this, the reduction of miR-449a may in part explain why levels of HDAC1 are frequently overexpressed in lung cancer (Table 1) [69,70,73].
2.7.2. K-Acetyltransferases (KATs; Formerly Acetyltransferases)
K-acetyltransferases (formerly known as either histone acetyltransferases or lysine acetyltransferases [85]) play a multitude of roles in the cell, and we have suggested that these enzymes play important roles in lung cancer [69,70]. For instance, in NSCLC the protein levels of K-acetyltransferase KAT3A (formerly CBP) and E2F-1 were found to be significantly higher in the tumor area than in the corresponding normal epithelium (p < 0.001) (Table 2, Figure 1)) [86]. Mutations within KAT3A have also been described in a small proportion of lung cancer patients [87]. KAT13B (or SRC-3) is also emerging as an important acetyltransferase whose expression is altered or important in cancer. The expression of KAT13B has been shown to be overexpressed in 27% of non-small cell lung cancer (NSCLC) patients correlating with poor disease-free (P = 0.0015) and overall (P = 0.0008) survival (Table 2, Figure 1) [88]. In breast cancer, a recently identified splice variant called SRC-3Delta4 has been found to act as an adaptor protein between EGFR and its downstream signaling molecule FAK to coordinately regulate EGF-induced cell migration, and overexpression of this KAT isoform leads to enhanced breast cancer metastasis to the lung [89]. It will be interesting to see if this splice isoform is overexpressed in NSCLC.
2.7.3. K-Methyltransferases (KMTs; Formerly Lysine Methyltransferases)
K-methyltransferases or KMTs (previously known as lysine methyltransferases [85]) function to add methyl groups to lysine residues as mono-, di- or tri- methylation [90]. Polymorphisms and haplotypes in KMTs have been associated with the risk of developing NSCLC. Polymorphisms and haplotypes associated with a reduced risk of NSCLC have been found in KMT6 (EZH2) and KMT8 (RIZ1) [91,92], while in contrast, polymorphisms in KMT1B (SUV39H2) are associated with an increased lung cancer risk (Table 2) [93].
Altered expression of KMTs has also been shown to be important in NSCLC (Figure 1).
In a lung cancer cell line model where bronchial epithelial (NHBE) cells were immortalized by overexpression of telomerase, SV40 large T antigen, and Ras, it was noted that several KMTs had high expression levels. These were KMT1A (SUV39H1), KMT1C (G9a), KMT1E (SETDB1), KMT4 (DOT1L) and KMT6 (EZH2) (Table 2) [68]. Of these, KMT1C (G9a) when expressed in NSCLC cells causes an aggressive phenotype promoting both invasion and metastasis by silencing expression of the cell adhesion molecule Ep-CAM [94], while overexpression of KMT6 (EZH2) has also been linked to both poor prognosis and cancer aggressiveness in NSCLC [95]. Interestingly, mIR-138 has recently been identified as a miRNA that targets KMT6 (EZH2) [96], and in a study of miRNA profiles for never-smoker lung cancers, this miRNA was a uniquely downregulated miRNA compared to tumors derived from smokers [97].
Menin, the product of the Multiple endocrine neoplasia type 1 (MEN1) gene has been shown to associate with various lysine methyltransferases [98], and mice mutated for Men1 develop NSCLC tumors [99]. In association with KMT6 (EZH2), menin has been shown to suppress lung adenocarcinoma cancer formation by repressing the growth factor pleiotrophin [100], a consequence of which is repression of lung cancer cell migration [101].
2.7.4. Arginine Methyltransferases
Histones can not only be methylated on lysines, they can also be methylated on arginine residues [102]. The enzymes involved are grouped into the protein arginine methyltransferase family (PRMTs) with 11 family members identified to date [103]. Various functions for this family of proteins have emerged including signal transduction, mRNA splicing, transcriptional control, protein translocation, and DNA repair [102]. The expression of various PRMTs has been examined in the mouse lung and for all members (PRMT1-7) examined, strong expression of their respective mRNAs was observed [104]. Immunohistochemical analysis identified strong homogeneous staining of PRMT1 in airway and alveolar type II epithelial cells. In contrast, PRMT2, 3, and 5 exhibited intermittent staining, and were localized in the cytosol of nonciliated airway epithelial cells and alveolar epithelial cells, and were notably absent in vascular smooth muscle and endothelial cells [104]. PRMT4 was present in the apical part of airway epithelial cells and in alveolar epithelial type II cells. Under hypoxia, a significant difference in PRMT2 protein expression was observed, whereas no significant expression differences for all other PRMT isoforms [104]. PRMT4 (also known as CARM1) has however, now been shown to be critical for the control of pulmonary epithelial cell proliferation and differentiation. During embryonic development, loss of CARM1 results in hyperproliferation of pulmonary epithelial cells (particularly alveolar type II cells), and the lungs of newborn mice have a substantially reduced airspace compared with their wild-type littermates. Due to this hyperproliferation lungs from mice lacking CARM1 have immature alveolar type II cells and an absence of alveolar type I cells [105]. Altered expression of this PRMT has been observed in prostate and colon cancer [106-108], but has yet to be fully explored in NSCLC. In this regard significantly upregulated expression of both PRMT1 and PRMT6 mRNA has been observed in NSCLC (Table 2, Figure 1) [109]. Furthermore, knockdown of PRMT1 and PRMT6 in three NSCLC cell lines was associated with a significant suppression of cell growth [109].
2.7.5. K-Demethylases (KDMs; Formerly Lysine Demethylases)
KDMs are a large family of proteins that catalyze the removal of mono-, di-, and tri- methyl marks on lysine residues in both histones and non-histone proteins [110,111]. Using cDNA microarray analysis, Hayami et al. identified KDM1A (formerly known as LSD1) as being elevated in bladder, lung and colorectal carcinomas (Table 2, Figure 1), and siRNA knockdown of KDM1 resulted in a decrease of various lung and bladder cancer cell lines, whereas overexpression promoted proliferation [112]. KDM5B (also known as JARID1B/PLU-1) has also been identified as being highly elevated in lung tumor tissues (Table 2) compared with corresponding non-neoplastic tissues and siRNA knockdown of KDM5B significantly suppressed the proliferation of cancer cells and increased the number of cells in sub-G1 phase [113].
Hypoxia has also been shown to play a role in KDM5A (JARID1A) activity in the lung bronchial epithelial cell line Beas-2B and NSCLC adenocarcinoma cell line A549. Under hypoxic conditions, total H3K4 demethylase activity is decreased/inhibited, and knockdown of the major H3K4 demethylase identified in Beas-2B, KDM-5, abrogated this effect [114].
Many K-Demethylases contain a specific JumonjiC (JmjC) domain essential for their demethylase activity [110]. A JmjC containing protein called Mineral Dust-Induced Gene (MDIG)/MYC-Induced Nuclear Antigen (MINA) has been shown to be overexpressed in NSCLC and promotes ribosomal RNA (rRNA) expression through demethylation of tri-methyl lysine 9 on histone H3 at the ribosomal RNA promoter (Figure 1) [115]. To our knowledge, this protein has yet to be assigned within the KDM nomenclature.
2.7.6. Arginine Demethylases
Few arginine demethylases have currently been identified. There is one report on the protein JMJD6 demonstrating that it functions to demethylate histone H3 at arginine 2 (H3R2) and histone H4 at arginine 3 (H4R3) [116]. JMJD6 was originally identified as Phosphatidyl Serine Receptor (PSR) [117], which in bronchial epithelial cells and alveolar cells is associated with the phagocytosis of apoptotic eosinophils [118, 119]. No data has yet emerged on the expression of JMJD6 in NSCLC, although the A549 NSCLC cell line has been shown to express JMJD6 [119].
Another mechanism by which arginine methylation is reversed is through a process known as demethylimination where deimination of the methylated arginine gives rise to citrulline [120]. The protein family responsible, peptidylarginine deiminase (PADI) enzymes currently comprises six members PADI1-6, of which PADI4 is capable of catalyzing the conversion of histone arginine methylation to histone citrullination [120]. Significant overexpression of PADI4 has been observed in NSCLC tumors (Table 2, Figure 1) [121], indicating that aberrant regulation of histone arginine methylation may be important in this disease. Furthermore PADI4 has also been shown to interact with HDAC1 [122], another histone modifying enzyme upregulated in NSCLC. The work by Fuks and colleagues suggest that PADI4 and HDAC1 collaborate to generate a repressive chromatin environment [122], indicating that aberrant repression of critical genes may be an important part of lung cancer tumorigenesis
2.8. Specialized Histone PTMs Associated with DNA Double Strand Breaks Caused by Cisplatin
One histone PTM associated with DNA damage repair is gamma histone H2AX (gamma-H2AX). Precancerous lesions of the lung were found to contain signs of a DNA damage response, which included the presence of histone H2AX. This has led to the suggestion by the authors that DNA replication stress is a significant factor in cancer development [123]. The gene Tumor Suppressor Candidate 4; (TUSC4), also known as NPRL2 has now been linked directly to cisplatin sensitivity. In a study of 40 NSCLC cell lines expression of NPRL2 was significantly and reciprocally correlated to cisplatin sensitivity [124], and exogenously expression of NPRL2 resulted in a 2- to 3-fold increase in induction of apoptosis of cells treated with cisplatin [124]. NPRL2 and cisplatin result in the regulation of key components of the DNA-damage checkpoint pathway by promoting (a) downstream gamma-H2AX formation in vitro and in vivo and (b) higher Chk1 and Chk2 kinase activity resulting in higher levels of G2/M arrest in tumor cells through elevated levels of cell cycle checkpoint [125].
2.9. Histone Modifying Enzymes and Cisplatin Resistance
In many solid tumors, various histone modifying enzymes have now been linked to resistance to cisplatin. In lung cancer these include the lysine acetyltransferases KAT13D (Clock) [126], KAT5 (Tip60) [127], KAT2B (PCAF) [128] and KAT13B (SRC-3) [88] (Table 3, Figure 1), while SIRT-1 expression has been linked to cisplatin resistance in epidermoid and hepatoma cells [129]. A recent study has linked both KAT5 (Tip60) and HDAC6 as important regulators of lung cancer cell responses to cisplatin (Table 3). The acetyltransferase Tip60 acetylates an important splicing factor SRSF2 on its lysine 52 residue promoting its proteasomal degradation, while HDAC6 abrogates this. In response to cisplatin an acetylation/phosphorylation signaling network regulates both the accumulation of SRSF2 and splicing of caspase-8 pre-mRNA and determines whether cells undergo apoptosis or G(2)/M cell cycle arrest [130]. Expression of CBP/p300-Interacting Transactivator, with GLU/ASP-Rich C-Terminal Domain, 2; (CITED2 ), has been shown to be involved with cisplatin resistance in cancer cell lines by a process dependent upon p53. Chao and colleagues demonstrated that knockdown of CITED2 sensitized cells in p53 positive cells, whereas H1299 cells which are p53 defective had negligible responses to cisplatin. Knockdown of CITED2 induced KAT3A-mediated p53 acetylation (Lys373) preventing ubiquitination and turnover of p53. This resulted in increased levels of the p53 target Bax, and was further increased following cisplatin treatment [131].
2.10. BRCA1 and the DNA Damage Response
The Breast Cancer 1 Gene (BRCA1) has two important functions (i) regulation of gene transcription and (ii) the response to DNA damage (DNA Repair) [132]. Indeed BRCA1 acts mainly as a tumor suppressor through transcriptionally regulating genes involved with DNA repair [133]. Loss of BRCA1 expression is a frequent event in NSCLC [134,135]. Studies have now shown that the loss of BRCA1 and BRCA2 expression can be due to epigenetic inactivation via DNA CpG methylation in 18-30% of tumors [134,136].
BRCA1 forms several complexes in response to DNA damage, and is emerging as a critical regulator of genome integrity through its ability to execute and coordinate various aspects of the DNA damage response [137].
BRCA1 has been shown to form a heterodimer with BARD1 to form an ubiquitin E3 ligase activity [138] that plays an essential role in response to DNA damage. Cisplatin has been shown to directly bind to BRCA1 and its transcriptional transactivation activity is dramatically diminished in the presence of multiple cisplatin-damaged DNA sites [139]. Furthermore, when complexed with BARD1, cisplatin treatment results in a significantly reduced E3 ligase activity [140].
One major multi-protein assembly with which BRCA1 has now been associated with is the Mi-2/nucleosome remodeling and deacetylase NuRD complex (Table 3). In response to DNA double strand breaks (DSBs) induced by ionizing radiation, the catalytic subunit of the NuRD complex CHD4, stimulates the formation of ubiquitin conjugates that facilitate the accrual of RNF168 and BRCA1 proteins to promote DSB repair [141,142].
2.11. BRCA1 and Sensitivity to Cisplatin
A clear indication that BRCA1 may be associated with sensitivity to cisplatin came from studies of breast cancer in mice. Cells deficient for BRCA1 were sensitive to cisplatin, while restoration of BRCA1 resulted in increased resistance, and xenografts of cells deficient for BRCA1 were more sensitive to cisplatin than those where BRCA1 had been restored [143-145].
In a study of ovarian cancer, of 115 primary sporadic ovarian carcinomas, 39 (34%) had low BRCA1 protein and 49 (42%) had low BRCA2 expression. Restoration of BRCA1 and BRCA2 mediates resistance to platinum chemotherapy in recurrent BRCA1 and BRCA2 mutated hereditary ovarian carcinomas [146].
In lung cancer the first clinical evidence that BRCA1 levels may predict response to cisplatin came from a study of patients treated with Gemcitabine/Cisplatin in the neoadjuvant setting. In this study patients whose tumors had low levels of BRCA1 mRNA had a better outcome than those whose tumors high levels of BRCA1 mRNA [135,147]. Wang et al. confirmed that BRCA1 expression levels in metastatic malignant effusions were negatively correlated with sensitivity to cisplatin (Table 3) [148]. Recently, in a prospective non-randomized phase II clinical trial, Rosell and colleagues tested the possibility that BRCA1 could be used to customize treatment of patients with NSCLC. Patients were segregated and treated based on EGFR mutation status and BRCA1 level. Patients with EGFR mutations received erlotinib, and those without EGFR mutations received chemotherapy with or without cisplatin based on their BRCA1 mRNA levels: low, cisplatin plus gemcitabine; intermediate, cisplatin plus docetaxel; high, docetaxel alone. In addition to BRCA1 the authors also examined its interacting partner proteins (RAP80 and Abraxas) for additional prognostic value. From this analysis it was found that patients with both low BRCA1 and low RAP80, had a median survival exceeding 26 months compared to 11 months for patients with low BRCA1 alone. RAP80 was a significant factor for survival in patients treated according to BRCA1 levels (hazard ratio, 1.3 [95% CI, 1–1.7]; P = 0.05) [149].
For patients with high BRCA1 levels, anti-tubulin-containing regimens have emerged as exciting contenders for therapeutic intervention strategies [133]. Indeed in a recent clinical study, NSCLC patients with high BRCA1 mRNA expression were found to benefit more from this type of treatment (8.7 vs. 13.0 months) [150].
2.12. BRCA1, K-Methyltransferases and Acquired Cisplatin Resistance
BRCA1-deficient mouse mammary tumor cells are selectively sensitive to an inhibitor of EZH2 [151]. EZH2 (also known as KMT6) is the catalytic subunit of Polycomb repressive complex 2 (PRC2), and is a highly conserved histone methyltransferase that targets lysine-27 of histone H3 [152,153]. A study on the expression of this protein in NSCLC found that patients who had high EZH2 expression in tumor cells had a poorer prognosis than patients who had low EZH2 expression in tumor cells for all pathologic stages of NSCLC (P = 0.001), and that high EZH2 expression was correlated significantly with nonadenocarcinoma histology (P = 0.001) [95] (Table 3). This may be due to the aberrant regulation of mIR-101 which has been shown to regulate expression of EZH2 [154] and this miRNA has been found to be downregulated in NSCLC particularly in the squamous cell subtype [155]. As overexpression of EZH2 has been shown to contribute to the development of acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo [156], then potentially NSCLC patients with either high BRCA1 or EZH2 levels might potentially benefit from treatments with poly(ADP-ribose) polymerase (PARP) inhibitors such as (DZNep), or could potentially be targeted to induce mIR-101.
2.13. BRCA1, the p53/p63/p73 Network, DNA Methylation and Cisplatin Resistance
It is well established that one of the transcription factors which BRCA1 associates with is the Tumor Protein p53 (p53) [157]. This protein plays important roles in regulating the cellular response to DNA damage [158], and levels of p53 has been shown to have prognostic value in NSCLC. In the JBR.10 trial which examined 482 patients with completely resected stage IB and II non-small-cell lung cancer (NSCLC) who received four cycles of adjuvant cisplatin plus vinorelbine or observation alone, patients with p53 protein overexpression had a significantly shortened survival [159]. However, wild-type p53 has also recently been shown to be required for the induction of COX-2 in response to cisplatin treatment in NSCLC cell lines [160]. High expression of this inflammatory enzyme has been shown to inhibit chemotherapy-induced apoptosis. This is in contrast to the findings of Shepherd and colleagues where p53 overexpression was a predictive for significantly greater benefit from adjuvant chemotherapy in completely resected NSCLC patients [159].
Other members of the p53 family also may play a role in cisplatin sensitivity. This family of proteins includes Tumor Protein 63 (p63) and Tumor Protein 73 (p73). An indication that these proteins may also be important in cisplatin sensitivity came from a study of “triple-negative” breast cancer tumors, which found that p63 controlled a pathway for p73-dependent cisplatin sensitivity [161]. A link between DNA CpG methylation and these proteins was recently shown in ovarian carcinoma [162]. BRCA1-deficient cells exhibited hypermethylation within a p73 regulatory region, which included the binding site for the p73 transcriptional repressor ZEB1, leading to the abrogation of ZEB1 binding and increased expression of transactivating p73 isoforms (TAp73). Cisplatin chemotherapy induced TAp73 target genes specifically in BRCA1-deficient cells, and knockdown of TAp73 in these cells caused chemoresistance while having little or no effect on BRCA1-expressing tumor cells. In primary ovarian carcinomas, ZEB1 binding site methylation and TAp73 expression correlated with BRCA1 status and with clinical response [162]. ZEB1 is also a master regulator of the epithelial-mesenchymal transition (EMT) and reports have demonstrated that ZEB1 is important for this process in lung cancer through its regulation of many EMT genes including E-cadherin [163-165], and knockdown of ZEB1 results in the suppression of anchorage-independent cell growth of lung cancer cells [166]. It is interesting to note that mIR-200 has been shown to target ZEB1 (Table 1) [45,167,168], and as levels of mIR-200c are known to be decreased in NSCLC [46], it may be important to determine the BRCA1 status of these tumors.
2.14. Epigenetic Targeting Therapies and Reversal of Platinum Based Resistance?
A pleiotropic agent which can act as an HDACi (Phenylbutyrate) has been shown to sensitize head and neck cancers to cisplatin by interfering with the Fanconi anemia and BRCA (FA/BRCA) pathway [169].
ZEB1 regulates E-cadherin expression via recruitment of HDACs and several studies have shown that HDACi can both induce E-cadherin and downregulate ZEB1 indicating a potential mechanism to target ZEB1 mediated effects in NSCLC [163,170,171]
Activation of Transcription Factor 3 (ATF-3) [172], and Activation of Transcription Factor 4 (ATF-4) have both been shown to regulate cisplatin resistance [173]. In this regard ATF-4 has been shown to associate with the lysine acetyltransferase KAT13D to regulate this resistance [126], and downregulation of this acetyltransferase confers sensitivity to cisplatin. However, it has also been shown that the HDACi M344 increased the levels of ATF-3 in A549 cells and enhanced the cytotoxic effects of cisplatin in this cancer cell line [174]
In a recent phase II randomized, double-blinded, placebo-controlled study evaluated the efficacy of vorinostat in combination with carboplatin and paclitaxel in patients with advanced-stage NSCLC. The results indicated a response rate for vorinostat of 34% with vorinostat versus placebo 12.5% (P = 0.02). There was also a trend although not significant toward improvement in both median progression-free survival (6.0 months vs. 4.1 months; P = 0.48) and overall survival (13.0 months vs. 9.7 months; P = 0.17) in the vorinostat arm [175].
Curcumin a lysine acetyltransferase inhibitor has also been shown to promote apoptosis in an NSCLC cell line model of multi-drug resistance through downregulation of mIR-186 [176].
As discussed in previous sections, several genes linked to cisplatin resistance in NSCLC have been shown to be repressed or silenced by DNA CpG methylation and cell treatments with DNMTi have been able to reactivate their expression.
3. Conclusions
It is clear from the above sections that epigenetics and the cellular machinery involved in regulating epigenetic regulation of gene expression play important roles in NSCLC tumorigenesis and increasing evidence is demonstrating a clear link between epigenetics and cisplatin resistance in this disease. As we continue to unravel the intricacies of the epigenome, we may be able to more effectively target diseases such as NSCLC by identifying those patients who may be able to benefit from platinum based chemotherapies, and perhaps resensitize patients to chemotherapy using epigenetic targeting. Given the relatively small benefits of platinum based chemotherapy, it is incumbent on medical oncologists and translational scientists to identify those patients most likely to benefit from therapy prior to starting treatment. It is clear from the data presented in this review that there is an immediate potential for testing some of the observed genes/miRNAs within the clinical setting to examine their utility in predicting response to cisplatin based therapy. This may lead to the development of a panel of markers or diagnostic tests that will allow cisplatin to be used only in those patients likely to benefit from therapy without exposing those unlikely to benefit to potential side-effects.

{kind=link}
| miRNA | Target | Altered in NSCLC | Ref. |
|---|---|---|---|
| miR-29a | DNMT1, -3A, -3B | downregulated | [25] |
| mIR-29b | DNMT1, -3A, -3B | downregulated | [25] |
| mIR-29c | DNMT1, -3A, -3B | downregulated | [25] |
| mIR-101 | KMT6 | downregulated | [155] |
| mIR-138 | KMT6 | Reduced in tumors of never-smokers | [97] |
| mIR-143 | DNMT3A | downregulated | [30] |
| mIR-152 | DNMT1 | Reduced in docetaxol resistant patients | [31] |
| mIR-181a | Bax/Bcl-2 | NSCLC cell line model Downregulated in NSCLC | [44] [30] |
| mIR-181b | Bax/Bcl2 | NSCLC cell line model | [43] |
| miR-200c | ZEB1 | Downregulated in NSCLC restoration of expression increases sensitivity to cisplatin | [46] |
| mIR-449a | SIRT1, HDAC1 | downregulated | [84] |
| mIR-630 | Blocks DNA Damage Response | NSCLC cell line model | [44] |
| Gene | Comments | Reference |
|---|---|---|
| DNMT1 | Elevated in NSCLC, prognostic | [22-24] |
| DNMT3A | Elevated in NSCLC, prognostic | [22-24] |
| DNMT3B | Elevated in NSCLC, prognostic | [22-24] |
| HDAC1 | Elevated in NSCLC | [69,70,73] |
| HDAC2 | Elevated in NSCLC | [69,70,73] |
| HDAC3 | Elevated in NSCLC, linked to poor prognosis | [69,70,73,74] |
| HDAC4 | Reduced in NSCLC, associated with poor prognosis | [75] |
| HDAC5 | Reduced in NSCLC, associated with poor prognosis | [75] |
| HDAC6 | Reduced in NSCLC, associated with poor prognosis | [75] |
| HDAC7 | Reduced in NSCLC, associated with poor prognosis | [75] |
| HDAC9 | Reduced in NSCLC, associated with poor prognosis | [75] |
| HDAC10 | Reduced in NSCLC, associated with poor prognosis | [75] |
| SIRT1 | Reduced in NSCLC, associated with poor prognosis | [76] |
| mSin3A | Reduced in NSCLC | [78] |
| BRG1 | lost or mutated in a proportion of NSCLC | [69] |
| BRM | lost or mutated in a proportion of NSCLC | [69] |
| MTA-1 | Elevated in NSCLC | [79] |
| KAT3A | Elevated in NSCLC, mutated in a small proportion | [86,87] |
| KAT13B | Elevated in 27% of NSCLC, prognostic | [88] |
| KMT1B | Polymorphisms associated with increased risk of NSCLC | [93] |
| KMT6 | Polymorphisms associated with reduced risk of NSCLC Overexpression linked to poor prognosis in NSCLC | [92] [95] |
| KMT8 | Polymorphisms associated with reduced risk of NSCLC | [91] |
| PRMT1 | Upregulated mRNA in NSCLC | [109] |
| PRMT6 | Upregulated mRNA in NSCLC | [109] |
| KDM1A | Elevated in NSCLC | [112] |
| KDM5B | Elevated in NSCLC | [113] |
| MDIG/MINA | Putative KDM, Elevated in NSCLC | [115] |
| PADI4 | Elevated in NSCLC | [121] |
References
- Mascaux, C.; Peled, N.; Garg, K.; Kato, Y.; Wynes, M.W.; Hirsch, F.R. Early detection and screening of lung cancer. Expert Rev. Mol. Diagn. 2010, 10, 799–815. [Google Scholar]
- Jemal, A.; Siegel, R.; Ward, E.; Hao, Y.; Xu, J.; Thun, M.J. Cancer statistics, 2009. CA Cancer J. Clin. 2009, 59, 225–249. [Google Scholar]
- Sato, M.; Shames, D.S.; Gazdar, A.F.; Minna, J.D. A translational view of the molecular pathogenesis of lung cancer. J. Thorac. Oncol. 2007, 2, 327–343. [Google Scholar]
- Allingham-Hawkins, D.; Lea, A.; Levine, S. ERCC1 Expression Analysis to Guide Therapy in Non-Small Cell Lung Cancer. PLoS Curr. 2010, 2, RRN1202. [Google Scholar]
- Zimmerman, A.L.; Wu, S. MicroRNAs, cancer and cancer stem cells. Cancer Lett. 2011, 300, 10–19. [Google Scholar]
- Du, L.; Schageman, J.J.; Irnov; Girard, L.; Hammond, S.M.; Minna, J.D.; Gazdar, A.F.; Pertsemlidis, A. MicroRNA expression distinguishes SCLC from NSCLC lung tumor cells and suggests a possible pathological relationship between SCLCs and NSCLCs. J. Exp. Clin. Cancer Res. 2010, 29, 75. [Google Scholar]
- Bishop, J.A.; Benjamin, H.; Cholakh, H.; Chajut, A.; Clark, D.P.; Westra, W.H. Accurate classification of non-small cell lung carcinoma using a novel microRNA-based approach. Clin. Cancer Res. 2010, 16, 610–619. [Google Scholar]
- Landi, M.T.; Zhao, Y.; Rotunno, M.; Koshiol, J.; Liu, H.; Bergen, A.W.; Rubagotti, M.; Goldstein, A.M.; Linnoila, I.; Marincola, F.M.; Tucker, M.A.; Bertazzi, P.A.; Pesatori, A.C.; Caporaso, N.E.; McShane, L.M.; Wang, E. MicroRNA expression differentiates histology and predicts survival of lung cancer. Clin. Cancer Res. 2010, 16, 430–441. [Google Scholar]
- Lebanony, D.; Benjamin, H.; Gilad, S.; Ezagouri, M.; Dov, A.; Ashkenazi, K.; Gefen, N.; Izraeli, S.; Rechavi, G.; Pass, H.; Nonaka, D.; Li, J.; Spector, Y.; Rosenfeld, N.; Chajut, A.; Cohen, D.; Aharonov, R.; Mansukhani, M. Diagnostic assay based on hsa-miR-205 expression distinguishes squamous from nonsquamous non-small-cell lung carcinoma. J. Clin. Oncol. 2009, 27, 2030–2037. [Google Scholar]
- Shen, J.; Todd, N.W.; Zhang, H.; Yu, L.; Lingxiao, X.; Mei, Y.; Guarnera, M.; Liao, J.; Chou, A.; Lu, C.L.; Jiang, Z.; Fang, H.; Katz, R.L.; Jiang, F. Plasma microRNAs as potential biomarkers for non-small-cell lung cancer. Lab. Invest. 2010. [Google Scholar] [CrossRef]
- Chen, X.; Ba, Y.; Ma, L.; Cai, X.; Yin, Y.; Wang, K.; Guo, J.; Zhang, Y.; Chen, J.; Guo, X.; Li, Q.; Li, X.; Wang, W.; Zhang, Y.; Wang, J.; Jiang, X.; Xiang, Y.; Xu, C.; Zheng, P.; Zhang, J.; Li, R.; Zhang, H.; Shang, X.; Gong, T.; Ning, G.; Wang, J.; Zen, K.; Zhang, J.; Zhang, C.Y. Characterization of microRNAs in serum: a novel class of biomarkers for diagnosis of cancer and other diseases. Cell Res. 2008, 18, 997–1006. [Google Scholar]
- Yanaihara, N.; Caplen, N.; Bowman, E.; Seike, M.; Kumamoto, K.; Yi, M.; Stephens, R.M.; Okamoto, A.; Yokota, J.; Tanaka, T.; Calin, G.A.; Liu, C.G.; Croce, C.M.; Harris, C.C. Unique microRNA molecular profiles in lung cancer diagnosis and prognosis. Cancer Cell 2006, 9, 189–198. [Google Scholar]
- Raponi, M.; Dossey, L.; Jatkoe, T.; Wu, X.; Chen, G.; Fan, H.; Beer, D.G. MicroRNA classifiers for predicting prognosis of squamous cell lung cancer. Cancer Res. 2009, 69, 5776–5783. [Google Scholar]
- Du, L.; Pertsemlidis, A. microRNAs and lung cancer: tumors and 22-mers. Cancer Metastasis Rev. 2010, 29, 109–122. [Google Scholar]
- Fabbri, M.; Calin, G.A. Epigenetics and miRNAs in human cancer. Adv. Genet. 2010, 70, 87–99. [Google Scholar]
- Iorio, M.V.; Piovan, C.; Croce, C.M. Interplay between microRNAs and the epigenetic machinery: An intricate network. Biochim. Biophys. Acta 2010, 1799, 694–701. [Google Scholar]
- Damiani, L.A.; Yingling, C.M.; Leng, S.; Romo, P.E.; Nakamura, J.; Belinsky, S.A. Carcinogen-induced gene promoter hypermethylation is mediated by DNMT1 and causal for transformation of immortalized bronchial epithelial cells. Cancer Res. 2008, 68, 9005–9014. [Google Scholar]
- Heller, G.; Zielinski, C.C.; Zochbauer-Muller, S. Lung cancer: from single-gene methylation to methylome profiling. Cancer Metastasis Rev. 2010, 29, 95–107. [Google Scholar]
- Sharma, S.; Kelly, T.K.; Jones, P.A. Epigenetics in cancer. Carcinogenesis 2010, 31, 27–36. [Google Scholar]
- Kulis, M.; Esteller, M. DNA methylation and cancer. Adv. Genet 2010, 70, 27–56. [Google Scholar]
- Jones, P.A.; Baylin, S.B. The epigenomics of cancer. Cell 2007, 128, 683–692. [Google Scholar]
- Lin, R.K.; Hsu, H.S.; Chang, J.W.; Chen, C.Y.; Chen, J.T.; Wang, Y.C. Alteration of DNA methyltransferases contributes to 5′CpG methylation and poor prognosis in lung cancer. Lung Cancer 2007, 55, 205–213. [Google Scholar]
- Kim, H.; Kwon, Y.M.; Kim, J.S.; Han, J.; Shim, Y.M.; Park, J.; Kim, D.H. Elevated mRNA levels of DNA methyltransferase-1 as an independent prognostic factor in primary nonsmall cell lung cancer. Cancer 2006, 107, 1042–1049. [Google Scholar]
- Vallbohmer, D.; Brabender, J.; Yang, D.; Schneider, P.M.; Metzger, R.; Danenberg, K.D.; Holscher, A.H.; Danenberg, P.V. DNA methyltransferases messenger RNA expression and aberrant methylation of CpG islands in non-small-cell lung cancer: association and prognostic value. Clin. Lung Cancer. 2006, 8, 39–44. [Google Scholar]
- Fabbri, M.; Garzon, R.; Cimmino, A.; Liu, Z.; Zanesi, N.; Callegari, E.; Liu, S.; Alder, H.; Costinean, S.; Fernandez-Cymering, C.; Volinia, S.; Guler, G.; Morrison, C.D.; Chan, K.K.; Marcucci, G.; Calin, G.A.; Huebner, K.; Croce, C.M. MicroRNA-29 family reverts aberrant methylation in lung cancer by targeting DNA methyltransferases 3A and 3B. Proc. Natl. Acad. Sci. USA 2007, 104, 15805–15810. [Google Scholar]
- Garzon, R.; Liu, S.; Fabbri, M.; Liu, Z.; Heaphy, C.E.; Callegari, E.; Schwind, S.; Pang, J.; Yu, J.; Muthusamy, N.; Havelange, V.; Volinia, S.; Blum, W.; Rush, L.J.; Perrotti, D.; Andreeff, M.; Bloomfield, C.D.; Byrd, J.C.; Chan, K.; Wu, L.C.; Croce, C.M.; Marcucci, G. MicroRNA-29b induces global DNA hypomethylation and tumor suppressor gene reexpression in acute myeloid leukemia by targeting directly DNMT3A and 3B and indirectly DNMT1. Blood 2009, 113, 6411–6418. [Google Scholar]
- Pass, H.I.; Goparaju, C.; Ivanov, S.; Donington, J.; Carbone, M.; Hoshen, M.; Cohen, D.; Chajut, A.; Rosenwald, S.; Dan, H.; Benjamin, S.; Aharonov, R. hsa-miR-29c* is linked to the prognosis of malignant pleural mesothelioma. Cancer Res. 2010, 70, 1916–1924. [Google Scholar]
- Braconi, C.; Huang, N.; Patel, T. MicroRNA-dependent regulation of DNA methyltransferase-1 and tumor suppressor gene expression by interleukin-6 in human malignant cholangiocytes. Hepatology 2010, 51, 881–890. [Google Scholar]
- Ng, E.K.; Tsang, W.P.; Ng, S.S.; Jin, H.C.; Yu, J.; Li, J.J.; Rocken, C.; Ebert, M.P.; Kwok, T.T.; Sung, J.J. MicroRNA-143 targets DNA methyltransferases 3A in colorectal cancer. Br. J. Cancer 2009, 101, 699–706. [Google Scholar]
- Gao, W.; Yu, Y.; Cao, H.; Shen, H.; Li, X.; Pan, S.; Shu, Y. Deregulated expression of miR-21, miR-143 and miR-181a in non small cell lung cancer is related to clinicopathologic characteristics or patient prognosis. Biomed. Pharmacother. 2010, 64, 399–408. [Google Scholar]
- Xie, L.; Chen, X.; Wang, L.; Qian, X.; Wang, T.; Wei, J.; Yu, L.; Ding, Y.; Zhang, C.; Liu, B. Cell-free miRNAs may indicate diagnosis and docetaxel sensitivity of tumor cells in malignant effusions. BMC Cancer 2010, 10, 591. [Google Scholar]
- Nyce, J.W. Drug-induced DNA hypermethylation: a potential mediator of acquired drug resistance during cancer chemotherapy. Mutat. Res. 1997, 386, 153–161. [Google Scholar]
- Koul, S.; McKiernan, J.M.; Narayan, G.; Houldsworth, J.; Bacik, J.; Dobrzynski, D.L.; Assaad, A.M.; Mansukhani, M.; Reuter, V.E.; Bosl, G.J.; Chaganti, R.S.; Murty, V.V. Role of promoter hypermethylation in Cisplatin treatment response of male germ cell tumors. Mol. Cancer 2004, 3, 16. [Google Scholar]
- Zhang, P.; Wang, J.; Gao, W.; Yuan, B.Z.; Rogers, J.; Reed, E. CHK2 kinase expression is down-regulated due to promoter methylation in non-small cell lung cancer. Mol. Cancer 2004, 3, 14. [Google Scholar]
- Okamoto, J.; Hirata, T.; Chen, Z.; Zhou, H.M.; Mikami, I.; Li, H.; Yagui-Beltran, A.; Johansson, M.; Coussens, L.M.; Clement, G.; Shi, Y.; Zhang, F.; Koizumi, K.; Shimizu, K.; Jablons, D.; He, B. EMX2 is epigenetically silenced and suppresses growth in human lung cancer. Oncogene 2010, 29, 5969–5975. [Google Scholar]
- Beleford, D.; Liu, Z.; Rattan, R.; Quagliuolo, L.; Boccellino, M.; Baldi, A.; Maguire, J.; Staub, J.; Molina, J.; Shridhar, V. Methylation induced gene silencing of HtrA3 in smoking-related lung cancer. Clin. Cancer Res. 2010, 16, 398–409. [Google Scholar]
- Cao, L.; Petrusca, D.N.; Satpathy, M.; Nakshatri, H.; Petrache, I.; Matei, D. Tissue transglutaminase protects epithelial ovarian cancer cells from cisplatin-induced apoptosis by promoting cell survival signaling. Carcinogenesis 2008, 29, 1893–1900. [Google Scholar]
- Park, K.S.; Kim, H.K.; Lee, J.H.; Choi, Y.B.; Park, S.Y.; Yang, S.H.; Kim, S.Y.; Hong, K.M. Transglutaminase 2 as a cisplatin resistance marker in non-small cell lung cancer. J. Cancer Res. Clin. Oncol. 2010, 136, 493–502. [Google Scholar]
- Ibanez de Caceres, I.; Cortes-Sempere, M.; Moratilla, C.; Machado-Pinilla, R.; Rodriguez-Fanjul, V.; Manguan-Garcia, C.; Cejas, P.; Lopez-Rios, F.; Paz-Ares, L.; de CastroCarpeno, J.; Nistal, M.; Belda-Iniesta, C.; Perona, R. IGFBP-3 hypermethylation-derived deficiency mediates cisplatin resistance in non-small-cell lung cancer. Oncogene 2010, 29, 1681–1690. [Google Scholar]
- Ramirez, J.L.; Rosell, R.; Taron, M.; Sanchez-Ronco, M.; Alberola, V.; de Las Penas, R.; Sanchez, J.M.; Moran, T.; Camps, C.; Massuti, B.; Sanchez, J.J.; Salazar, F.; Catot, S. 14-3-3sigma methylation in pretreatment serum circulating DNA of cisplatin-plus-gemcitabine-treated advanced non-small-cell lung cancer patients predicts survival: The Spanish Lung Cancer Group. J. Clin. Oncol. 2005, 23, 9105–9112. [Google Scholar]
- Bruno, M.D.; Korfhagen, T.R.; Liu, C.; Morrisey, E.E.; Whitsett, J.A. GATA-6 activates transcription of surfactant protein A. J. Biol. Chem. 2000, 275, 1043–1049. [Google Scholar]
- Salazar, F.; Molina, M.A.; Sanchez-Ronco, M.; Moran, T.; Ramirez, J.L.; Sanchez, J.M.; Stahel, R.; Garrido, P.; Cobo, M.; Isla, D.; Bertran-Alamillo, J.; Massuti, B.; Cardenal, F.; Manegold, C.; Lianes, P.; Trigo, J.M.; Sanchez, J.J.; Taron, M.; Rosell, R. First-line therapy and methylation status of CHFR in serum influence outcome to chemotherapy versus EGFR tyrosine kinase inhibitors as second-line therapy in stage IV non-small-cell lung cancer patients. Lung Cancer 2011, 72, 84–91. [Google Scholar]
- Zhu, W.; Shan, X.; Wang, T.; Shu, Y.; Liu, P. miR-181b modulates multidrug resistance by targeting BCL2 in human cancer cell lines. Int. J. Cancer 2010, 127, 2520–2529. [Google Scholar]
- Galluzzi, L.; Morselli, E.; Vitale, I.; Kepp, O.; Senovilla, L.; Criollo, A.; Servant, N.; Paccard, C.; Hupe, P.; Robert, T.; Ripoche, H.; Lazar, V.; Harel-Bellan, A.; Dessen, P.; Barillot, E.; Kroemer, G. miR-181a and miR-630 regulate cisplatin-induced cancer cell death. Cancer Res. 2010, 70, 1793–1803. [Google Scholar]
- Gregory, P.A.; Bert, A.G.; Paterson, E.L.; Barry, S.C.; Tsykin, A.; Farshid, G.; Vadas, M.A.; Khew-Goodall, Y.; Goodall, G.J. The miR-200 family and miR-205 regulate epithelial to mesenchymal transition by targeting ZEB1 and SIP1. Nat. Cell Biol. 2008, 10, 593–601. [Google Scholar]
- Ceppi, P.; Mudduluru, G.; Kumarswamy, R.; Rapa, I.; Scagliotti, G.V.; Papotti, M.; Allgayer, H. Loss of miR-200c expression induces an aggressive, invasive, and chemoresistant phenotype in non-small cell lung cancer. Mol. Cancer Res. 2010, 8, 1207–1216. [Google Scholar]
- Advani, A.S.; Gibson, S.E.; Douglas, E.; Jin, T.; Zhao, X.; Kalaycio, M.; Copelan, E.; Sobecks, R.; Sekeres, M.; Sungren, S.; Hsi, E.D. Histone H4 acetylation by immunohistochemistry and prognosis in newly diagnosed adult acute lymphoblastic leukemia (ALL) patients. BMC Cancer 2010, 10, 387. [Google Scholar]
- Muller-Tidow, C.; Klein, H.U.; Hascher, A.; Isken, F.; Tickenbrock, L.; Thoennissen, N.; Agrawal-Singh, S.; Tschanter, P.; Disselhoff, C.; Wang, Y.; Becker, A.; Thiede, C.; Ehninger, G.; zur Stadt, U.; Koschmieder, S.; Seidl, M.; Muller, F.U.; Schmitz, W.; Schlenke, P.; McClelland, M.; Berdel, W.E.; Dugas, M.; Serve, H. Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood 2010, 116, 3564–3571. [Google Scholar]
- Elsheikh, S.E.; Green, A.R.; Rakha, E.A.; Powe, D.G.; Ahmed, R.A.; Collins, H.M.; Soria, D.; Garibaldi, J.M.; Paish, C.E.; Ammar, A.A.; Grainge, M.J.; Ball, G.R.; Abdelghany, M.K.; Martinez-Pomares, L.; Heery, D.M.; Ellis, I.O. Global histone modifications in breast cancer correlate with tumor phenotypes, prognostic factors, and patient outcome. Cancer Res. 2009, 69, 3802–3809. [Google Scholar]
- Wei, Y.; Xia, W.; Zhang, Z.; Liu, J.; Wang, H.; Adsay, N.V.; Albarracin, C.; Yu, D.; Abbruzzese, J.L.; Mills, G.B.; Bast, R.C., Jr.; Hortobagyi, G.N.; Hung, M.C. Loss of trimethylation at lysine 27 of histone H3 is a predictor of poor outcome in breast, ovarian, and pancreatic cancers. Mol. Carcinog. 2008, 47, 701–706. [Google Scholar]
- Fraga, M.F.; Ballestar, E.; Villar-Garea, A.; Boix-Chornet, M.; Espada, J.; Schotta, G.; Bonaldi, T.; Haydon, C.; Ropero, S.; Petrie, K.; Iyer, N.G.; Perez-Rosado, A.; Calvo, E.; Lopez, J.A.; Cano, A.; Calasanz, M.J.; Colomer, D.; Piris, M.A.; Ahn, N.; Imhof, A.; Caldas, C.; Jenuwein, T.; Esteller, M. Loss of acetylation at Lys16 and trimethylation at Lys20 of histone H4 is a common hallmark of human cancer. Nat. Genet. 2005, 37, 391–400. [Google Scholar]
- Park, Y.S.; Jin, M.Y.; Kim, Y.J.; Yook, J.H.; Kim, B.S.; Jang, S.J. The global histone modification pattern correlates with cancer recurrence and overall survival in gastric adenocarcinoma. Ann. Surg. Oncol. 2008, 15, 1968–1976. [Google Scholar]
- Liu, B.L.; Cheng, J.X.; Zhang, X.; Wang, R.; Zhang, W.; Lin, H.; Xiao, X.; Cai, S.; Chen, X.Y.; Cheng, H. Global histone modification patterns as prognostic markers to classify glioma patients. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 2888–2896. [Google Scholar]
- Cai, M.Y.; Hou, J.H.; Rao, H.L.; Luo, R.Z.; Li, M.; Pei, X.Q.; Lin, M.C.; Guan, X.Y.; Kung, H.F.; Zeng, Y.X.; Xie, D. High expression of H3K27me3 in human hepatocellular carcinomas correlates closely with vascular invasion and predicts patients worse prognosis. Mol. Med. 2011, 17, 12–20. [Google Scholar]
- Manuyakorn, A.; Paulus, R.; Farrell, J.; Dawson, N.A.; Tze, S.; Cheung-Lau, G.; Hines, O.J.; Reber, H.; Seligson, D.B.; Horvath, S.; Kurdistani, S.K.; Guha, C.; Dawson, D.W. Cellular histone modification patterns predict prognosis and treatment response in resectable pancreatic adenocarcinoma: results from RTOG 9704. J. Clin. Oncol. 2010, 28, 1358–1365. [Google Scholar]
- Ellinger, J.; Kahl, P.; von der Gathen, J.; Rogenhofer, S.; Heukamp, L.C.; Gutgemann, I.; Walter, B.; Hofstadter, F.; Buttner, R.; Muller, S.C.; Bastian, P.J.; von Ruecker, A. Global levels of histone modifications predict prostate cancer recurrence. Prostate 2010, 70, 61–69. [Google Scholar]
- Seligson, D.B.; Horvath, S.; Shi, T.; Yu, H.; Tze, S.; Grunstein, M.; Kurdistani, S.K. Global histone modification patterns predict risk of prostate cancer recurrence. Nature 2005, 435, 1262–1266. [Google Scholar]
- Tzao, C.; Tung, H.J.; Jin, J.S.; Sun, G.H.; Hsu, H.S.; Chen, B.H.; Yu, C.P.; Lee, S.C. Prognostic significance of global histone modifications in resected squamous cell carcinoma of the esophagus. Mod. Pathol. 2009, 22, 252–260. [Google Scholar]
- He, L.R.; Liu, M.Z.; Li, B.K.; Rao, H.L.; Liao, Y.J.; Guan, X.Y.; Zeng, Y.X.; Xie, D. Prognostic impact of H3K27me3 expression on locoregional progression after chemoradiotherapy in esophageal squamous cell carcinoma. BMC Cancer 2009, 9, 461. [Google Scholar]
- I, H.; Ko, E.; Kim, Y.; Cho, E.Y.; Han, J.; Park, J.; Kim, K.; Kim, D.H.; Shim, Y.M. Association of global levels of histone modifications with recurrence-free survival in stage IIB and III esophageal squamous cell carcinomas. Cancer Epidemiol. Biomarkers Prev. 2010, 19, 566–573. [Google Scholar]
- Zhen, L.; Gui-lan, L.; Ping, Y.; Jin, H.; Ya-li, W. The expression of H3K9Ac, H3K14Ac, and H4K20TriMe in epithelial ovarian tumors and the clinical significance. Int. J. Gynecol. Cancer 2010, 20, 82–86. [Google Scholar]
- Minardi, D.; Lucarini, G.; Filosa, A.; Milanese, G.; Zizzi, A.; Di Primio, R.; Montironi, R.; Muzzonigro, G. Prognostic role of global dna-methylation and histone acetylation in pT1a clear cell renal carcinoma in partial nephrectomy specimens. J. Cell Mol. Med. 2009, 13, 2115–2121. [Google Scholar]
- Seligson, D.B.; Horvath, S.; McBrian, M.A.; Mah, V.; Yu, H.; Tze, S.; Wang, Q.; Chia, D.; Goodglick, L.; Kurdistani, S.K. Global levels of histone modifications predict prognosis in different cancers. Am. J. Pathol. 2009, 174, 1619–1628. [Google Scholar]
- Mosashvilli, D.; Kahl, P.; Mertens, C.; Holzapfel, S.; Rogenhofer, S.; Hauser, S.; Buttner, R.; Von Ruecker, A.; Muller, S.C.; Ellinger, J. Global histone acetylation levels: Prognostic relevance in patients with renal cell carcinoma. Cancer Sci. 2010, 101, 2664–2669. [Google Scholar]
- Ellinger, J.; Kahl, P.; Mertens, C.; Rogenhofer, S.; Hauser, S.; Hartmann, W.; Bastian, P.J.; Buttner, R.; Muller, S.C.; von Ruecker, A. Prognostic relevance of global histone H3 lysine 4 (H3K4) methylation in renal cell carcinoma. Int. J. Cancer 2010, 127, 2360–2366. [Google Scholar]
- Barlesi, F.; Giaccone, G.; Gallegos-Ruiz, M.I.; Loundou, A.; Span, S.W.; Lefesvre, P.; Kruyt, F.A.; Rodriguez, J.A. Global histone modifications predict prognosis of resected non small-cell lung cancer. J. Clin. Oncol. 2007, 25, 4358–4364. [Google Scholar]
- Van Den Broeck, A.; Brambilla, E.; Moro-Sibilot, D.; Lantuejoul, S.; Brambilla, C.; Eymin, B.; Khochbin, S.; Gazzeri, S. Loss of Histone H4K20 Trimethylation Occurs in Preneoplasia and Influences Prognosis of Non-Small Cell Lung Cancer. Clin. Cancer Res. 2008, 14, 7237–7245. [Google Scholar]
- Watanabe, H.; Soejima, K.; Yasuda, H.; Kawada, I.; Nakachi, I.; Yoda, S.; Naoki, K.; Ishizaka, A. Deregulation of histone lysine methyltransferases contributes to oncogenic transformation of human bronchoepithelial cells. Cancer Cell Int. 2008, 8, 15. [Google Scholar]
- Lawless, M.W.; O'Byrne, K.J.; Gray, S.G. Oxidative stress induced lung cancer and COPD: Opportunities for epigenetic therapy. J. Cell. Mol. Med. 2009, 13, 2800–2821. [Google Scholar]
- Lawless, M.W.; O'Byrne, K.J.; Gray, S.G. Targeting oxidative stress in cancer. Expert Opin. Ther. Targets 2010, 14, 1225–1245. [Google Scholar]
- Gray, S.G.; Ekstrom, T.J. The human histone deacetylase family. Exp. Cell Res. 2001, 262, 75–83. [Google Scholar]
- Ledent, V.; Vervoort, M. Comparative genomics of the class 4 histone deacetylase family indicates a complex evolutionary history. BMC Biol. 2006, 4, 24. [Google Scholar]
- Lawless, M.W.; Norris, S.; O'Byrne, K.J.; Gray, S.G. Targeting histone deacetylases for the treatment of disease. J. Cell. Mol. Med. 2009, 13, 826–852. [Google Scholar]
- Minamiya, Y.; Ono, T.; Saito, H.; Takahashi, N.; Ito, M.; Motoyama, S.; Ogawa, J. Strong expression of HDAC3 correlates with a poor prognosis in patients with adenocarcinoma of the lung. Tumor Biol. 2010, 31, 533–539. [Google Scholar]
- Osada, H.; Tatematsu, Y.; Saito, H.; Yatabe, Y.; Mitsudomi, T.; Takahashi, T. Reduced expression of class II histone deacetylase genes is associated with poor prognosis in lung cancer patients. Int. J. Cancer 2004, 112, 26–32. [Google Scholar]
- Tseng, R.C.; Lee, C.C.; Hsu, H.S.; Tzao, C.; Wang, Y.C. Distinct HIC1-SIRT1-p53 loop deregulation in lung squamous carcinoma and adenocarcinoma patients. Neoplasia 2009, 11, 763–770. [Google Scholar]
- Yang, X.J.; Seto, E. The Rpd3/Hda1 family of lysine deacetylases: From bacteria and yeast to mice and men. Nat. Rev. Mol. Cell Biol. 2008, 9, 206–218. [Google Scholar]
- Suzuki, H.; Ouchida, M.; Yamamoto, H.; Yano, M.; Toyooka, S.; Aoe, M.; Shimizu, N.; Date, H.; Shimizu, K. Decreased expression of the SIN3A gene, a candidate tumor suppressor located at the prevalent allelic loss region 15q23 in non-small cell lung cancer. Lung Cancer 2008, 59, 24–31. [Google Scholar]
- Sasaki, H.; Moriyama, S.; Nakashima, Y.; Kobayashi, Y.; Yukiue, H.; Kaji, M.; Fukai, I.; Kiriyama, M.; Yamakawa, Y.; Fujii, Y. Expression of the MTA1 mRNA in advanced lung cancer. Lung Cancer 2002, 35, 149–154. [Google Scholar]
- Lawless, M.W.; O'Byrne, K.J.; Gray, S.G. Oxidative stress induced lung cancer and COPD: opportunities for epigenetic therapy. J. Cell Mol. Med. 2009, 13, 2800–2821. [Google Scholar]
- Yang, X.; Feng, M.; Jiang, X.; Wu, Z.; Li, Z.; Aau, M.; Yu, Q. miR-449a and miR-449b are direct transcriptional targets of E2F1 and negatively regulate pRb-E2F1 activity through a feedback loop by targeting CDK6 and CDC25A. Genes Dev. 2009, 23, 2388–2393. [Google Scholar]
- Lizé, M.; Pilarski, S.; Dobbelstein, M. E2F1-inducible microRNA 449a/b suppresses cell proliferation and promotes apoptosis. Cell Death Differ. 2010, 17, 452–458. [Google Scholar]
- Noonan, E.J.; Place, R.F.; Pookot, D.; Basak, S.; Whitson, J.M.; Hirata, H.; Giardina, C.; Dahiya, R. miR-449a targets HDAC-1 and induces growth arrest in prostate cancer. Oncogene 2009, 28, 1714–1724. [Google Scholar]
- Liang, Y. An expression meta-analysis of predicted microRNA targets identifies a diagnostic signature for lung cancer. BMC Med. Genomics 2008, 1, 61. [Google Scholar]
- Allis, C.D.; Berger, S.L.; Cote, J.; Dent, S.; Jenuwien, T.; Kouzarides, T.; Pillus, L.; Reinberg, D.; Shi, Y.; Shiekhattar, R.; Shilatifard, A.; Workman, J.; Zhang, Y. New nomenclature for chromatin-modifying enzymes. Cell 2007, 131, 633–636. [Google Scholar]
- Gorgoulis, V.G.; Zacharatos, P.; Mariatos, G.; Kotsinas, A.; Bouda, M.; Kletsas, D.; Asimacopoulos, P.J.; Agnantis, N.; Kittas, C.; Papavassiliou, A.G. Transcription factor E2F-1 acts as a growth-promoting factor and is associated with adverse prognosis in non-small cell lung carcinomas. J. Pathol. 2002, 198, 142–156. [Google Scholar]
- Kishimoto, M.; Kohno, T.; Okudela, K.; Otsuka, A.; Sasaki, H.; Tanabe, C.; Sakiyama, T.; Hirama, C.; Kitabayashi, I.; Minna, J.D.; Takenoshita, S.; Yokota, J. Mutations and deletions of the CBP gene in human lung cancer. Clin. Cancer Res. 2005, 11, 512–519. [Google Scholar]
- Cai, D.; Shames, D.S.; Raso, M.G.; Xie, Y.; Kim, Y.H.; Pollack, J.R.; Girard, L.; Sullivan, J.P.; Gao, B.; Peyton, M.; Nanjundan, M.; Byers, L.; Heymach, J.; Mills, G.; Gazdar, A.F.; Wistuba, I.; Kodadek, T.; Minna, J.D. Steroid receptor coactivator-3 expression in lung cancer and its role in the regulation of cancer cell survival and proliferation. Cancer Res. 2010, 70, 6477–6485. [Google Scholar]
- Long, W.; Yi, P.; Amazit, L.; LaMarca, H.L.; Ashcroft, F.; Kumar, R.; Mancini, M.A.; Tsai, S.Y.; Tsai, M.J.; O'Malley, B.W. SRC-3Delta4 mediates the interaction of EGFR with FAK to promote cell migration. Mol. Cell. 2010, 37, 321–332. [Google Scholar]
- Albert, M.; Helin, K. Histone methyltransferases in cancer. Semin. Cell Dev. Biol. 2010, 21, 209–220. [Google Scholar]
- Yoon, K.A.; Park, S.; Hwangbo, B.; Shin, H.D.; Cheong, H.S.; Shin, H.R.; Lee, J.S. Genetic polymorphisms in the Rb-binding zinc finger gene RIZ and the risk of lung cancer. Carcinogenesis 2007, 28, 1971–1977. [Google Scholar]
- Yoon, K.A.; Gil, H.J.; Han, J.; Park, J.; Lee, J.S. Genetic polymorphisms in the polycomb group gene EZH2 and the risk of lung cancer. J. Thorac Oncol. 2010, 5, 10–16. [Google Scholar]
- Yoon, K.A.; Hwangbo, B.; Kim, I.J.; Park, S.; Kim, H.S.; Kee, H.J.; Lee, J.E.; Jang, Y.K.; Park, J.G.; Lee, J.S. Novel polymorphisms in the SUV39H2 histone methyltransferase and the risk of lung cancer. Carcinogenesis 2006, 27, 2217–2222. [Google Scholar]
- Chen, M.W.; Hua, K.T.; Kao, H.J.; Chi, C.C.; Wei, L.H.; Johansson, G.; Shiah, S.G.; Chen, P.S.; Jeng, Y.M.; Cheng, T.Y.; Lai, T.C.; Chang, J.S.; Jan, Y.H.; Chien, M.H.; Yang, C.J.; Huang, M.S.; Hsiao, M.; Kuo, M.L. H3K9 histone methyltransferase G9a promotes lung cancer invasion and metastasis by silencing the cell adhesion molecule Ep-CAM. Cancer Res. 2010, 70, 7830–7840. [Google Scholar]
- Kikuchi, J.; Kinoshita, I.; Shimizu, Y.; Kikuchi, E.; Konishi, J.; Oizumi, S.; Kaga, K.; Matsuno, Y.; Nishimura, M.; Dosaka-Akita, H. Distinctive expression of the polycomb group proteins Bmi1 polycomb ring finger oncogene and enhancer of zeste homolog 2 in nonsmall cell lung cancers and their clinical and clinicopathologic significance. Cancer 2010, 116, 3015–3024. [Google Scholar]
- Kisliouk, T.; Yosefi, S.; Meiri, N. MiR-138 inhibits EZH2 methyltransferase expression and methylation of histone H3 at lysine 27, and affects thermotolerance acquisition. Eur. J. Neurosci. 2011, 33, 224–235. [Google Scholar]
- Seike, M.; Goto, A.; Okano, T.; Bowman, E.D.; Schetter, A.J.; Horikawa, I.; Mathe, E.A.; Jen, J.; Yang, P.; Sugimura, H.; Gemma, A.; Kudoh, S.; Croce, C.M.; Harris, C.C. MiR-21 is an EGFR-regulated anti-apoptotic factor in lung cancer in never-smokers. Proc. Natl. Acad. Sci. USA 2009, 106, 12085–12090. [Google Scholar]
- Wu, X.; Hua, X. Menin, histone h3 methyltransferases, and regulation of cell proliferation: current knowledge and perspective. Curr. Mol. Med. 2008, 8, 805–815. [Google Scholar]
- Pei, X.H.; Bai, F.; Smith, M.D.; Xiong, Y. p18Ink4c collaborates with Men1 to constrain lung stem cell expansion and suppress non-small-cell lung cancers. Cancer Res. 2007, 67, 3162–3170. [Google Scholar]
- Gao, S.B.; Feng, Z.J.; Xu, B.; Wu, Y.; Yin, P.; Yang, Y.; Hua, X.; Jin, G.H. Suppression of lung adenocarcinoma through menin and polycomb gene-mediated repression of growth factor pleiotrophin. Oncogene 2009, 28, 4095–4104. [Google Scholar]
- Feng, Z.J.; Gao, S.B.; Wu, Y.; Xu, X.F.; Hua, X.; Jin, G.H. Lung cancer cell migration is regulated via repressing growth factor PTN/RPTP beta/zeta signaling by menin. Oncogene 2010, 29, 5416–5426. [Google Scholar]
- Bedford, M.T.; Clarke, S.G. Protein arginine methylation in mammals: Who, what, and why. Mol. Cell 2009, 33, 1–13. [Google Scholar]
- Wolf, S.S. The protein arginine methyltransferase family: An update about function, new perspectives and the physiological role in humans. Cell Mol. Life Sci. 2009, 66, 2109–2121. [Google Scholar]
- Yildirim, A.O.; Bulau, P.; Zakrzewicz, D.; Kitowska, K.E.; Weissmann, N.; Grimminger, F.; Morty, R.E.; Eickelberg, O. Increased protein arginine methylation in chronic hypoxia: role of protein arginine methyltransferases. Am. J. Respir. Cell Mol. Biol. 2006, 35, 436–443. [Google Scholar]
- O'Brien, K.B.; Alberich-Jorda, M.; Yadav, N.; Kocher, O.; Diruscio, A.; Ebralidze, A.; Levantini, E.; Sng, N.J.; Bhasin, M.; Caron, T.; Kim, D.; Steidl, U.; Huang, G.; Halmos, B.; Rodig, S.J.; Bedford, M.T.; Tenen, D.G.; Kobayashi, S. CARM1 is required for proper control of proliferation and differentiation of pulmonary epithelial cells. Development 2010, 137, 2147–2156. [Google Scholar]
- Hong, H.; Kao, C.; Jeng, M.H.; Eble, J.N.; Koch, M.O.; Gardner, T.A.; Zhang, S.; Li, L.; Pan, C.X.; Hu, Z.; MacLennan, G.T.; Cheng, L. Aberrant expression of CARM1, a transcriptional coactivator of androgen receptor, in the development of prostate carcinoma and androgen-independent status. Cancer 2004, 101, 83–89. [Google Scholar]
- Majumder, S.; Liu, Y.; Ford, O.H., 3rd; Mohler, J.L.; Whang, Y.E. Involvement of arginine methyltransferase CARM1 in androgen receptor function and prostate cancer cell viability. Prostate 2006, 66, 1292–1301. [Google Scholar]
- Kim, Y.R.; Lee, B.K.; Park, R.Y.; Nguyen, N.T.; Bae, J.A.; Kwon, D.D.; Jung, C. Differential CARM1 expression in prostate and colorectal cancers. BMC Cancer 2010, 10, 197. [Google Scholar]
- Yoshimatsu, M.; Toyokawa, G.; Hayami, S.; Unoki, M.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Maehara, Y.; Ponder, B.A.; Nakamura, Y.; Hamamoto, R. Dysregulation of PRMT1 and PRMT6, Type I arginine methyltransferases, is involved in various types of human cancers. Int. J. Cancer 2011, 128, 562–573. [Google Scholar]
- Mosammaparast, N.; Shi, Y. Reversal of histone methylation: Biochemical and molecular mechanisms of histone demethylases. Annu. Rev. Biochem. 2010, 79, 155–179. [Google Scholar]
- Pedersen, M.T.; Helin, K. Histone demethylases in development and disease. Trends Cell Biol. 2010, 20, 662–671. [Google Scholar]
- Hayami, S; Kelly, JD; Cho, HS; Yoshimatsu, M; Unoki, M; Tsunoda, T; Field, HI; Neal, DE; Yamaue, H; Ponder, BA; Nakamura, Y; Hamamoto, R. Overexpression of LSD1 contributes to human carcinogenesis through chromatin regulation in various cancers. Int. J. Cancer. 2011, 128, 574–86. [Google Scholar]
- Hayami, S.; Yoshimatsu, M.; Veerakumarasivam, A.; Unoki, M.; Iwai, Y.; Tsunoda, T.; Field, H.I.; Kelly, J.D.; Neal, D.E.; Yamaue, H.; Ponder, B.A.; Nakamura, Y.; Hamamoto, R. Overexpression of the JmjC histone demethylase KDM5B in human carcinogenesis: involvement in the proliferation of cancer cells through the E2F/RB pathway. Mol. Cancer 2010, 9, 59. [Google Scholar]
- Zhou, X.; Sun, H.; Chen, H.; Zavadil, J.; Kluz, T.; Arita, A.; Costa, M. Hypoxia induces trimethylated H3 lysine 4 by inhibition of JARID1A demethylase. Cancer Res. 2010, 70, 4214–4221. [Google Scholar]
- Lu, Y.; Chang, Q.; Zhang, Y.; Beezhold, K.; Rojanasakul, Y.; Zhao, H.; Castranova, V.; Shi, X.; Chen, F. Lung cancer-associated JmjC domain protein mdig suppresses formation of tri-methyl lysine 9 of histone H3. Cell Cycle 2009, 8, 2101–2109. [Google Scholar]
- Chang, B.; Chen, Y.; Zhao, Y.; Bruick, R.K. JMJD6 is a histone arginine demethylase. Science 2007, 318, 444–447. [Google Scholar]
- Fadok, V.A.; Bratton, D.L.; Rose, D.M.; Pearson, A.; Ezekewitz, R.A.; Henson, P.M. A receptor for phosphatidylserine-specific clearance of apoptotic cells. Nature 2000, 405, 85–90. [Google Scholar]
- Sexton, D.W.; Al-Rabia, M.; Blaylock, M.G.; Walsh, G.M. Phagocytosis of apoptotic eosinophils but not neutrophils by bronchial epithelial cells. Clin. Exp. Allergy 2004, 34, 1514–1524. [Google Scholar]
- Sexton, D.W.; Blaylock, M.G.; Walsh, G.M. Human alveolar epithelial cells engulf apoptotic eosinophils by means of integrin- and phosphatidylserine receptor-dependent mechanisms: a process upregulated by dexamethasone. J. Allergy Clin. Immunol. 2001, 108, 962–969. [Google Scholar]
- Klose, R.J.; Zhang, Y. Regulation of histone methylation by demethylimination and demethylation. Nat. Rev. Mol. Cell Biol. 2007, 8, 307–318. [Google Scholar]
- Chang, X.; Han, J. Expression of peptidylarginine deiminase type 4 (PAD4) in various tumors. Mol. Carcinog. 2006, 45, 183–196. [Google Scholar]
- Denis, H.; Deplus, R.; Putmans, P.; Yamada, M.; Metivier, R.; Fuks, F. Functional connection between deimination and deacetylation of histones. Mol. Cell Biol. 2009, 29, 4982–4993. [Google Scholar]
- Gorgoulis, V.G.; Vassiliou, L.V.; Karakaidos, P.; Zacharatos, P.; Kotsinas, A.; Liloglou, T.; Venere, M.; Ditullio, R.A., Jr.; Kastrinakis, N.G.; Levy, B.; Kletsas, D.; Yoneta, A.; Herlyn, M.; Kittas, C.; Halazonetis, T.D. Activation of the DNA damage checkpoint and genomic instability in human precancerous lesions. Nature 2005, 434, 907–913. [Google Scholar]
- Ueda, K.; Kawashima, H.; Ohtani, S.; Deng, W.G.; Ravoori, M.; Bankson, J.; Gao, B.; Girard, L.; Minna, J.D.; Roth, J.A.; Kundra, V.; Ji, L. The 3p21.3 tumor suppressor NPRL2 plays an important role in cisplatin-induced resistance in human non-small-cell lung cancer cells. Cancer Res. 2006, 66, 9682–9690. [Google Scholar]
- Jayachandran, G.; Ueda, K.; Wang, B.; Roth, J.A.; Ji, L. NPRL2 sensitizes human non-small cell lung cancer (NSCLC) cells to cisplatin treatment by regulating key components in the DNA repair pathway. PLoS One 2010, 5, e11994. [Google Scholar]
- Igarashi, T.; Izumi, H.; Uchiumi, T.; Nishio, K.; Arao, T.; Tanabe, M.; Uramoto, H.; Sugio, K.; Yasumoto, K.; Sasaguri, Y.; Wang, K.Y.; Otsuji, Y.; Kohno, K. Clock and ATF4 transcription system regulates drug resistance in human cancer cell lines. Oncogene 2007, 26, 4749–4760. [Google Scholar]
- Miyamoto, N.; Izumi, H.; Noguchi, T.; Nakajima, Y.; Ohmiya, Y.; Shiota, M.; Kidani, A.; Tawara, A.; Kohno, K. Tip60 is regulated by circadian transcription factor clock and is involved in cisplatin resistance. J. Biol. Chem. 2008, 283, 18218–18226. [Google Scholar]
- Hirano, G.; Izumi, H.; Kidani, A.; Yasuniwa, Y.; Han, B.; Kusaba, H.; Akashi, K.; Kuwano, M.; Kohno, K. Enhanced expression of PCAF endows apoptosis resistance in cisplatin-resistant cells. Mol. Cancer Res. 2010, 8, 864–872. [Google Scholar]
- Liang, X.J.; Finkel, T.; Shen, D.W.; Yin, J.J.; Aszalos, A.; Gottesman, M.M. SIRT1 contributes in part to cisplatin resistance in cancer cells by altering mitochondrial metabolism. Mol. Cancer Res. 2008, 6, 1499–1506. [Google Scholar]
- Edmond, V.; Moysan, E.; Khochbin, S.; Matthias, P.; Brambilla, C.; Brambilla, E.; Gazzeri, S.; Eymin, B. Acetylation and phosphorylation of SRSF2 control cell fate decision in response to cisplatin. EMBO J. 2011, 30, 510–523. [Google Scholar]
- Wu, Z.Z.; Sun, N.K.; Chao, C.C. Knockdown of CITED2 using short-hairpin RNA sensitizes cancer cells to cisplatin through stabilization of p53 and enhancement of p53-dependent apoptosis. J. Cell Physiol. 2010. [Google Scholar] [CrossRef]
- Yang, E.S.; Xia, F. BRCA1 16 years later: DNA damage-induced BRCA1 shuttling. FEBS J. 2010, 277, 3079–3085. [Google Scholar]
- Price, M.; Monteiro, A.N. Fine tuning chemotherapy to match BRCA1 status. Biochem. Pharmacol. 2010, 80, 647–653. [Google Scholar]
- Lee, M.N.; Tseng, R.C.; Hsu, H.S.; Chen, J.Y.; Tzao, C.; Ho, W.L.; Wang, Y.C. Epigenetic inactivation of the chromosomal stability control genes BRCA1, BRCA2, and XRCC5 in non-small cell lung cancer. Clin. Cancer Res. 2007, 13, 832–838. [Google Scholar]
- Rosell, R.; Skrzypski, M.; Jassem, E.; Taron, M.; Bartolucci, R.; Sanchez, J.J.; Mendez, P.; Chaib, I.; Perez-Roca, L.; Szymanowska, A.; Rzyman, W.; Puma, F.; Kobierska-Gulida, G.; Farabi, R.; Jassem, J. BRCA1: A novel prognostic factor in resected non-small-cell lung cancer. PLoS One 2007, 2, e1129. [Google Scholar]
- Wang, Y.; Zhang, D.; Zheng, W.; Luo, J.; Bai, Y.; Lu, Z. Multiple gene methylation of nonsmall cell lung cancers evaluated with 3-dimensional microarray. Cancer 2008, 112, 1325–1336. [Google Scholar]
- Huen, M.S.; Sy, S.M.; Chen, J. BRCA1 and its toolbox for the maintenance of genome integrity. Nat. Rev. Mol. Cell Biol. 2010, 11, 138–148. [Google Scholar]
- Baer, R.; Ludwig, T. The BRCA1/BARD1 heterodimer, a tumor suppressor complex with ubiquitin E3 ligase activity. Curr. Opin. Genet. Dev. 2002, 12, 86–91. [Google Scholar]
- Ratanaphan, A.; Wasiksiri, S.; Canyuk, B.; Prasertsan, P. Cisplatin-damaged BRCA1 exhibits altered thermostability and transcriptional transactivation. Cancer Biol. Ther. 2009, 8, 890–898. [Google Scholar]
- Atipairin, A.; Canyuk, B.; Ratanaphan, A. The RING heterodimer BRCA1-BARD1 is a ubiquitin ligase inactivated by the platinum-based anticancer drugs. Breast Cancer Res. Treat. 2010, 126, 203–209. [Google Scholar]
- Smeenk, G.; Wiegant, W.W.; Vrolijk, H.; Solari, A.P.; Pastink, A.; van Attikum, H. The NuRD chromatin-remodeling complex regulates signaling and repair of DNA damage. J. Cell. Biol. 2010, 190, 741–749. [Google Scholar]
- Larsen, D.H.; Poinsignon, C.; Gudjonsson, T.; Dinant, C.; Payne, M.R.; Hari, F.J.; Danielsen, J.M.; Menard, P.; Sand, J.C.; Stucki, M.; Lukas, C.; Bartek, J.; Andersen, J.S.; Lukas, J. The chromatin-remodeling factor CHD4 coordinates signaling and repair after DNA damage. J. Cell. Biol. 2010, 190, 731–740. [Google Scholar]
- Bhattacharyya, A.; Ear, U.S.; Koller, B.H.; Weichselbaum, R.R.; Bishop, D.K. The breast cancer susceptibility gene BRCA1 is required for subnuclear assembly of Rad51 and survival following treatment with the DNA cross-linking agent cisplatin. J. Biol.Chem. 2000, 275, 23899–23903. [Google Scholar]
- Tassone, P.; Tagliaferri, P.; Perricelli, A.; Blotta, S.; Quaresima, B.; Martelli, M.L.; Goel, A.; Barbieri, V.; Costanzo, F.; Boland, C.R.; Venuta, S. BRCA1 expression modulates chemosensitivity of BRCA1-defective HCC1937 human breast cancer cells. Br. J. Cancer 2003, 88, 1285–1291. [Google Scholar]
- Tassone, P.; Di Martino, M.T.; Ventura, M.; Pietragalla, A.; Cucinotto, I.; Calimeri, T.; Bulotta, A.; Neri, P.; Caraglia, M.; Tagliaferri, P. Loss of BRCA1 function increases the antitumor activity of cisplatin against human breast cancer xenografts in vivo. Cancer Biol. Ther. 2009, 8, 648–653. [Google Scholar]
- Swisher, E.M.; Gonzalez, R.M.; Taniguchi, T.; Garcia, R.L.; Walsh, T.; Goff, B.A.; Welcsh, P. Methylation and protein expression of DNA repair genes: Association with chemotherapy exposure and survival in sporadic ovarian and peritoneal carcinomas. Mol. Cancer 2009, 8, 48. [Google Scholar]
- Taron, M.; Rosell, R.; Felip, E.; Mendez, P.; Souglakos, J.; Ronco, M.S.; Queralt, C.; Majo, J.; Sanchez, J.M.; Sanchez, J.J.; Maestre, J. BRCA1 mRNA expression levels as an indicator of chemoresistance in lung cancer. Hum. Mol. Genet. 2004, 13, 2443–2449. [Google Scholar]
- Wang, L.; Wei, J.; Qian, X.; Yin, H.; Zhao, Y.; Yu, L.; Wang, T.; Liu, B. ERCC1 and BRCA1 mRNA expression levels in metastatic malignant effusions is associated with chemosensitivity to cisplatin and/or docetaxel. BMC Cancer 2008, 8, 97. [Google Scholar]
- Rosell, R.; Perez-Roca, L.; Sanchez, J.J.; Cobo, M.; Moran, T.; Chaib, I.; Provencio, M.; Domine, M.; Sala, M.A.; Jimenez, U.; Diz, P.; Barneto, I.; Macias, J.A.; de Las Penas, R.; Catot, S.; Isla, D.; Sanchez, J.M.; Ibeas, R.; Lopez-Vivanco, G.; Oramas, J.; Mendez, P.; Reguart, N.; Blanco, R.; Taron, M. Customized treatment in non-small-cell lung cancer based on EGFR mutations and BRCA1 mRNA expression. PLoS One 2009, 4, e5133. [Google Scholar]
- Su, C.; Zhou, S.; Zhang, L.; Ren, S.; Xu, J.; Zhang, J.; Lv, M.; Zhang, J.; Zhou, C. ERCC1, RRM1 and BRCA1 mRNA expression levels and clinical outcome of advanced non-small cell lung cancer. Med. Oncol. 2010. [Google Scholar] [CrossRef]
- Puppe, J.; Drost, R.; Liu, X.; Joosse, S.A.; Evers, B.; Cornelissen-Steijger, P.; Nederlof, P.; Yu, Q.; Jonkers, J.; van Lohuizen, M.; Pietersen, A.M. BRCA1-deficient mammary tumor cells are dependent on EZH2 expression and sensitive to Polycomb Repressive Complex 2-inhibitor 3-deazaneplanocin A. Breast Cancer Res. 2009, 11, R63. [Google Scholar]
- Kuzmichev, A.; Nishioka, K.; Erdjument-Bromage, H.; Tempst, P.; Reinberg, D. Histone methyltransferase activity associated with a human multiprotein complex containing the Enhancer of Zeste protein. Genes Dev. 2002, 16, 2893–2905. [Google Scholar]
- Cao, R.; Wang, L.; Wang, H.; Xia, L.; Erdjument-Bromage, H.; Tempst, P.; Jones, R.S.; Zhang, Y. Role of histone H3 lysine 27 methylation in Polycomb-group silencing. Science 2002, 298, 1039–1043. [Google Scholar]
- Varambally, S.; Cao, Q.; Mani, R.S.; Shankar, S.; Wang, X.; Ateeq, B.; Laxman, B.; Cao, X.; Jing, X.; Ramnarayanan, K.; Brenner, J.C.; Yu, J.; Kim, J.H.; Han, B.; Tan, P.; Kumar-Sinha, C.; Lonigro, R.J.; Palanisamy, N.; Maher, C.A.; Chinnaiyan, A.M. Genomic loss of microRNA-101 leads to overexpression of histone methyltransferase EZH2 in cancer. Science 2008, 322, 1695–1699. [Google Scholar]
- Yang, Y.; Li, X.; Yang, Q.; Wang, X.; Zhou, Y.; Jiang, T.; Ma, Q.; Wang, Y.J. The role of microRNA in human lung squamous cell carcinoma. Cancer Genet. Cytogenet. 2010, 200, 127–133. [Google Scholar]
- Hu, S.; Yu, L.; Li, Z.; Shen, Y.; Wang, J.; Cai, J.; Xiao, L.; Wang, Z. Overexpression of EZH2 contributes to acquired cisplatin resistance in ovarian cancer cells in vitro and in vivo. Cancer Biol. Ther. 2010, 10, 788–795. [Google Scholar]
- Murray, M.M.; Mullan, P.B.; Harkin, D.P. Role played by BRCA1 in transcriptional regulation in response to therapy. Biochem. Soc. Trans. 2007, 35, 1342–1346. [Google Scholar]
- Yoshida, K.; Miki, Y. The cell death machinery governed by the p53 tumor suppressor in response to DNA damage. Cancer Sci. 2010, 101, 831–835. [Google Scholar]
- Tsao, M.S.; Aviel-Ronen, S.; Ding, K.; Lau, D.; Liu, N.; Sakurada, A.; Whitehead, M.; Zhu, C.Q.; Livingston, R.; Johnson, D.H.; Rigas, J.; Seymour, L.; Winton, T.; Shepherd, F.A. Prognostic and predictive importance of p53 and RAS for adjuvant chemotherapy in non small-cell lung cancer. J. Clin. Oncol. 2007, 25, 5240–5247. [Google Scholar]
- Duarte, M.L.; de Moraes, E.; Pontes, E.; Schluckebier, L.; de Moraes, J.L.; Hainaut, P.; Ferreira, C.G. Role of p53 in the induction of cyclooxygenase-2 by cisplatin or paclitaxel in non-small cell lung cancer cell lines. Cancer Lett. 2009, 279, 57–64. [Google Scholar]
- Leong, C.O.; Vidnovic, N.; DeYoung, M.P.; Sgroi, D.; Ellisen, L.W. The p63/p73 network mediates chemosensitivity to cisplatin in a biologically defined subset of primary breast cancers. J. Clin. Invest. 2007, 117, 1370–1380. [Google Scholar]
- Ibrahim, N.; He, L.; Leong, C.O.; Xing, D.; Karlan, B.Y.; Swisher, E.M.; Rueda, B.R.; Orsulic, S.; Ellisen, L.W. BRCA1-associated epigenetic regulation of p73 mediates an effector pathway for chemosensitivity in ovarian carcinoma. Cancer Res. 2010, 70, 7155–7165. [Google Scholar]
- Witta, S.E.; Gemmill, R.M.; Hirsch, F.R.; Coldren, C.D.; Hedman, K.; Ravdel, L.; Helfrich, B.; Dziadziuszko, R.; Chan, D.C.; Sugita, M.; Chan, Z.; Baron, A.; Franklin, W.; Drabkin, H.A.; Girard, L.; Gazdar, A.F.; Minna, J.D.; Bunn, P.A., Jr. Restoring E-cadherin expression increases sensitivity to epidermal growth factor receptor inhibitors in lung cancer cell lines. Cancer Res. 2006, 66, 944–950. [Google Scholar]
- Dohadwala, M.; Yang, S.C.; Luo, J.; Sharma, S.; Batra, R.K.; Huang, M.; Lin, Y.; Goodglick, L.; Krysan, K.; Fishbein, M.C.; Hong, L.; Lai, C.; Cameron, R.B.; Gemmill, R.M.; Drabkin, H.A.; Dubinett, S.M. Cyclooxygenase-2-dependent regulation of E-cadherin: prostaglandin E(2) induces transcriptional repressors ZEB1 and snail in non-small cell lung cancer. Cancer Res. 2006, 66, 5338–5345. [Google Scholar]
- Gemmill, R.M.; Roche, J.; Potiron, V.A.; Nasarre, P.; Mitas, M.; Coldren, C.D.; Helfrich, B.A.; Garrett-Mayer, E.; Bunn, P.A.; Drabkin, H.A. ZEB1-responsive genes in non-small cell lung cancer. Cancer Lett. 2011, 300, 66–78. [Google Scholar]
- Takeyama, Y.; Sato, M.; Horio, M.; Hase, T.; Yoshida, K.; Yokoyama, T.; Nakashima, H.; Hashimoto, N.; Sekido, Y.; Gazdar, A.F.; Minna, J.D.; Kondo, M.; Hasegawa, Y. Knockdown of ZEB1, a master epithelial-to-mesenchymal transition (EMT) gene, suppresses anchorage-independent cell growth of lung cancer cells. Cancer Lett. 2010, 296, 216–224. [Google Scholar]
- Korpal, M.; Lee, E.S.; Hu, G.; Kang, Y. The miR-200 family inhibits epithelial-mesenchymal transition and cancer cell migration by direct targeting of E-cadherin transcriptional repressors ZEB1 and ZEB2. J. Biol. Chem. 2008, 283, 14910–14914. [Google Scholar]
- Park, S.M.; Gaur, A.B.; Lengyel, E.; Peter, M.E. The miR-200 family determines the epithelial phenotype of cancer cells by targeting the E-cadherin repressors ZEB1 and ZEB2. Genes Dev. 2008, 22, 894–907. [Google Scholar]
- Burkitt, K.; Ljungman, M. Phenylbutyrate interferes with the Fanconi anemia and BRCA pathway and sensitizes head and neck cancer cells to cisplatin. Mol. Cancer 2008, 7, 24. [Google Scholar]
- Kakihana, M.; Ohira, T.; Chan, D.; Webster, R.B.; Kato, H.; Drabkin, H.A.; Gemmill, R.M. Induction of E-cadherin in lung cancer and interaction with growth suppression by histone deacetylase inhibition. J. Thorac. Oncol. 2009, 4, 1455–1465. [Google Scholar]
- Moody, T.W.; Switzer, C.; Santana-Flores, W.; Ridnour, L.A.; Berna, M.; Thill, M.; Jensen, R.T.; Sparatore, A.; Del Soldato, P.; Yeh, G.C.; Roberts, D.D.; Giaccone, G.; Wink, D.A. Dithiolethione modified valproate and diclofenac increase E-cadherin expression and decrease proliferation of non-small cell lung cancer cells. Lung Cancer 2010, 68, 154–160. [Google Scholar]
- St Germain, C.; Niknejad, N.; Ma, L.; Garbuio, K.; Hai, T.; Dimitroulakos, J. Cisplatin induces cytotoxicity through the mitogen-activated protein kinase pathways and activating transcription factor 3. Neoplasia 2010, 12, 527–538. [Google Scholar]
- Tanabe, M.; Izumi, H.; Ise, T.; Higuchi, S.; Yamori, T.; Yasumoto, K.; Kohno, K. Activating transcription factor 4 increases the cisplatin resistance of human cancer cell lines. Cancer Res. 2003, 63, 8592–8595. [Google Scholar]
- St Germain, C.; O'Brien, A.; Dimitroulakos, J. Activating Transcription Factor 3 regulates in part the enhanced tumour cell cytotoxicity of the histone deacetylase inhibitor M344 and cisplatin in combination. Cancer Cell Int. 2010, 10, 32. [Google Scholar]
- Ramalingam, S.S.; Maitland, M.L.; Frankel, P.; Argiris, A.E.; Koczywas, M.; Gitlitz, B.; Thomas, S.; Espinoza-Delgado, I.; Vokes, E.E.; Gandara, D.R.; Belani, C.P. Carboplatin and Paclitaxel in combination with either vorinostat or placebo for first-line therapy of advanced non-small-cell lung cancer. J. Clin. Oncol. 2010, 28, 56–62. [Google Scholar]
- Zhang, J.; Zhang, T.; Ti, X.; Shi, J.; Wu, C.; Ren, X.; Yin, H. Curcumin promotes apoptosis in A549/DDP multidrug-resistant human lung adenocarcinoma cells through an miRNA signaling pathway. Biochem. Biophys. Res. Commun. 2010, 399, 1–6. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
O’Byrne, K.J.; Barr, M.P.; Gray, S.G. The Role of Epigenetics in Resistance to Cisplatin Chemotherapy in Lung Cancer. Cancers 2011, 3, 1426-1453. https://doi.org/10.3390/cancers3011426
O’Byrne KJ, Barr MP, Gray SG. The Role of Epigenetics in Resistance to Cisplatin Chemotherapy in Lung Cancer. Cancers. 2011; 3(1):1426-1453. https://doi.org/10.3390/cancers3011426
Chicago/Turabian StyleO’Byrne, Kenneth J., Martin P. Barr, and Steven G. Gray. 2011. "The Role of Epigenetics in Resistance to Cisplatin Chemotherapy in Lung Cancer" Cancers 3, no. 1: 1426-1453. https://doi.org/10.3390/cancers3011426
APA StyleO’Byrne, K. J., Barr, M. P., & Gray, S. G. (2011). The Role of Epigenetics in Resistance to Cisplatin Chemotherapy in Lung Cancer. Cancers, 3(1), 1426-1453. https://doi.org/10.3390/cancers3011426