Analysis of Somatic Mutations in Cancer: Molecular Mechanisms of Activation in the ErbB Family of Receptor Tyrosine Kinases


Abstract
: The ErbB/EGFR/HER family of kinases consists of four homologous receptor tyrosine kinases which are important regulatory elements in many cellular processes, including cell proliferation, differentiation, and migration. Somatic mutations in, or over-expression of, the ErbB family is found in many cancers and is correlated with a poor prognosis; particularly, clinically identified mutations found in non-small-cell lung cancer (NSCLC) of ErbB1 have been shown to increase its basal kinase activity and patients carrying these mutations respond remarkably to the small tyrosine kinase inhibitor gefitinib. Here, we analyze the potential effects of the currently catalogued clinically identified mutations in the ErbB family kinase domains on the molecular mechanisms of kinase activation. Recently, we identified conserved networks of hydrophilic and hydrophobic interactions characteristic to the active and inactive conformation, respectively. Here, we show that the clinically identified mutants influence the kinase activity in distinctive fashion by affecting the characteristic interaction networks.1. Introduction
Lung cancer is the number one cause of death (157,000 in 2010) in America and accounts for almost as many deaths as the next three cancers—breast/prostate (40,000/32,000 in 2010), colon (51,000 in 2010) and pancreas (36,000 in 2010)—in both genders combined (American Cancer Society). About 85–90% of lung cancers are non-small cell lung cancers (NSCLCs), and present a poor prognosis in comparison to breast/prostate and colon cancers. Recent advances in cancer therapeutics targeting the EGFR/ErbB family of Receptor Tyrosine Kinases (RTKs) have shown promise in providing an alternate therapy with efficacy at least equivalent to that of chemotherapy in mutant forms of EGFR [1,2]. Here we shall focus on the intracellular kinase domain of the ErbB family receptors as a target for cancer therapy, while reviewing the overall RTK function and regulation.
1.1. RTK Structure and Signaling
The ErbB family kinases are a set of four homologous RTKs: EGFR/ErbB1/HER1, ErbB2/HER2, ErbB3/HER3, and ErbB4/HER4. RTKs are single pass transmembrane proteins important in intercellular signaling, by translating extracellular signals (ligands or growth factors) into activation of specific cell signaling cascades (reviewed in [3,4]). In humans, there are currently 58 known RTKs divided into 20 families. RTKs are composed of an extracellular ligand binding domain, a single transmembrane helix, a juxtamembrane domain, a cytoplasmic kinase domain and a C-terminal tail containing multiple phosphorylatable tyrosines. Activation of the extracellular domain by ligands triggers receptor dimerization and trans-phosphorylation of both the cytoplasmic kinase domains as well as the C-terminal tail. Dimerization and phosphorylation of the kinase domains activates the kinase domain which then phosphorylates the tyrosines in the C-terminal tail. The phosphorylated tyrosines serve as a docking site for downstream signaling molecules containing Src homology domain 2 and/or phosphotyrosine-binding domains and relay the signal into the subsequent steps of the cascade. The cell signaling pathways RTKs modulate involve crucial cellular processes such as cell proliferation, differentiation, metabolism, migration, and apoptosis.
The ErbB signaling network presents a “bow-tie” architecture, where multiple inputs and outputs are linked through a set of core processes [5]. The ErbB kinases are capable of binding a variety of ligands resulting in homo and heterodimerization, improving the flexibility and robustness in the ErbB signaling network by increasing response to extracellular signaling as well as allowing for cross-talk between ErbB kinase dimers, compensating for any reduced signaling of an individual ErbB member in a given cell type. For example, ErbB2 does not currently have a known ligand and ErbB3 is a pseudo kinase, missing key residues in the active site, greatly reducing its kinase efficacy [6]. However, ErbB2/ErbB3 heterodimers are extremely relevant in signaling and their overexpression is correlated with oncogenic transformation in breast cancers [7,8]. While response to extracellular signals occurs only through the four ErbB family kinases, which is then channeled through a conserved, relatively small collection of core biochemical interactions, the ErbB network expands again through transcription factors and positive as well as negative feedback mechanisms, eventually leading to the important cell signaling pathways of proliferation, differentiation, etc.
1.2. Temporal Regulation of RTK Signaling at the Cell Surface
Following the activation of RTKs, there are pathways which modulate the length of time the kinase is active on the cell surface, mainly regulated by receptor-mediated endocytosis and cellular phosphatase activity. Upon ligand induced activation, RTKs are internalized which removes the active RTK as well as the ligand from the cell surface (reviewed in [9-11]). The predominant pathway for endocytosis for RTKs is clathrin-mediated endocytosis, where the RTKs are rapidly endocytosed through clathrin-coated pits. One of the members of the ErbB family, ErbB4, has an alternate method of internalization by means of a proteolytic cleavage [12], which constitutes a biochemical switch and is involved in proper cardiac and neural development [13,14].
Protein Tyrosine Phosphatases (PTPs) oppose RTK activity, by removing the phosphate group on phosphotyrosines. The balance of the interplay between the RTKs and PTPs act as a major switch controlling the full activation of RTKs and thereby the cell fate decisions [15]. Prior to RTK activation, PTPs are in constant activity to reduce any residual phosphorylation from cross-talk. Given significant ligand, the RTKs inhibit/over-ride local PTP activity and have enhanced signal propagation [16]. In some cases, ligand binding also causes recruitment of PTPs that bind to target RTKs, dephosphorylates them, stabilizes the inactive form at the cell surface and inhibits further signaling [17]. The bivalent relationship between PTPs and RTKs thus forms a versatile regulatory unit in signaling.
Adding to activation and regulation of RTKs is the role of recently discovered cytoplasmic proteins cytohesins in EGFR [18] (and the proteins Dok7 in MuSK [19]). These proteins modulate RTK activity in both positive and negative fashion dependent on concentration. Increased amounts of such proteins activate the RTKs without any ligand binding events, while lacking the proteins prevents the activation of the RTKs even with a ligand binding event. It is an emerging view that cytohesins are important in the scheme of ErbB in dimerization and activation, although their specific role as an extra layer of control in the cell is unclear.
1.3. Regulation, Structure and Auto-inhibition of RTKs
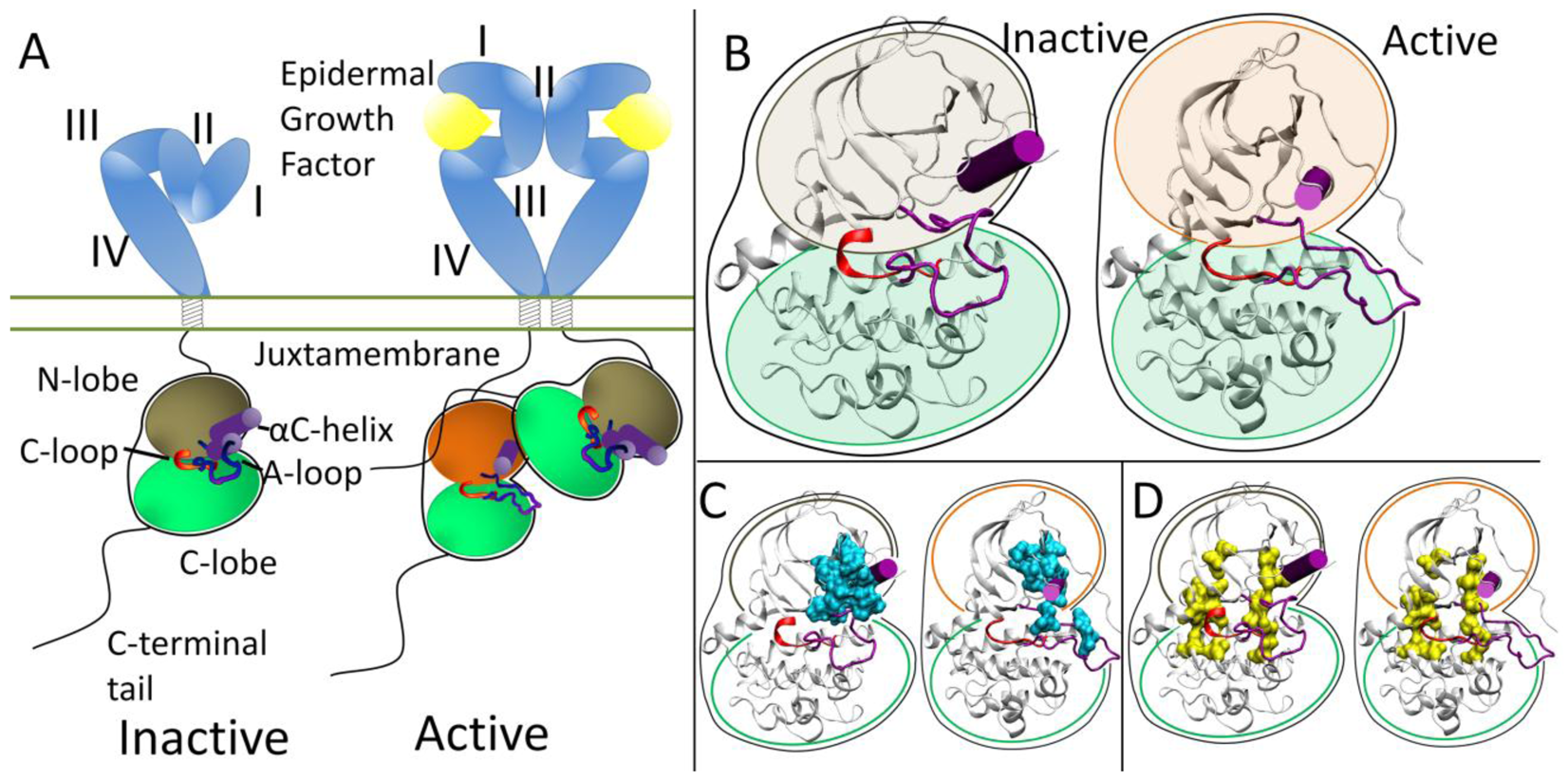
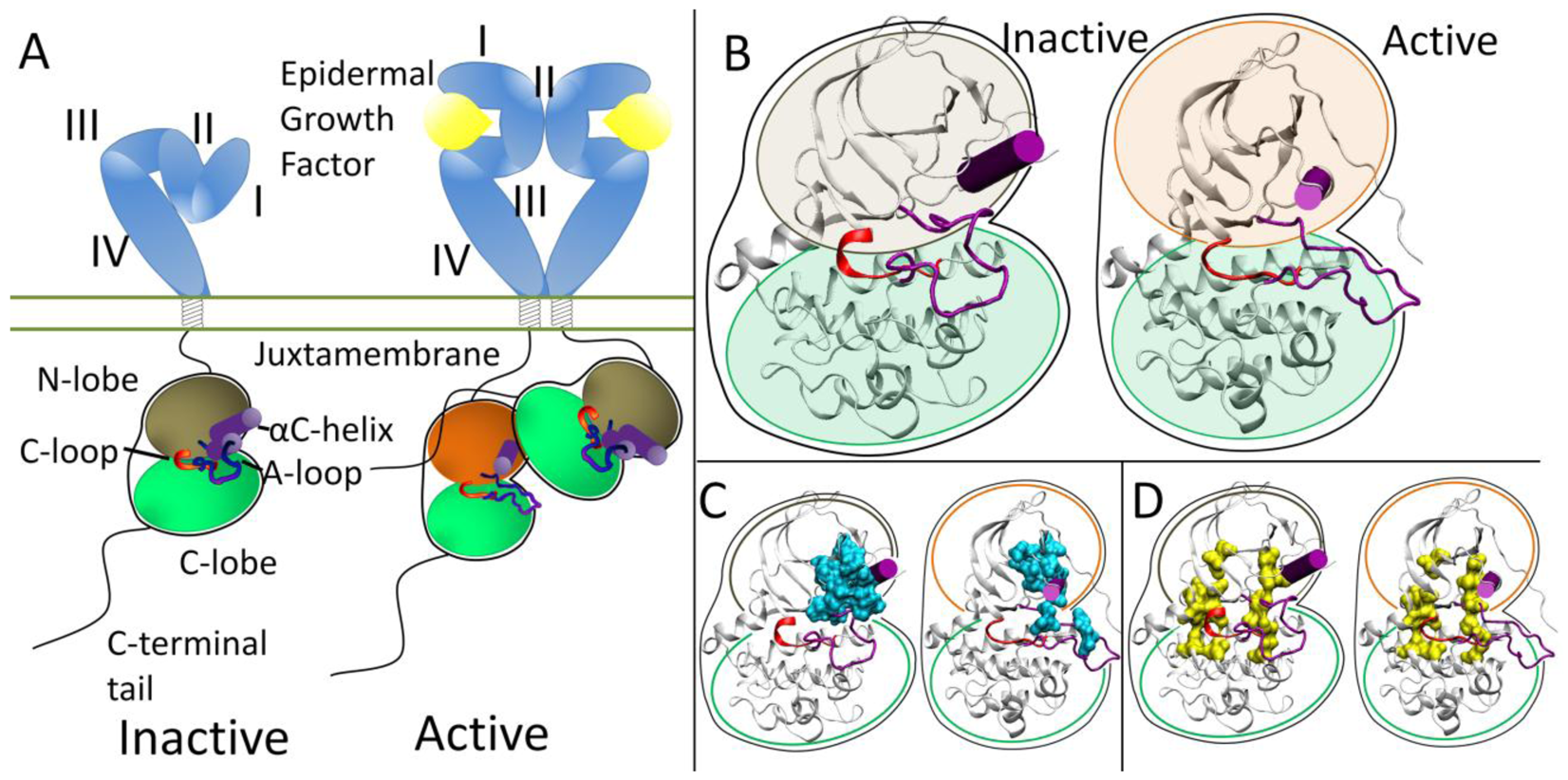
At the protein level, the extracellular domains are locked into an auto-inhibitory state preventing dimerization and are released with a ligand binding event. The specifics of how ligand binding facilitates dimerization for each RTK falls in the spectrum of “ligand mediated” dimerization, where the ligands bridge the two receptors without the receptors making direct contact, and “receptor mediated” dimerization, where the ligands make no direct contribution to the dimer interface (all mechanisms are reviewed in [20]). The ErbB family represents the extreme of “receptor mediated” dimerization [21,22]. The ErbB extracellular domain consists of four domains, with auto-inhibitory interaction between domains II and IV in a tethered conformation (Figure 1A) [23-26]. Ligands for the ErbB family are bivalent and bind to Domains I and III which cause a conformational change breaking the tethered conformation and exposing a dimerization arm in domain II allowing the dimerization arm to contact another ErbB RTK molecule.
Each RTK kinase domain is cis-autoinhibited in a characteristic fashion (reviewed in [20]) with activation mechanisms being unveiled as each RTK is being studied more in-depth. In RTK signaling, the intracellular kinase domain catalyzes transfer of the γ-phosphate of ATP to tyrosines on both the RTK itself and in other target substrates (reviewed in [3]). Regulation of the RTK kinase domain is thought to involve contributions from several conserved subregions: the catalytic loop (C-loop), the activation loop (A-loop), the glycine-rich nucleotide binding loop (P-loop), and the αC-helix, which together define the active site in the cleft between the β strand-rich N-lobe and the helical C-lobe. The catalytic loop residues directly participate in phosphoryl transfer. The A-loop and the αC-helix (Figure 1B) modulate the activity of the kinase domain by regulating accessibility of the active site to binding and coordinating both ATP and the substrate tyrosine. The ∼20 amino acid A-loop in ErbB kinases contains one phosphorylatable tyrosine (Y845 in EGFR, Y877 in ErbB2, Y850 in ErbB4, note: there is an alternate numbering scheme for the ErbB family, depending on whether a signaling segment is included; for example in EGFR, Y845 would be equivalent to Y869 in the alternate numbering scheme). The αC-helix and P-loop must be positioned correctly to coordinate the ATP and the substrate tyrosine for effective phosphoryl transfer.
Recent structural studies have revealed highly conserved hydrophobic “spines” within kinases that are considered important for defining their catalytic state [27,28], shown in Figure 1D. The regulatory spine (R-spine) consists of four hydrophobic side chains (M742, L753, H811, F832 in EGFR) anchored by an aspartic acid in the αF-helix (D872 in EGFR). The R-spine spans several key regulatory subdomains, and coordinates the motion of the N- and C-lobes of the kinase [27]. The catalytic spine (C-spine) involves eight hydrophobic side-chains (V702, A719, L774, V819, L820, V821, T879, L883 in EGFR) that help support and coordinate the adenine ring of ATP in the active state [28]. Similarly, in the inactive state there is a small hydrophobic ‘core’ formed between the αC-helix and the A-loop, which maintains the kinase in the inactive conformation (Figure 1C). Disruption of this hydrophobic core by single point mutations has been shown to activate EGFR [29-33].
Many of the kinase domains are inhibited through steric hindrances of a protein segment blocking off the active site and greatly reducing the efficacy of the kinase. Dimerization puts two kinase domains in close proximity to each other and although the kinase efficiency is greatly reduced, it is theorized that each kinase still has enough activity to phosphorylate its dimer partner. Phosphorylation of the protein segment prevents the protein segment from binding into the active site and allows the kinase to fully function. For the Ins [34] and FGFR [35] family of kinases, the A-loop serves as the inhibitory segment, while the juxtamembrane domain serves the same autoinhibitory role as the A-loop in MuSK [36], Flt3 [37], KIT [38] and the Eph [39] family; in the Tie2 [40] kinase, a segment of the C-terminal tail acts as the auto-inhibitor.
The ErbB family (and also Ret) has a different method of inactivation as phosphorylation of the A-loop or any other protein segment does activate the kinase; rather than a steric hindrance of the active site, there are collective auto-inhibitory interactions preventing the proper coordination between key loops in the kinase [41,42]. Activation of the kinase domain is also achieved through dimerization, though in this case, the dimer interface itself serves as the activating mechanism. In the ErbB family, the dimer interface is asymmetric similar to that of the cyclin dependent kinases and cyclin; the C-lobe of one kinase, the “activator”, contacts the N-lobe of the other kinase, the “receiver,” with the asymmetric dimer contacts causing a conformational change towards the active state through allosteric methods (Figure 1A) [43,44]. The αC-helix in the inactive ErbB kinase is rotated out, preventing key interactions from forming. Introduction of the activating asymmetric dimer interface forces the αC-helix to sample a different conformational space biasing towards the active state. Furthermore, the juxtamembrane domain in EGFR serves as latch to facilitate the asymmetric dimer interface between kinase domains [45-47].
1.4. Kinase Domain Mutations in Cancer and their Therapeutic Importance
Deregulation and mutation of RTKs have been correlated with cancer almost immediately after their discovery and purification in the early 1980s. The v-erbb oncogene in the avian erythroblastosis virus that was capable of inducing acute leukemia encoded a constituently active form of the homologous ErbB kinase protein [48]. With the increased study upon RTKs, the correlation between deregulation of RTKs and a variety of ailments and particularly in cancer has only grown stronger. Deregulation of RTKs in cancers can occur at several points: (1) increased ligand production through enhanced local autocrine activation; (2) specific gene translocations to produce kinase fusions with altered signaling profiles; (3) RTK overexpression at the cell surface; (4) mutation of the RTK protein to modulate activity; (5) disregulation of phosphatase and endocytosis mechanisms to increase RTK signal propagation.
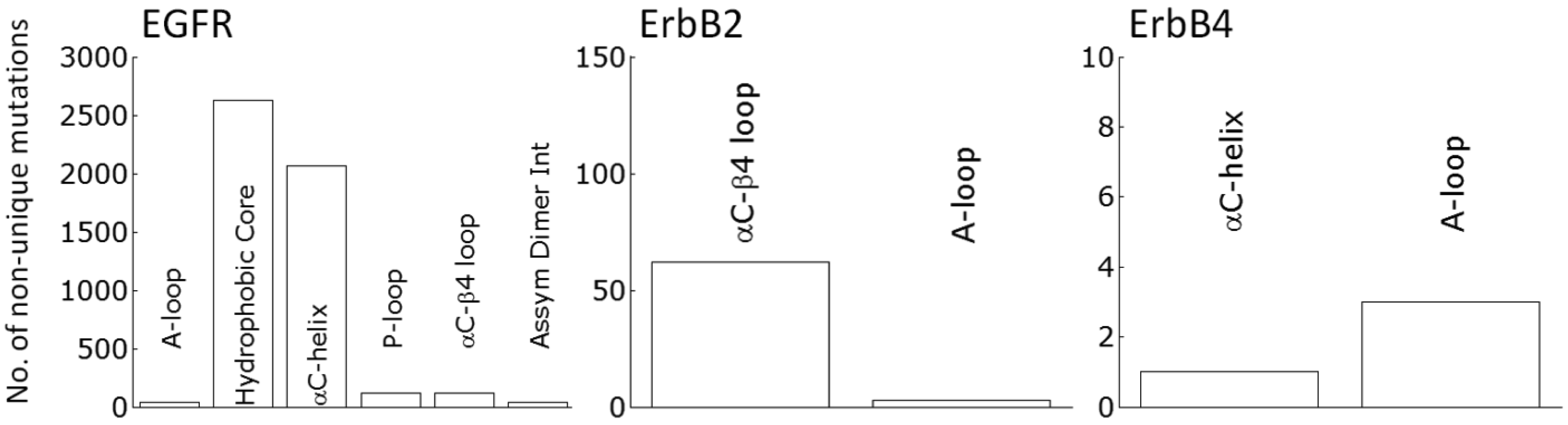
Clinically identified activating RTK kinase domain mutations have been discovered throughout many cancers (Tables 1 and 2). The results from Tables 1 and 2 are curated from the Catalog of Somatic Mutations In Cancer (COSMIC [91]), which has a much more thorough listing for all the mutations and all cancers. The oncogenic mutations cluster near the characteristic aspects of kinase activation (Tables 1 and 2). In Kit the predominant clinically identified activating mutations are focused on the juxtamembrane domain and the A-loop, both of which alter how the juxtamembrane domain serves as a steric hindrance to the active site. Whereas in the FGFR family, the kinase mutations are around the A-loop which serves as the inactivating segment. EGFR is cis-inhibited through autoinhibitory interactions centered around the rotation of the αC-helix and is released by the asymmetric dimer interface and the activating mutations observed in cancers for EGFR are dominated by two mutations accounting for ∼4500 of the 5000 or so total mutations (Figure 2): a point mutation (L834R) within the hydrophobic core as well as a small in frame deletion at least involving residues 747 to 751, at the tip of the αC-helix. The ErbB2 RTK is prevented from forming heterodimers through association with HSP90 through the uniquely hydrophobic αC-β4 region [92,93], which is where the majority of the activating mutations present (Figure 2). ErbB4 is not as well studied as EGFR and ErbB2; however it has recently come under scrutiny as a potential therapeutic target. Nevertheless, there is debate as to whether ErbB4 functions as an oncogenic promoter [61] or as protection from oncogenic transformation [94].
The increased kinase activity increases the dependency of tumor upon the RTK, which becomes “oncogenically addicted [123]” and inhibition of the RTK is a viable route for cancer therapeutics. EGFR and one of its small molecule inhibitor, Gefitinib, is a canonical example of RTKs, cancer and targeted therapeutics. The initial discovery of Gefitinib in 1994 was met with much excitement as a potential cancer therapeutic since it would be a low-dose targeted oral cancer therapeutic. In two phase II clinical trials of Gefitinib for advanced non-small cell lung cancer after progression of the cancer with chemotherapy, patients overall showed symptom improvement rates around 40% and 1-year survival rates of 25–35% [124,125]. The favorable results from the phase II trials gained Gefitinib FDA approval in 2003 prior to phase III clinical trials. However, the phase III clinical trials of Gefitinib versus placebo as a second-line therapy did not show any statistical significance in survival in the overall population, but there was a therapeutic benefit to the sub-group of Asian non-smokers [126]. Examination of the tumors revealed sets of mutations in the EGFR tyrosine kinase domain [31-33]. The sub-set of the tumors harboring these EGFR mutations are exceptionally sensitive to inhibition through Gefitinib, so much so that Gefitinib has equal to or greater efficacy than standard chemotherapy treatments in EGFR mutation positive patients [1,2]. There are several other small molecule tyrosine kinase inhibitors (TKIs) as well as antibodies already approved by the FDA and in use in the clinical settings (Table 3).
Given the importance of the ErbB family in cancers, it is necessary to understand their activation mechanisms at the molecular level to help design higher specificity therapeutics. This is especially important recently as, after sustained use of TKIs, the cancers tend to adapt through resistance mutants. In EGFR, the main mutation seen after extended treatment with Gefitinib is the T766M mutation [127]; the T766M resistance mutation was correctly predicted in EGFR through homology with resistance mutations seen in BCR-ABL, which was verified in vitro [128] and discovered several years later in patients [129-131]. Computational methodologies offer a powerful, quantitative, and complimentary alternative for the study of intracellular kinase domains which, if utilized correctly, can predict resistance mutations [132]. Here we review our recent results investigating the hydrophilic and hydrophobic networks using molecular dynamics simulation techniques as well as signal network models to help differentiate the conformational states across the ErbB family and prove the importance of understanding somatic mutations in the ErbB family.
2. Results and Discussion
2.1. The Inactive and Active Conformations Have Distinct, Characteristic Protein Motions
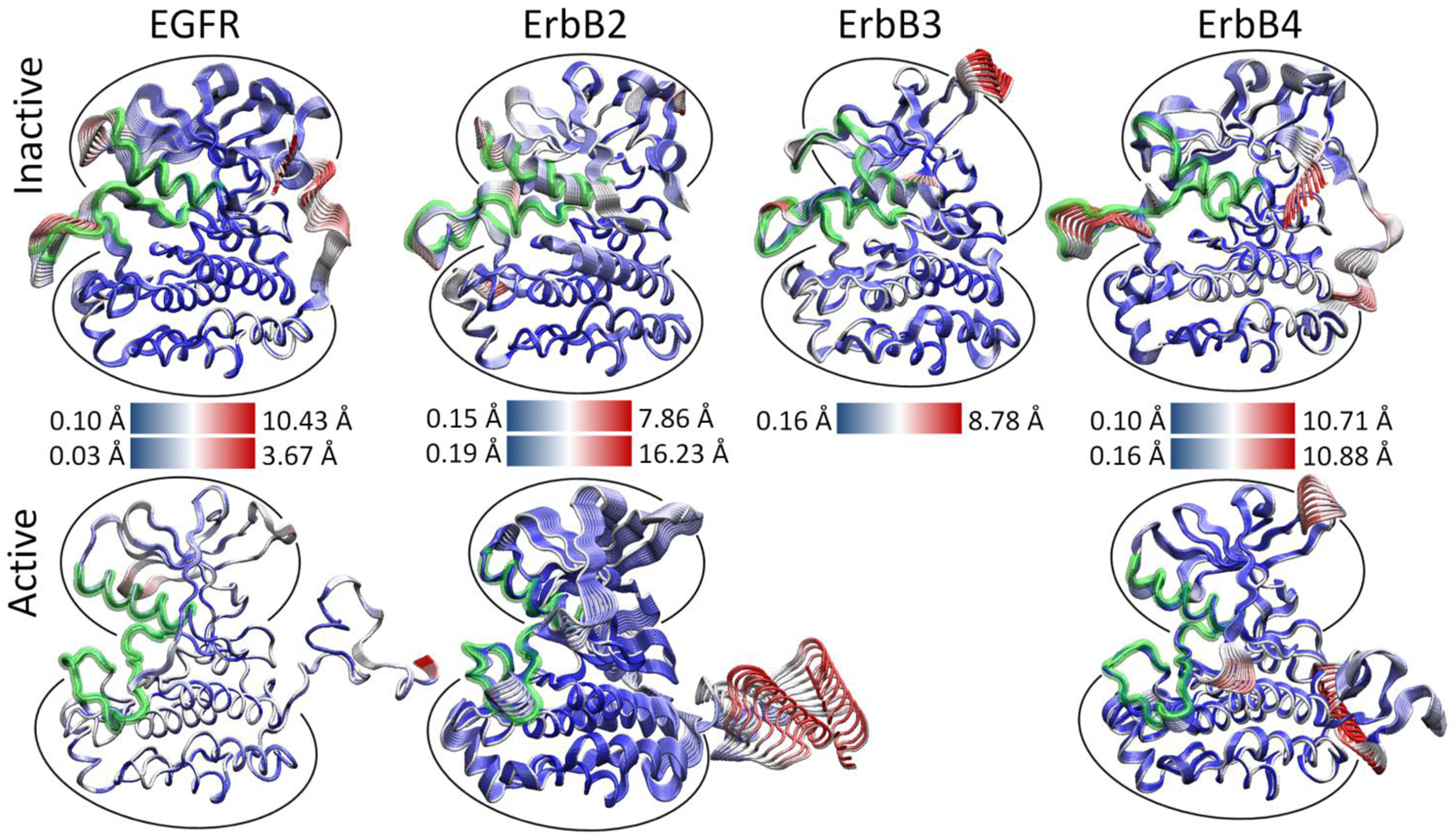
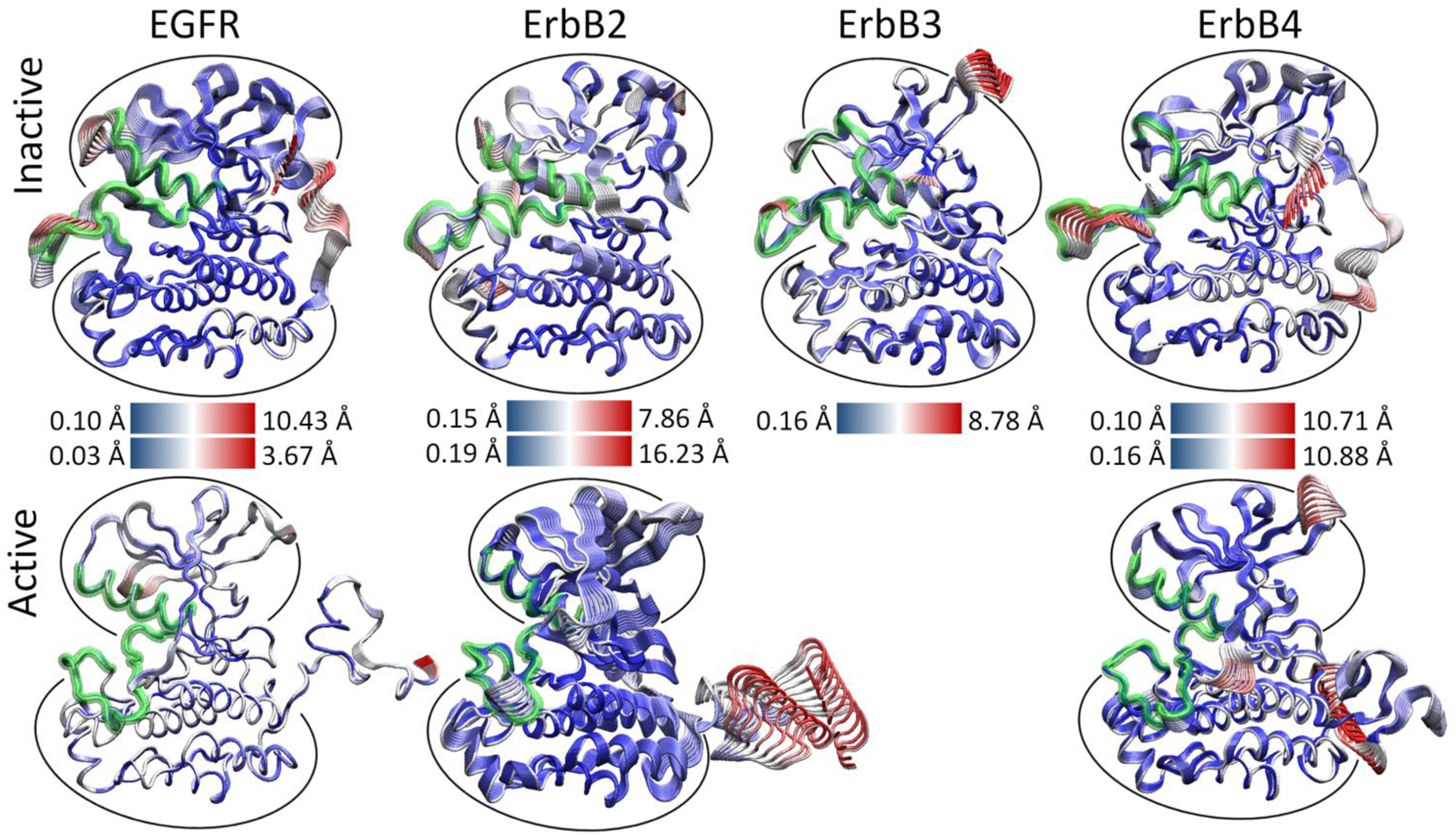
We hypothesized that the atomic fluctuations in the inactive and active forms of the ErbB family kinases would differ dramatically, as conformational rearrangement of the kinase domain is expected to correlate with significant changes in the dynamical behavior of the protein. Root-mean-squared deviation (RMSD) analysis of the overall kinase motion as well as individual sub-domain motions in our molecular dynamics (MD) simulations (see Section 4) showed all of the kinase systems were stable over the course of the simulation. Principal component analysis (PCA) of the kinase systems was performed for the Cα atoms of the entire kinase domain, but the majority of the fluctuations for the first few eigenmodes focused either on the free floating N-terminal and C-terminal tails or an active site consisting of sub-domains critical for catalysis, including the A-, C-, and P-loops and the αC helix [133]. We observed that the first three eigenmodes account for the majority of the atomic fluctuations in each trajectory, and the top 10 eigenmodes account for 75–85% of the total variation in each system.
As evident, there is a stark difference between the inactive and active conformations of the ErbB family. The active conformations have only low-level fluctuations within the kinase with the majority focused on the freely moving N-terminal and C-terminal tails (Figure 3). The inactive not only has motion in the tails, but also has significant motion in the active site: A-loop, C-loop, P-loop and αC-helix. A focused look at the PCA of the active site of the ErbB kinases revealed that motions occurring within the active sites of the inactive and active kinase monomers differ significantly, particularly in the A-loop [133-135]. The inactive ErbB kinase monomers (except ErbB3) exhibit large-amplitude motion in both the αC-helix and the A-loop, with smaller fluctuations in the P-loop and C-loop. We attribute the larger amplitudes to a more flexible protein segment; this is consistent with the observation that part of the activation loop is unresolved in almost all of the crystal structures of ErbB kinases to date. In contrast, the active ErbB monomers demonstrate a uniform level of motion across all four subdomains of the active site with only low-amplitude fluctuations and show no significant local deformations. This implies that the motions are more tightly coordinated across the active site in the active conformation than in the inactive conformation. The inactive ErbB3 kinase monomer also exhibits coordination of its active site loops without much local deformation, more similar to the active motion than the inactive motion, though its protein conformation is distinctly not in the active conformation.
Thus, for the three canonical kinase members of the ErbB family, which have a high-degree of sequence similarity, not only are the principal motions conserved across the systems, but the characteristic differences between the inactive and active kinase conformations are maintained in character. This finding suggests large (and possibly similar) differences in the internal network of interactions between the two activity states of each kinase.
2.2. Hydrophilic Interaction Networks Between Catalytically Important Sub-domains Help Coordinate Motions in the Active Kinases and Sequester Key Residues in the Inactive States
We find that several interactions are conserved across the canonical kinase members of the ErbB family in the active state (EGFR numbering is used in this discussion: see Table 4). Two salt bridges: E734-K851 and E738-K721, three H-bonds: L834-R812, K836-V810, and L838-R808, as well as the interaction D813-R817 which is a salt bridge in EGFR and ErbB4, but an H-bond in ErbB2. The E738-K721 salt bridge is highly conserved across all active kinases and helps coordinate the α and β phosphates of ATP bound in the active site. The E734-K851 salt bridge connects the A-loop and the αC-helix, coordinating the movements of these two sub-domains and dampening larger fluctuations. Similarly, three conserved H-bonds link the A-loop and the C-loop, coupling the motions of these two loops. These can be regarded as “fastening” H-bonds that maintain the N-terminal side of the A-loop open in its active state—the alternative (in the inactive state) being steric hindrance to the binding of ATP and peptide substrates.
In contrast with the case for the active configurations, few intramolecular interactions in the inactive kinase conformation are conserved across the ErbB family (Table 4). The inactive kinases do share an autoinhibitory pattern in which key residues required for kinase activation are ‘sequestered’, mainly breaking up the E738-K721 salt bridge. Similar to the situation described for Lck [136,137], E738 of EGFR is salt bridged to K836 in a manner that sequesters this glutamate and prevents it from forming the highly conserved E738-K721 salt bridge [134] required for full activation. In ErbB2, ErbB3 and ErbB4, the other half of the highly conserved salt bridge is sequestered; namely, the homologous K721 interacts with the D831 side chain, and this interaction in turn prevents K721 from forming the coordinating salt bridge.
The αC-β4 loop in the ErbB2 kinase is a hydrophobically unique region compared to the other ErbB family kinases and is important in association with the molecular chaperone HSP90 to prevent heterodimerization with the other ErbB members [92,138]. The ErbB2 kinase domain shares 83% sequence identity with EGFR; in the αC-β4 loop, however, five of the eight residues in ErbB2 differ from those in EGFR. In particular, the polar residues in the αC-β4 loop of EGFR are replaced by nonpolar residues in ErbB2, which form a hydrophobic patch that interacts with another segment of hydrophobic residues positioned in the A-loop. The hydrophobic patch in ErbB2 includes the following residues: V773, M774, G776, V777, G778, and V782 in the αC-β4 loop, and I861, T862, F864, L866, and L869 in the A-loop.
In ErbB2 only the S783-I861 interaction is shared by the inactive and active conformations and links the αC-β4 loop to the A-loop [134]. EGFR and ErbB4, by contrast, reveal a stronger hydrogen bonding network in the αC-β4 loop. The active EGFR and ErbB4 systems contain 10 and eight hydrogen bonds, respectively. The active ErbB2 system lacks these hydrogen bonds because hydrophobic interactions, rather than hydrophilic contacts, predominate in the αC-β4 region (see Section 2.4 for a hydrophobic analysis).
The interaction-networks highlight key conserved residues and interactions that are characteristic of each conformational state. Overall, in the active conformation, the six conserved interactions tightly couple the N-lobe to the αC-helix, the αC-helix to the A-loop, and the A-loop to the C-loop. The nature of this interaction network suggests that interactions among the nearest key subdomains in the ErbB family help to maintain the proximity of important catalytic residues, and position them so they can contribute directly to kinase activity. The conserved interaction network in the inactive conformations of the ErbB family kinases is less extensive (in EGFR, E738-K836 and D831-K721), but appears to serve a crucial role in sequestering key catalytic residues and thus preventing activity. The ErbB3 kinase hydrophilic interaction patterns fall in-line with those of the inactive kinase while presenting a more coordinated active site. Therefore we hypothesize that other interactions, mainly hydrophobic (see Section 2.4) help distinguish the canonical ErbB kinases from ErbB3.
2.3. Dimerization Disrupts the Inactivating Interaction Networks
We also analyzed fluctuations in the active and inactive EGFR [135], inactive ErbB2 [134] and inactive ErbB4 [134,135] kinase within the context of the asymmetric dimer described by Zhang et al. [42]. The fluctuations recorded for active EGFR in this context are very similar to those seen in the active EGFR monomer, with the conserved interactions described above being mostly preserved. In contrast, in the inactive dimers there is substantial motion of the αC-helix that is much greater than seen in the inactive monomer system. Even in the short timescale of the dimer trajectories, we observe a rearrangement of the αC-helix position towards the active conformation. Consistent with the allosteric activation mechanism proposed by Zhang et al. [42], several interactions in the inactivating interaction network surrounding the A-loop and the αC-helix are indeed disrupted in the inactive EGFR dimer trajectory, including Y740-S744, L834-D813, H846-R865, and K851-R812 interactions. Some interactions (e.g., E738-K836) are still present, although the population statistics indicate that their survival percentage (fraction present in the trajectory) has decreased significantly. The ErbB2 and ErbB4 inactive dimers demonstrate a similar loss of interactions surrounding the αC-helix and the A-loop. For ErbB4, a list of interactions disrupted upon dimerization includes: E739-R841, D742-R841, E743-R817, G838-R817, G855-E730, and K856-E844. Similar to the E738-K836 salt bridge in EGFR, the E743-R841 salt bridge in ErbB4 shows a marked decrease in survival time from >90% in the monomer trajectory to ∼70% in the dimer trajectory. Overall the introduction of the asymmetric dimer interface to the inactive ErbB kinases results in a significant weakening of the interactions in the inactivating interaction-network (discussed above) which sequesters key side-chains in the inactive state.
2.4. Hydrophobic Interaction Networks Help Differentiate between Active and Inactive Conformations
Hydrophobic interactions appear to provide context-specific contributions to stability of the active and inactive conformations of ErbB kinases. To investigate the effect of hydrophobic interactions on the ErbB kinase conformations, we analyzed the hydrophobicity as well as the solvent accessible surface area (SASA) of relevant hydrophobic sub-regions, namely, the C-spine, R-spine, hydrophobic core, and the αC-β4 region [133,135]. The four regions have a high percentage of hydrophobic side chains; however, some minor differences between members of the ErbB family exist, particularly in the αC-β4 region. The hydrophobicity of the sub-region is a non-additive quantity, dependent not only upon the primary structure but also on the surrounding environment. Garde et al. [139,140] have recently proposed an approach for quantifying the hydrophobicity of heterogeneous surfaces using water density fluctuations, according to which increased normalized water density fluctuations are used as a signature of a more hydrophobic surface. We have normalized the water density fluctuations so that 1 is indicative of a neutral region and plotted them against the SASA. By splitting the graph into four quadrants (Figure 4A) we can see quadrant I represents a hydrophilically favorable region with low hydrophobicity and high SASA, while quadrant IV represents a hydrophobically favorable region with high hydrophobicity and low SASA. Quadrant II represents a fragile or perturbation sensitive region with high hydrophobicity but also with a high SASA.
The active conformations in the C-spine show a clear delineation between the active and inactive conformations regardless of dimer or mutational state (Figure 4A). The active conformations minimize the SASA (mean of 500 Å2) in comparison to the inactive (mean value of 700 Å2). In addition, in the transition between the inactive to active states, the active conformations increase in hydrophobicity and settle into the hydrophobically favorable region, implying the active conformations have a better formed C-spine, correlating with the observation that the C-spine helps in coordinating the loops in the active conformation. The SASA of the ErbB3 C-spine falls within range of the inactive EGFR and ErbB4 systems, reflecting that, there is no corresponding ‘fully-active’ state for ErbB3 as for the other ErbB kinases. This inability to ‘fully’ activate can be attributed to the lack of the crucial hydrogen bonding network identified earlier, which is required to stabilize the active-like kinase conformation. The hydrophobicity plots of the R-spine show a similar difference between the active and inactive conformations but not as drastically as the C-spine (Figure 4B). For each system the active systems expose less surface area than the inactive systems, but only for each system locally (dotted lines on Figure 4). The SASA of the ErbB3 R-spine deviates from the values for the inactive EGFR and ErbB4 systems, and instead demonstrates low SASA (high hydrophobicity). This result can be rationalized by the increased hydrophobicity of the R-spine, which includes segments of the truncated αC helix in ErbB3.
With respect to the hydrophobicity plots of the hydrophobic core, the monomer EGFR and ErbB4 inactive systems are situated within the perturbation sensitive region with their respective active conformations lying outside of the perturbation sensitive region (Figure 4C). For ErbB2 the reverse is true, which is consistent with the sensitivity of the inactive conformation to hydrophobic perturbation, especially for EGFR and ErbB4 but not for ErbB2. Therefore single point mutations would be expected to disrupt local conformations contributing to hydrophobicity. Notably, mutations of hydrophobic residues in the hydrophobic core are reported for EGFR and ErbB4 in clinical studies [31-33], whereas, for ErbB2, such mutations are found surrounding the αC-β4 region [57,58]. By contrast, an analogous mutation in ErbB3 abolishes ATP-binding and phosphorylation activity [141], indicating that hydrophobic interactions in the core promote ErbB3 activity, rather than maintain an autoinhibited state as they do in EGFR and ErbB4.
So far all the hydrophobic plots have shown preferential hydrophobic interactions in the active systems versus the inactive systems. However, examination of the asymmetric dimer interface reveals a clear delineation in hydrophobicity in preference for the inactive systems as well as dimerization reducing this hydrophobic benefit implying an activating stimulus (Figure 4D). The asymmetric dimer interface consists largely of hydrophobic side-chains in the N-lobe of the receiver kinase (L680, I682, L736, L758, and V762) and the C-lobe of the activator kinase (I917, Y920, M921, V924, and M928) [42]. The active monomer systems trend to the perturbation sensitive quadrant, while the inactive monomer systems show a reduced SASA in comparison to the active systems, removing them from the perturbation sensitive quadrant. This decrease for the inactive monomers may imply a preference for the inactive state in the monomer context. Notably, the dimeric systems record much lower SASA, pushing all the dimer systems into the hydrophobically favorable quadrant, implying that hydrophobic interactions provide a dominant driving force for dimerization; interestingly, for the EGFR dimer, the inactive state is very similar to the active and hence the preference for the inactive conformation is not implied in the context of the dimer. Thus, dimerization also provides a stimulus for activation.
Interestingly, all ErbB2 monomer systems present a very high hydrophobicity (black bordered circles on Figure 4) across all of the hydrophobic subregions, while the ErbB2 dimer has a reduced hydrophobicity in comparison to the monomers (green circles on Figure 4). This is consistent with the notion that hydrophobicity is particularly important in the context of ErbB2, owing to its hydrophobic interaction with HSP90, particularly the αC-β4 region which is an unstructured span between the αC-helix and the β4 sheet in RTKs. From a sequence perspective, only in ErbB2 is the αC-β4 region predominantly hydrophobic and exceptionally so. Both the inactive and active conformations of the ErbB2 monomer systems reflect the trend by singling out the ErbB2 monomer systems as particularly hydrophobic in the αC-β4 region (Figure 4E). The mean SASA for the αC-β4 region in the ErbB2 systems is also consistently lower than in other members of the ErbB family. As discussed in [134], this unique feature of ErbB2 is thought to be responsible for its preferential association with the molecular chaperone Hsp90.
Thus, the analyses for the spine regions and the hydrophobic core collectively lead to the remarkable prediction that while the αC-β4 region is perturbation sensitive for the inactive conformation of ErbB2, the hydrophobic core has the same effect for EGFR and ErbB4. Indeed, this correlates well with clinical studies, where activating point mutations in the hydrophobic core have been found in EGFR and ErbB4 but those in the αC-β4 region are found in ErbB2, suggesting that the hydrophobic analysis enables the context-specific identification of fragile sub-regions.
2.5. A-Loop Phosphorylation in EGFR and ErbB2 Reveals Extra Domain Coupling
The role of A-loop phosphorylation in the ErbB family kinases is controversial. Phosphorylation of EGFR on Y845 (Y877 in ErbB2 and Y850 in ErbB4) has been observed experimentally, though does not seem to be required for catalytic activity, as a Y845F mutation is fully active [142]. In contrast, multiple studies have reported the importance of Y877 phosphorylation for kinase activity [143,144], though the precise mechanism of phosphorylation is not clear. Hence it is possible that phosphorylation of Y877 potentiates ErbB2 kinase activity.
To investigate the effects of A-loop phosphorylation in the ErbB family, we simulated EGFR [135] and ErbB2 Y845-phosphorylated and Y877-phosphorylated systems [134], respectively, both in the inactive and active conformation. In the active A-loop phosphorylated systems, we discovered that they mostly preserved the same set of a network of hydrophilic interactions seen in the unphosphorylated systems, but also gain several more fastening interactions that maintain the C-terminal side of the A-loop in the open conformation: the ErbB2 systems gain Y877-F899, A879-R897 as seen in [134]; the EGFR phosphorylated systems have the homologous fastening interactions in Y845-Y867 as well as A879-R865. All of the phosphorylated systems (active and inactive systems) share a common interaction in Y845-R812, which strengthens the coordination between the A-loop and C-loop. The inactive systems do see a shift in interaction patterns, but no subsequent weakening of the sequestration interactions. Therefore we hypothesize that while phosphorylation incrementally strengthens the coordination of the active site, it is not a strong enough stimulus to cause activation.
To quantify the effect of A-loop phosphorylation on ErbB2 activity, we computed the Helmholtz free energy difference between the Y877-unphosphorylated and Y877-phosphorylated states in the NVT ensemble using the FEP method [134]. Four different simulations were performed for comparison, including the phosphorylation transformation of Y877 to pY877 in the unphosphorylated structures (inactive and active) as well as the respective pY877 to Y877 de-phosphorylation transformation in the phosphorylated systems. The transformation of Y877 to phosphorylated Y877 produced a free energy change ΔΔF value of −1.1 ± 1.4 kcal/mol [134]. The free energy values (ΔΔF values) suggest that phosphorylation of Y877 provides a small increase in stability of the active conformation relative to the inactive state, although it is insufficient to significantly lower the kinase activation barrier. The free energy studies support our hypothesis that phosphorylation does not cause activation, but does marginally increase coupling of key loops in the active site. Consequently, we suspect molecular stimuli in addition to A-loop phosphorylation are likely required for the complete conformational activation of the ErbB2 kinase.
2.6. Effects of Clinically Identified Somatic Mutations on the Interaction Networks
The inactive and active conformations are differentiated by the balance between the hydrophobic and hydrophilic interaction networks [135], with perturbation by A-loop phosphorylation not greatly altering these networks. Here we examine what happens to these networks with the perturbation by clinically identified activating mutations [135]. The predominant mutations in the clinically identified EGFR activating mutations, a small in-frame deletion at the start of the αC-helix (we used del 723–729 ins S), alter both the hydrophilic and hydrophobic interaction networks. The L834R mutation increases the coupling of the A-loop and C-loop (through the G833-H811 H-bond) and the A-loop to the C-lobe (through the R834-R865 interaction) not seen in other systems. The hydrophobic interaction networks are disrupted because of the switch of the hydrophilic Leu to a hydrophilic Arg, particularly in the R-spine and the hydrophobic core, shifting them into the perturbation sensitive region, indicating a stimulus for a conformational shift (Figure 4B and 4C). The deletion mutant directly alters the conformation of the αC-helix by shortening the connecting subdomain, thereby shifting it towards the active conformation. However because it is missing residues in the αC-helix, the dynamics and the potential extension of the αC-helix are significantly inhibited. In studies by Choi et al. [30], the deletion mutant had increased basal kinase activity, but under EGF stimulation it had less overall kinase activity compared to wildtype. In the monomer state, the deletion mutant causes a shift towards active, disrupting key interactions and potentially increasing activity; once the asymmetric dimer forms, the active conformation is destabilized, through the deformation of the αC-helix interfering with the dimer interface and lack of a completely formed helix.
Other EGFR mutations in NSCLC [30,31,49] (E685S, G695S, S744I and L837Q) plus a set of mutations in ErbB4 found in melanoma [61] (E836K, E872K and G936R) also alter the hydrophilic and hydrophobic interaction networks and allow us to propose a set of mechanisms for their increased basal kinase activity. For the L837Q mutation, the replacement of the hydrophobic Leu with the hydrophilic Gln would disrupt the hydrophobic core between the A-loop and the αC-helix, with the Q837 side-chain rotating away and inducing a local steric effect; similar to L834R the Q837 residue may cause increased coupling between the active site subdomains and also disrupt the interaction of K836 with E738. The S744I mutation in NSCLC is situated at the base of the αC-helix and alteration of hydrophobicity likely causes a transition of the αC-helix into its active position. Two of the mutations E685G and G695S are in the N-lobe, near the asymmetric dimer interface, and are likely to alter kinase activity by either increasing the dimerization affinity or by reconfiguring the EGFR RTK monomer by partially mimicking the formation of the asymmetric dimer interface. The G936R mutation in ErbB4 is located in the C-lobe, but is close to the asymmetric dimer interface and potentially increases the dimerization affinity between kinase domains. The E836K and E872K ErbB4 mutations are situated around the homologous small hydrophobic core and are poised to disrupt the hydrophobic interaction by the change from a negatively charged side chain to a positively charged chain, which could provide an activating stimulus.
Clinically identified mutations in ErbB2 are predominantly found in αC-β4 region (see Figure 2) [56,145], shown here to be uniquely hydrophobic in the ErbB family. The most common mutant is an in-frame insertion at residue G776 (G776YVMA). We hypothesize these mutations operate by weakening the hydrophobic interactions surrounding the αC-β4 loop and forming a hydrophilic interaction network similar to those we have observed in EGFR and ErbB4. Alteration of the hydrophobicity of the αC-β4 region would disrupt the binding to HSP90 [93] and allow ErbB2 to heterodimerize freely with other ErbB members [92] and thus, activity of ErbB2.
Another set of mutations in ErbB4 found in NSCLC are not activating mutations, but inactivate the kinase domain: G802dup and D861Y [94]. D861 is the start of the highly conserved DFG motif in RTKs (considered the coordinating aspartate), with mutation poised to disrupt proper ATP placement and reduce activity. The G802dup is spatially located near the P-loop and similarly affects ATP binding affinity. In ErbB4, two of the point mutations are located near the active site: E836 is located next to the C-loop, whereas E872 is situated in the A-loop. Considering their proximity to the active site, the mutations are also poised to alter substrate binding and phosphorylation; these mechanisms are discussed in a recent computational study in the context of EGFR [146].
2.7. Clinical Implications of Oncogenic EGFR Mutations from a Multiscale Model of ErbB Receptor Signaling
For cellular homeostasis, pro-survival signals are balanced by pro-apoptotic signals, with both being triggered and balanced by a variety of interacting intracellular pathways. Using a simplified model (Figure 5) for the effect of AKT activation on cell response, we showed that preferential AKT activation is conducive for the cell to rely on and be addicted to [123,147]) for generation of pro-survival signals [148]. Our simplified model illustrates a mechanism by which inhibition of the dominant source of pro-survival signals shifts the cellular state to one devoid of pro-survival signals, representing a perturbation sensitive point in the cellular signaling network which can account for a remarkable inhibitor sensitivity [149].
We hypothesized the mechanisms that lead to inhibitor hypersensitivity (Gefitinib for EGFR) attack these perturbation sensitive points of network hypersensitivity and fragility. Since preferential AKT activation is a hall mark of the hyper-sensitive mutants as well as the efficacy of the inhibitors, we determined, through a global sensitivity analysis [149,150], the combinations of model parameter perturbations that drive enhanced production of pAKT and pERK. The top components that produce maximum sensitivity in terms of changes to the pAKT and pERK levels were PI3K, Ras, Gab-1, MEK, Raf which have all been observed in several human cancers [151-155]. Moreover, it has been established in screened breast and colorectal cancer patients that the GAB-1, MEK, and Ras mutations are non-random and likely arise from selective evolutionary pressures that give the cancer cells a survival advantage [155].
With reference to the clinically identified EGFR mutants found in non-small cell lung cancer patients, mainly L834R and del L723-P729 ins S, we found the mutations had altered affinities for phosphorylation of specific tyrosines in the C-terminal tail: Y1068 and Y1173. The preferential binding characteristics of different cytosolic substrates to different phospho-tyrosine locations of the ErbB family kinases has been reported [156]. Thus, differences in the phosphorylation kinetics associated the different tyrosine sites of the cytoplasmic C-terminal tail of the EGFR kinase can induce differential patterns of downstream signaling leading to differences in the activation of cell signaling networks. The effect of altered affinities of the Y1068 and Y1173 sites to the catalytic domain of the EGFR is that the L834R under normal EGFR expression sees a ∼5-fold decrease in ERK activation and a smaller ∼15% decrease in AKT activation. The del L723-P729 ins S mutant however, shows sustained ERK as well as AKT activation relative to wildtype. For EGFR over-expressed cells, both ERK and AKT activation characteristics show relative insensitivity to EGFR as a result of signal saturation. Furthermore, the mutants can continue to signal even in the absence of the growth factor. In addition, the mutant signaling can be different due to changes in the ATP affinity. However, both these factors do not introduce any differential characteristics (in terms of preferring Y1068 to Y1173) and cause a differential in overall activation levels of ERK and AKT.
The perturbation of the phosphotyrosine kinetics of Y1068 and Y1173 through mutations (L834R and del L723-P729 ins S) as well as trafficking (which is not explicitly considered in our model) could be directly responsible for the differential signaling leading to preferential AKT activation [148]. The restoration of signaling has also been reported through a double mutation of L834R/T766M [128,157]. This double mutant increases receptor phosphorylation (Y1068 and Y1173) kinetics 100-fold [158] while simultaneously decreasing inhibitor affinity [157]. Another drug resistance mechanism related to Y1068 kinetics that circumvents Y1068 has been identified. In the presence of ErbB3, a branch of signaling analogous to that through Y1068 becomes available through ErbB hetero-dimerization, directly resulting in PI3K recruitment on ErbB3 and subsequent AKT activation, which is discussed below.
2.8. Systems Model of ErbB Signaling Defines a Mechanism for ErbB3-mediated TKI Resistance
Previous experimental studies have demonstrated that ErbB3 is a key mediator of resistance to various tyrosine kinase inhibitors (TKIs) currently in use [159-163]. Some postulated mechanisms include leaky ErbB2-catalyzed phosphorylation of ErbB3, e.g., incomplete inhibition of ErbB2 catalytic activity by the TKI [159,162,164]. Indeed, previous experimental studies have demonstrated that leaky ErbB2 phosphorylation of ErbB3 in TKI-bound ErbB2/3 heterodimers is amplified by additional resistance mechanisms, such as inhibition of cellular phosphatases by TKI-mediated production of reactive oxygen species (ROS), and increased expression of ErbB3 at the plasma membrane [159,165,166]. However, Shi et al. [141] have recently shown the assumed inactive ErbB3 pseudokinase is, in fact, a weakly active kinase with ∼1000 fold weaker phosphorylation than the canonical kinase members of the ErbB family [141]. Therefore, though the ErbB2 kinase is a viable route of resistance, here we consider ErbB3 catalytic activity in the ErbB signaling network as a potential TKI resistance mechanism [133].
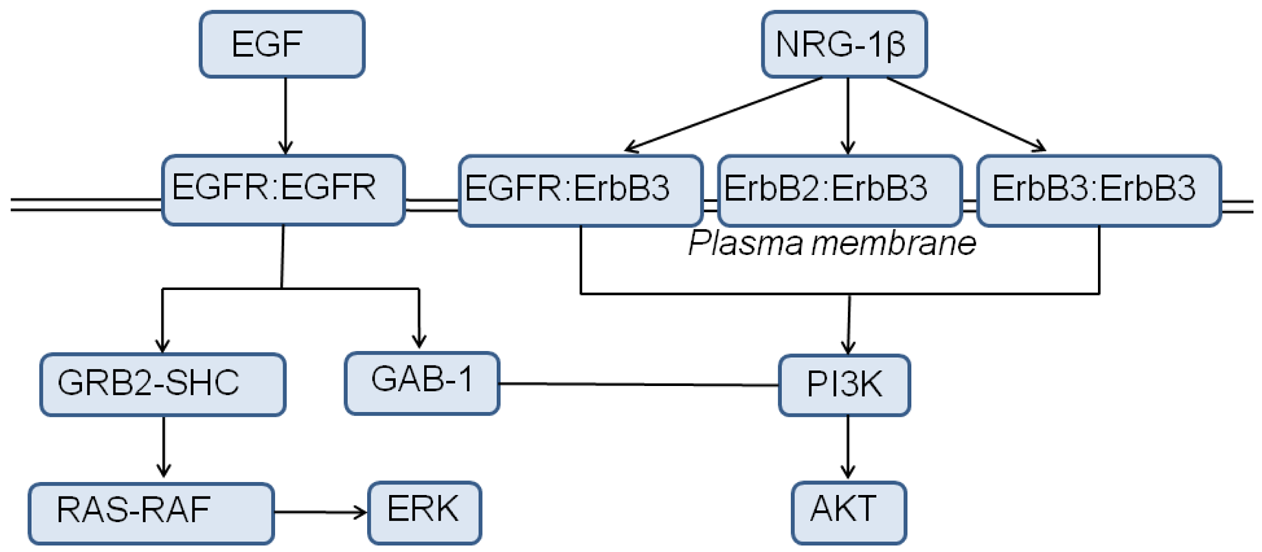
To translate our observations of the weak, yet robust, activity of the ErbB3 kinase into a physiologically relevant context and investigate the implications of ErbB3 activation for ErbB signaling dynamics, we constructed a systems-level model for ErbB3 based on that of Schoeberl et al. [167], with added ErbB3 phosphorylation rate constants derived from experiments [141] (Figure 5). We simulated stimulation of the ErbB signaling network through NRG-1β, which signals through the ErbB2/ErbB3/ErbB4 kinases (though ErbB4 kinase is omitted from the model since ErbB4 signaling is weak or absent in many cancer cell lines).
A parameter sensitivity analysis identified the key proteins that direct signaling in our model of the ErbB network with respect to AKT activation via stimulation of NRG-1β. ErbB3 and NRG-1β represent the most sensitive species in the signaling network, followed by ErbB2 concentration. EGFR is not a strong determinant of the extent of AKT phosphorylation, as expected from the weak ability of NRG-1β to elicit EGFR dimers. PTEN (the PIP3 phosphatase) and the ErbB phosphatase exhibited a negative sensitivity in the analysis, as these phosphatases negatively regulate the signaling network through dephosphorylation of key molecular species.
Upon incorporation of the TKI lapatinib, which inhibits EGFR and ErbB2 catalytic activity, into our model of ErbB3, the ErbB signaling network relies more heavily upon ErbB3 activity for AKT induction. With completely inhibited EGFR and ErbB2 signaling via introduction of lapatinib into the model, ErbB3 and AKT signals still persist at maximal inhibition by lapatinib. The results of pAKT sensitivity analysis of the lapatinib-treated model to those of the inhibitor-free model show that sensitivity to ErbB3 and NRG-1β increases, whereas sensitivity to EGFR and ErbB2 decreases, as lapatinib sequesters EGFR and ErbB2 molecules. The negative normalized sensitivity to the ErbB phosphatase also increases, as the pool of ErbB dimers has diminished due to sequestration of EGFR and ErbB2 by lapatinib. Thus a single alteration to the signaling model (in this case, addition of lapatinib) significantly redefines the most perturbation-sensitive nodes in the network.
Although the pAKT signal induced by ErbB3 phosphorylation in our in silico lapatinib-treated cell is relatively weak, in an actual physiological context, a tumor cell may employ several resistance mechanisms at once [159,162,168,169]. One of the sensitive nodes of NRG-1β-driven ErbB signaling is ErbB phosphatase levels, and simulations showed decreased activity of phosphatases resulted in an amplification of the level of AKT signaling induced by ErbB3. It has been demonstrated that in certain cases of TKI resistance, the tumor cell responds to the reduction in pAKT levels by upregulating vesicular transport of ErbB3 from the cytoplasm to the plasma membrane [159,166]. Our results show for a 2-fold increase in surface ErbB3 level, AKT signaling is similarly amplified. For 25 nM NRG-1β, the pAKT signal is restored to nearly 60% of its no-inhibitor control level, and pAKT levels are nearly 100% regained for 100 nM NRG-1β, effectively recreating drug resistance in silico.
Our data parallels the experimental studies performed by Sergina and colleagues [159], which describe ErbB3-mediated resistance and pAKT signaling in various TKI-treated tumor cell lines as well as in vivo. Thus our model demonstrates that even a weak level of ErbB3 signaling, as suggested by our previous results [141], are physiologically relevant in the context of an ErbB-driven tumor cell, and illustrates several routes through which ErbB3 signaling may be compounded by other previously postulated resistance mechanisms to generate TKI resistance.
3. Conclusions
Understanding the activation mechanisms of the ErbB kinases is important considering the crucial cell signaling pathways they control. Mutations of the ErbB kinases, and particularly EGFR, have been revealed in mutational screens of multiple cancers (see Tables 1 and 2). The regulation of the ErbB kinases is maintained through a flexible multiple-point network with multiple redundancies to ensure signal propagation. Here we reviewed a hierarchical multiscale model of the ErbB family kinases to investigate their activation mechanisms and the subsequent effects of kinase perturbation on their cell signaling network.
At the molecular level, the active versus the inactive conformations of the canonical ErbB kinases (EGFR, ErbB2, ErbB4) have a distinct fluctuation pattern, where the active conformations demonstrate low levels of fluctuations and coordination between the subdomains dictating the active site (A-loop, P-loop, αC-helix and C-loop), while the inactive conformations exhibit high-levels of fluctuations with little coordination in the active site. The similarity is carried through to the hydrophilic and hydrophobic interaction networks. The interaction networks also highlight perturbation sensitive points in EGFR which correlate well with clinically identified activating mutations in NSCLC. Simulation of the clinically identified EGFR mutations in the ErbB system model reveals enhanced dependence on the EGFR for AKT signaling; thus they are perturbation sensitive points at the cell signaling level and are sensitive to inhibition by TKIs.
The ErbB3 kinase presents a different case from the canonical ErbB kinase members, as it is missing key catalytic residues. Even though the ErbB3 kinase is distinctly in the inactive conformation with a correlated inactive hydrophilic interaction network, it exhibits a coordinated active site which seems to stem from increased hydrophobic interactions, in line with our recent studies [141] showing ErbB3 does have kinase activity, though ∼1000 fold less than the canonical ErbB kinases. At the cellular level, a weak level of ErbB3 activity may be sufficient to induce drug resistance in the context of an ErbB signaling-dependent tumor cell.
The multiscale model we present is useful in examining atomic level protein characteristics and carrying them over into differential activity of cell signaling networks [170]. The results we show have a striking correlation with highlighting perturbation sensitive regions with the regions mutated in clinically identified oncogenic kinase mutations: the hydrophobic core in the EGFR and ErbB4 as well as the αC-β4 region in ErbB2. We have similar correlation with observed EGFR kinase mutations (L834R and del 723–729 ins S) affecting cell signaling pathways so that inhibition of the mutant kinase is a viable therapeutic strategy as well as the observed mechanisms of resistance to EGFR kinase inhibition. Furthermore, by examining the “inactive” pseudokinase ErbB3 and characterizing its weak activity, we justify an alternate means of resistance to ErbB TKI inhibition and propose that targeting of ErbB3 may represent a superior therapeutic strategy for certain ErbB-driven cancers. Therefore, multiscale modeling is a tool that can not only link kinase domain fluctuations with signaling effects to help contextualize mutations found in oncogenic cell lines, but also refine therapeutic strategies to target key perturbation sensitive regions of kinase signaling, at the molecular level as well as the cellular level.
4. Models and Methods
Here we provide a brief summary of the hierarchical multiscale modeling scheme we have employed for describing ErbB signaling. Sections 2.1-2.6 use a fully atomistic molecular model of the kinase domains in a solvated, equilibrated system. We model the kinase domain receptor activation characteristics of the ErbB family RTKs using molecular dynamics (MD) simulations [133-135,148] with the ErbB2 and ErbB3 kinase domains created through homology modeling [133,134]. The EGFR, ErbB2, ErbB3 and ErbB4 inactive and active monomer systems were simulated for 10 ns apiece. The EGFR active dimer and ErbB2 inactive dimer systems were also simulated for 10 ns. The EGFR inactive wildtype dimer system was simulated for 30 ns while the EGFR L834R and del 723–729 ins S mutant dimer systems were simulated for 20 ns. Principal component analysis (PCA) was performed using CARMA to find the dominant fluctuations of each domain [133-135]. There are multiple measures of protein fluctuations (B-factor, RMSF, RMSD, etc. [171]), we choose to use RMSD here to track the specific protein deviation at each time point. Hydrophilic and hydrophobic interaction networks were characterized for each state as well [133-135,148]. The normalized water density fluctuations were calculated using the method by the Garde group [139,140]: VolN*(〈N2〉-〈N〉2)/〈N〉2, where VolN is the volume of interest and N is the number of water molecules in the volume of interest. The water density fluctuations are normalized by the water density fluctuations of bulk water far away from the protein.
Sections 2.7 and 2.8 use a cellular model of mass-action reactions. Signaling through EGFR is modeled by combining three published models and augmented by our own set of reactions [148]: phosphorylation and docking reactions are modeled according to [172]; the MAPK pathway reactions are modeled after [173]; Akt and PI3K activation are incorporated into the model as described in [174]. We resolve phosphorylation of the active ErbB1 dimer at tyrosine site Y1068, which, when phosphorylated, is a binding site for growth-factor-receptor bound-2 (Grb2) or Grb2-associated binding protein (GAB-1) proteins, or at tyrosine site Y1173, which, when phosphorylated, is a binding site for the Src-homology-2-containing (Shc) adaptor protein.
The ErbB3 systems model, which was based on the model by Schoeberl et al. [167], included reactions describing ligand-induced ErbB receptor homo- and heterodimerization, receptor internalization and degradation, constitutive dimerization, and activation of the PI3K-AKT signaling pathway [133]. The ErbB kinase inhibitor lapatinib was implemented in the model according to Schoeberl et al. [167]; lapatinib was assumed to inhibit activation but not dimerization or ligand binding of the EGFR and HER2 kinases.


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Lung | Colon | Skin | Breast | Prostate | Leukemia | |
|---|---|---|---|---|---|---|
| EGFR | PL: [31,32] αC: [31-33,49] αCβ4: [50,51] HC: [31-33,49] AD1: [49] | – | αCβ4: [52] | αC: [53] | αC: [54] | – |
| ErbB2 | αC: [55] αCβ4: [56-58] | αCβ4: [55] | – | αCβ4: [55] αC: [59] | – | – |
| ErbB4 | – | AL: [60] | AL: [61] | AL: [60] | – | – |
| PDGFRα | JM: [62] CT: [63] | – | – | – | – | AL: [64] |
| CSF1R/Fms | – | – | – | – | – | CT: [65,66] |
| Kit/SCFR | JM: [67] | – | JM: [68-71] αC: [69,71] AL: [69-71] | – | – | JM: [72,73] AL: [72-75] |
| Flt3/Flk2 | – | – | – | – | – | JM: [76-78] AL: [79,80] |
| VEGFR2/KDR | CT: [62] | – | TM: [81] AL: [81] | – | – | |
| FGFR1 | AL: [63] | – | – | – | – | – |
| FGFR2 | – | – | JM: [82]* αC: [82]* AL: [82]* | – | – | – |
| FGFR3 | – | – | AL: [83-85] | – | – | AL: [86-88] |
| FGFR4 | AL: [62] | – | – | – | – | – |
| Met | JM: [62,89] | αC: [90] | – | – | – | – |
| EphA1-8,10 | AL: [62,63] CT: [62] | – | – | – | – | – |
| LTK | AL: [62] CT: [62] | – | – | – | – | – |
| Ovary | Kidney | Thyroid | Gastro Intestinal | Neuro Blastoma | |
|---|---|---|---|---|---|
| EGFR | – | αC: [95,96] | αC: [97] AL: [97] AD1: [98] | – | – |
| ErbB2 | αCβ4: [99-101] | – | – | – | – |
| ErbB4 | – | – | – | αC: [60] | – |
| PDGFRα | – | – | – | AL: [102-104] JM: [102-104] | – |
| Kit/SCFR | AL: [105,106] | – | – | JM: [107-110] | – |
| Met | – | – | JM [111,112] | – | – |
| Ret | – | – | AD2: [113-116] JM: [116,117] AL: [118] | – | – |
| ALK | – | – | – | – | AL: [119-122] αC: [120-122] |
| Name | Target | Company | Class |
|---|---|---|---|
| Bevacizumab (Avastin) | VEGF | Genentech Imclone/Bristol-Meyers | Monoclonal antibody |
| Cetuximab (Erbitux) | EGFR | Squib | Monoclonal antibody |
| Panitumumab (Vectibix) | EGFR | Amgen | Monoclonal antibody |
| Ranibizumab (Lucentis) | VEGF | Genentech | Monoclonal antibody |
| Trastuzumab (Herceptin) | Erb2 | Genentech | Monoclonal antibody |
| Pegaptanib (Macugen) | VEGF | OSI/Pfizer | RNA Aptamer |
| Dasatinib (Sprycel) | Src/Bcr-Abl | Bristol-Meyers Squib | Small molecule |
| Erlotinib (Tarceva) | EGFR | Genentech/OSI | Small molecule |
| Gefitinib (Iressa) | EGFR | AstraZeneca | Small molecule |
| Imatinib (Gleevec) | Bcr-Abl | Novartis | Small molecule |
| Lapatinib (Tykerb) | EGFR/Erb2 | GSK | Small molecule |
| Nilotinib (Tasigna) | Bcr-Abl VEGFR1/2/3 | Novartis | Small molecule |
| Pazopanib (Votrient) | PDGFR/c-kit | GlaxoSmithKline | Small molecule |
| Sorafenib (Nexavar) | RAF/VEGFR2/PDGFRB VEGFR2/PDGFRB | Onyx/Bayer | Small molecule |
| Sunitinib (Sutent) | c-kit/FLT3 | Pfizer | Small molecule |
| EGFR active | ErbB2 active | ErbB4 active | EGFR inactive | ErbB2 inactive | ErbB3 Inactive | ErbB4 inactive | ||
|---|---|---|---|---|---|---|---|---|
| aC-helix A-loop interactions | ||||||||
| – – | – – | – – | – – | – – | – – | E739,R841 | ||
| E734,K851 | E766,K883 | E739,K856 | – – | – – | – – | – – | ||
| D737,K836 | D769,R868 | – – | – – | – – | – – | D742,R841 | ||
| E738,F832 | – – | E743,F837 | – – | – – | – – | – – | ||
| – – | – – | – – | E738,K836 | – – | – – | E743,R841 | ||
| A-loop C-loop interactions | ||||||||
| – – | – – | – – | – – | G865,V842 | – – | – – | ||
| – – | – – | – – | – – | – – | – – | G838,R817 | ||
| L834,R812 | L866,R844 | L839,R817 | – – | – – | – – | – – | ||
| – – | – – | – – | L834,D813 | – – | – – | – – | ||
| K836,V810 | R868,V842 | R841,V815 | – – | – – | – – | – – | ||
| – – | – – | – – | – – | – – | D838,R814 | – – | ||
| E848,R812 | – – | – – | – – | – – | – – | – – | ||
| – – | – – | – – | K851,R812 | – – | – – | – – | ||
| C-loop C-loop interactions | ||||||||
| – – | H843,D845 | – – | – – | – – | H813,N815 | – – | ||
| – – | – – | – – | R812,D813 | R844,D845 | – – | – – | ||
| D813,R817 | D845,R849 | D818,R822 | – – | – – | – – | – – | ||
| D813,N818 | – – | – – | – – | – – | N815,N820 | – – | ||
| A815,N818 | A847,N850 | A820,N823 | A815,N818 | A847,N850 | A817,N820 | – – | ||
| – – | A848,V851 | – – | – – | – – | – – | – – | ||
| aC-helix interactions | ||||||||
| – – | A763,S760 | – – | – – | – – | – – | – – | ||
| – – | E766,R756 | – – | – – | – – | – – | – – | ||
| E738,K721 | E770,K753 | E743,K726 | – – | – – | – – | – – | ||
| – – | – – | – – | M742,L753 | M774,L785 | – – | M747,L758 | ||
| A743,L679 | – – | A748,Q684 | – – | – – | – – | – – | ||
| – – | – – | – – | – – | – – | – – | A748,R757 | ||
| A-loop interactions | ||||||||
| – – | – – | D836,K726 | – – | D863,K753 | D833,K723 | D836,K726 | ||
| – – | – – | D836,T835 | – – | – – | – – | – – | ||
| L838,R808 | L870,R840 | L843,R813 | – – | – – | – – | – – | ||
| – – | D871,R840 | – – | – – | – – | – – | – – | ||
| – – | – – | – – | – – | – – | D844,K853 | – – | ||
| – – | – – | K848,T873 | – – | – – | – – | – – | ||
| K843,D932 | – – | K848,D937 | – – | – – | – – | – – | ||
| – – | E876,R898 | – – | – – | – – | – – | – – | ||
| – – | – – | E849,K871 | – – | – – | – – | – – | ||
| Y845,Y867 | – – | Y850,F872 | – – | – – | – – | – – | ||
| – – | – – | A852,R870 | – – | – – | – – | – – | ||
| – – | D880,R897 | D853,R870 | E848,R865 | D880,R897 | – – | D853,R870 | ||
| – – | – – | – – | – – | – – | – – | G855,E730 | ||
| – – | – – | – – | – – | K883,E757 | – – | – – | ||
| – – | – – | – – | – – | – – | – – | K856,E844 | ||
Acknowledgments
We thank the Lemmon lab for insightful discussions and perspectives into RTK function. We acknowledge financial support from NSF grants CBET-0853389, CBET-0853539, and NIH grants NIBIB-1R01EB006818, NHLBI-1R01HL087036. Computational resources were provided in part by the National Partnership for Advanced Computational Infrastructure (NPACI) under the allocation grant MRAC MCB060006. S.E.T. received support through an NSF Graduate Research Fellowship, a GAANN fellowship from the Department of Education and the National Institutes of Health under Ruth L. Kirschstein National Research Service Award 2T32HL007954 from the NIH-NHLBI.
References
- Niho, S.; Ichinose, Y.; Tamura, T.; Yamamoto, N.; Tsuboi, M.; Nakagawa, K.; Shinkai, T.; Jiang, H.; Nishiwaki, Y.; Fukuoka, M. Results of a randomized phase III study to compare the overall survival of gefitinib (IRESSA) versus docetaxel in Japanese patients with non-small cell lung cancer who failed one or two chemotherapy regimens. J. Clin. Oncol. (Meeting Abstracts) 2007, 25, LBA7509. [Google Scholar]
- Douillard, J.-Y.; Kim, E.; Hirsh, V.; Mok, T.; Socinski, M.; Gervais, R.; Wu, Y.-L.; Li, L.; Sellers, M.; Lowe, E. Gefitinib (IRESSA) versus docetaxel in patients with locally advanced or metastatic non-small-cell lung cancer pre-treated with platinum-based chemotherapy: a randomized, open-label Phase III study (INTEREST): PRS-02. J. Thorac. Oncol. 2007, 2, S305–S306. [Google Scholar]
- Hubbard, S.R.; Till, J.H. Protein Tyrosine Kinase Structure and Function. Annu. Rev. Biochem. 2000, 69, 373–398. [Google Scholar]
- Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2000, 103, 211–225. [Google Scholar]
- Citri, A.; Yarden, Y. EGF-ERBB signalling: towards the systems level. Nat. Rev. Mol. Cell Biol. 2006, 7, 505–516. [Google Scholar]
- Boudeau, J.; Miranda-Saavedra, D.; Barton, G.J.; Alessi, D.R. Emerging roles of pseudokinases. Trends Cell Biol. 2006, 16, 443–452. [Google Scholar]
- Alimandi, M.; Romano, A.; Curia, M.C.; Muraro, R.; Fedi, P.; Aaronson, S.A.; Di Fiore, P.P.; Kraus, M.H. Cooperative signaling of ErbB3 and ErbB2 in neoplastic transformation and human mammary carcinomas. Oncogene 1995, 10, 1813–1821. [Google Scholar]
- Holbro, T.; Beerli, R.R.; Maurer, F.; Koziczak, M.; Barbas, C.F., 3rd; Hynes, N.E. The ErbB2/ErbB3 heterodimer functions as an oncogenic unit: ErbB2 requires ErbB3 to drive breast tumor cell proliferation. Proc. Natl. Acad. Sci. USA 2003, 100, 8933–8938. [Google Scholar]
- Sorkin, A.; von Zastrow, M. Endocytosis and signalling: intertwining molecular networks. Nat. Rev. Mol. Cell Biol. 2009, 10, 609–622. [Google Scholar]
- Doherty, G.J.; McMahon, H.T. Mechanisms of Endocytosis. Annu. Rev. Biochem. 2009, 78, 857–902. [Google Scholar]
- Ramanan, V.; Agrawal, N.J.; Liu, J.; Engles, S.; Toy, R.; Radhakrishnan, R. Systems Biology and Physical Biology of Clathrin-Mediated Endocytosis: An Integrative Experimental and Theoretical Perspective. Integr. Biol. 2011. Submitted. [Google Scholar]
- Vecchi, M.; Baulida, J.; Carpenter, G. Selective Cleavage of the Heregulin Receptor ErbB-4 by Protein Kinase C Activation. J. Biol. Chem. 1996, 271, 18989–18995. [Google Scholar]
- Gassmann, M. Aberrant neural and cardiac development in mice lacking the ErbB4 neuregulin receptor. Nature 1995, 378, 390–394. [Google Scholar]
- Hahn, C.G.; Wang, H.Y.; Cho, D.S.; Talbot, K.; Gur, R.E.; Berrettini, W.H.; Bakshi, K.; Kamins, J.; Borgmann-Winter, K.E.; Siegel, S.J.; Gallop, R.J.; Arnold, S.E. Altered neuregulin 1-erbB4 signaling contributes to NMDA receptor hypofunction in schizophrenia. Nat. Med. 2006, 12, 824–828. [Google Scholar]
- Tonks, N.K. Protein tyrosine phosphatases: from genes, to function, to disease. Nat. Rev. Mol. Cell Biol. 2006, 7, 833–846. [Google Scholar]
- Reynolds, A.R.; Tischer, C.; Verveer, P.J.; Rocks, O.; Bastiaens, P.I.H. EGFR activation coupled to inhibition of tyrosine phosphatases causes lateral signal propagation. Nat. Cell Biol. 2003, 5, 447–453. [Google Scholar]
- Tarcic, G.; Boguslavsky, S.K.; Wakim, J.; Kiuchi, T.; Liu, A.; Reinitz, F.; Nathanson, D.; Takahashi, T.; Mischel, P.S.; Ng, T.; Yarden, Y. An Unbiased Screen Identifies DEP-1 Tumor Suppressor as a Phosphatase Controlling EGFR Endocytosis. Curr. Biol. 2009, 19, 1788–1798. [Google Scholar]
- Bill, A.; Schmitz, A.; Albertoni, B.; Song, J.-N.; Heukamp, L.C.; Walrafen, D.; Thorwirth, F.; Verveer, P.J.; Zimmer, S.; Meffert, L.; Schreiber, A.; Chatterjee, S.; Thomas, R.K.; Ullrich, R.T.; Lang, T.; Famulok, M. Cytohesins Are Cytoplasmic ErbB Receptor Activators. Cell 2010, 143, 201–211. [Google Scholar]
- Inoue, A.; Setoguchi, K.; Matsubara, Y.; Okada, K.; Sato, N.; Iwakura, Y.; Higuchi, O.; Yamanashi, Y. Dok-7 Activates the Muscle Receptor Kinase MuSK and Shapes Synapse Formation. Sci. Signal. 2009, 2, ra7. [Google Scholar]
- Lemmon, M.A.; Schlessinger, J. Cell Signaling by Receptor Tyrosine Kinases. Cell 2010, 141, 1117–1134. [Google Scholar]
- Garrett, T.P.J.; McKern, N.M.; Lou, M.; Elleman, T.C.; Adams, T.E.; Lovrecz, G.O.; Zhu, H.-J.; Walker, F.; Frenkel, M.J.; Hoyne, P.A.; Jorissen, R.N.; Nice, E.C.; Burgess, A.W.; Ward, C.W. Crystal Structure of a Truncated Epidermal Growth Factor Receptor Extracellular Domain Bound to Transforming Growth Factor [alpha]. Cell 2002, 110, 763–773. [Google Scholar]
- Ogiso, H.; Ishitani, R.; Nureki, O.; Fukai, S.; Yamanaka, M.; Kim, J.-H.; Saito, K.; Sakamoto, A.; Inoue, M.; Shirouzu, M.; Yokoyama, S. Crystal Structure of the Complex of Human Epidermal Growth Factor and Receptor Extracellular Domains. Cell 2002, 110, 775–787. [Google Scholar]
- Bouyain, S.; Longo, P.A.; Li, S.; Ferguson, K.M.; Leahy, D.J. The extracellular region of ErbB4 adopts a tethered conformation in the absence of ligand. Proc. Natl. Acad. Sci. USA 2005, 102, 15024–15029. [Google Scholar]
- Burgess, A.W.; Cho, H.S.; Eigenbrot, C.; Ferguson, K.M.; Garrett, T.P.J.; Leahy, D.J.; Lemmon, M.A.; Sliwkowski, M.X.; Ward, C.W.; Yokoyama, S. An open-and-shut case? Recent insights into the activation of EGF/ErbB receptors. Mol. Cell 2003, 12, 541–552. [Google Scholar]
- Cho, H.S.; Leahy, D.J. Structure of the extracellular region of HER3 reveals an interdomain tether. Science 2002, 297, 1330–1333. [Google Scholar]
- Ferguson, K.M.; Berger, M.B.; Mendrola, J.M.; Cho, H.-S.; Leahy, D.J.; Lemmon, M.A. EGF Activates Its Receptor by Removing Interactions that Autoinhibit Ectodomain Dimerization. Mol. Cell 2003, 11, 507–517. [Google Scholar]
- Kornev, A.P.; Haste, N.M.; Taylor, S.S.; Ten Eyck, L.F. Surface comparison of active and inactive protein kinases identifies a conserved activation mechanism. Proc. Natl. Acad. Sci. USA 2006, 103, 17783–17788. [Google Scholar]
- Kornev, A.P.; Taylor, S.S.; Ten Eyck, L.F. A helix scaffold for the assembly of active protein kinases. Proc. Natl. Acad. Sci. USA 2008, 105, 14377–14382. [Google Scholar]
- Stamos, J.; Sliwkowski, M.X.; Eigenbrot, C. Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor. J. Biol. Chem. 2002, 277, 46265–46272. [Google Scholar]
- Choi, S.H.; Mendrola, J.M.; Lemmon, M.A. EGF-independent activation of cell-surface EGF receptors harboring mutations found in gefitinib-sensitive lung cancer. Oncogene 2007, 26, 1567–1576. [Google Scholar]
- Lynch, T.J.; Bell, D.W.; Sordella, R.; Gurubhagavatula, S.; Okimoto, R.A.; Brannigan, B.W. Activating mutations in the epidermal growth factor receptor underlying responsiveness of non-small-cell lung cancer to gefitinib. N. Engl. J. Med. 2004, 350, 2129–2139. [Google Scholar]
- Paez, J.G.; Janne, P.A.; Lee, J.C.; Tracy, S.; Greulich, H.; Gabriel, S. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304, 1497–1500. [Google Scholar]
- Pao, W.; Miller, V.; Zakowski, M.; Doherty, J.; Politi, K.; Sarkaria, I. EGF receptor gene mutations are common in lung cancers from ‘never smokers’ and are associated with sensitivity of tumors to gefitinib and erlotinib. Proc. Natl Acad. Sci. USA 2004, 101, 13306–13311. [Google Scholar]
- Hubbard, S.R.; Wei, L.; Ellis, L.; Hendrickson, W.A. Crystal structure of the tyrosine kinase domain of the human insulin receptor. Nature 1994, 372, 746–754. [Google Scholar]
- Mohammadi, M.; Schlessinger, J.; Hubbard, S.R. Structure of the FGF receptor tyrosine kinase domain reveals a novel autoinhibitory mechanism. Cell 1996, 86, 577–587. [Google Scholar]
- Till, J.H.; Becerra, M.; Watty, A.; Lu, Y.; Ma, Y.; Neubert, T.A.; Burden, S.J.; Hubbard, S.R. Crystal Structure of the MuSK Tyrosine Kinase: Insights into Receptor Autoregulation. Structure 2002, 10, 1187–1196. [Google Scholar]
- Griffith, J.; Black, J.; Faerman, C.; Swenson, L.; Wynn, M.; Lu, F.; Lippke, J.; Saxena, K. The structural basis for autoinhibition of FLT3 by the juxtamembrane domain. Mol. Cell 2004, 13, 169–178. [Google Scholar]
- Mol, C.D.; Dougan, D.R.; Schneider, T.R.; Skene, R.J.; Kraus, M.L.; Scheibe, D.N.; Snell, G.P.; Zou, H.; Sang, B.C.; Wilson, K.P. Structural basis for the autoinhibition and STI-571 inhibition of c-Kit tyrosine kinase. J. Biol. Chem. 2004, 279, 31655–31663. [Google Scholar]
- Wybenga-Groot, L.E.; Baskin, B.; Ong, S.H.; Tong, J.F.; Pawson, T.; Sicheri, F. Structural basis for autoinhibition of the EphB2 receptor tyrosine kinase by the unphosphorylated juxtamembrane region. Cell 2001, 106, 745–757. [Google Scholar]
- Shewchuk, L.M.; Hassell, A.M.; Ellis, B.; Holmes, W.D.; Davis, R.; Horne, E.L.; Kadwell, S.H.; McKee, D.D.; Moore, J.T. Structure of the Tie2 RTK Domain: Self-Inhibition by the Nucleotide Binding Loop, Activation Loop, and C-Terminal Tail. Structure 2000, 8, 1105–1113. [Google Scholar]
- Knowles, P.P.; Murray-Rust, J.; Kjær, S.; Scott, R.P.; Hanrahan, S.; Santoro, M.; Ibáñez, C.F.; McDonald, N.Q. Structure and Chemical Inhibition of the RET Tyrosine Kinase Domain. J. Biol. Chem. 2006, 281, 33577–33587. [Google Scholar]
- Zhang, X.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An Allosteric Mechanism for Activation of the Kinase Domain of Epidermal Growth Factor Receptor. Cell 2006, 125, 1137–1149. [Google Scholar]
- Zhang, X.W.; Gureasko, J.; Shen, K.; Cole, P.A.; Kuriyan, J. An allosteric mechanism for activation of the kinase domain of epidermal growth factor receptor. Cell 2006, 125, 1137–1149. [Google Scholar]
- Qiu, C.; Tarrant, M.K.; Choi, S.H.; Sathyamurthy, A.; Bose, R.; Banjade, S.; Pal, A.; Bornmann, W.G.; Lemmon, M.A.; Cole, P.A.; Leahy, D.J. Mechanism of Activation and Inhibition of the HER4/ErbB4 Kinase. Structure 2008, 16, 460–467. [Google Scholar]
- Jura, N.; Endres, N.F.; Engel, K.; Deindl, S.; Das, R.; Lamers, M.H.; Wemmer, D.E.; Zhang, X.; Kuriyan, J. Mechanism for Activation of the EGF Receptor Catalytic Domain by the Juxtamembrane Segment. Cell 2009, 137, 1293–1307. [Google Scholar]
- Red Brewer, M.; Choi, S.H.; Alvarado, D.; Moravcevic, K.; Pozzi, A.; Lemmon, M.A.; Carpenter, G. The Juxtamembrane Region of the EGF Receptor Functions as an Activation Domain. Mol. Cell 2009, 34, 641–651. [Google Scholar]
- Thiel, K.W.; Carpenter, G. Epidermal growth factor receptor juxtamembrane region regulates allosteric tyrosine kinase activation. Proc. Natl. Acad. Sci. USA 2007, 104, 19238–19243. [Google Scholar]
- Downward, J.; Yarden, Y.; Mayes, E.; Scrace, G.; Totty, N.; Stockwell, P.; Ullrich, A.; Schlessinger, J.; Waterfield, M.D. Close similarity of epidermal growth factor receptor and v-erb-B oncogene protein sequences. Nature 1984, 307, 521–527. [Google Scholar]
- Sordella, R.; Bell, D.W.; Haber, D.A.; Settleman, J. Gefitinib-Sensitizing EGFR Mutations in Lung Cancer Activate Anti-Apoptotic Pathways. Science 2004, 305, 1163–1167. [Google Scholar]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Kuwano, H.; Takahashi, T.; Mitsudomi, T. Mutations of the Epidermal Growth Factor Receptor Gene in Lung Cancer: Biological and Clinical Implications. Cancer Res. 2004, 64, 8919–8923. [Google Scholar]
- Huang, S.-F.; Liu, H.-P.; Li, L.-H.; Ku, Y.-C.; Fu, Y.-N.; Tsai, H.-Y.; Chen, Y.-T.; Lin, Y.-F.; Chang, W.-C.; Kuo, H.-P.; Wu, Y.-C.; Chen, Y.-R.; Tsai, S.-F. High Frequency of Epidermal Growth Factor Receptor Mutations with Complex Patterns in Non-Small Cell Lung Cancers Related to Gefitinib Responsiveness in Taiwan. Clin. Cancer Res. 2004, 10, 8195–8203. [Google Scholar]
- Willmore-Payne, C.; Holden, J.A.; Layfield, L.J. Detection of EGFR- and HER2-activating mutations in squamous cell carcinoma involving the head and neck. Mod. Pathol. 2006, 19, 634–640. [Google Scholar]
- Da Silva, L.; Simpson, P.; Smart, C.; Cocciardi, S.; Waddell, N.; Lane, A.; Morrison, B.; Vargas, A.; Healey, S.; Beesley, J.; Pakkiri, P.; Parry, S.; Kurniawan, N.; Reid, L.; Keith, P.; Faria, P.; Pereira, E.; Skalova, A.; Bilous, M.; Balleine, R.; Do, H.; Dobrovic, A.; Fox, S.; Franco, M.; Reynolds, B.; Khanna, K.; Cummings, M.; Chenevix-Trench, G.; Lakhani, S. HER3 and downstream pathways are involved in colonization of brain metastases from breast cancer. Breast Cancer Res. 2010, 12, R46. [Google Scholar]
- Cho, K.S.; Lee, J.S.; Cho, N.H.; Park, K.; Ham, W.S.; Choi, Y.D. Gene amplification and mutation analysis of epidermal growth factor receptor in hormone refractory prostate cancer. Prostate 2008, 68, 803–808. [Google Scholar]
- Lee, J.W.; Soung, Y.H.; Seo, S.H.; Kim, S.Y.; Park, C.H.; Wang, Y.P.; Park, K.; Nam, S.W.; Park, W.S.; Kim, S.H.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Somatic mutations of ERBB2 kinase domain in gastric, colorectal, and breast carcinomas. Clin. Cancer Res. 2006, 12, 57–61. [Google Scholar]
- Stephens, P.; Hunter, C.; Bignell, G.; Edkins, S.; Davies, H.; Teague, J.; Stevens, C.; O'Meara, S.; Smith, R.; Parker, A.; Barthorpe, A.; Blow, M.; Brackenbury, L.; Butler, A.; Clarke, O.; Cole, J.; Dicks, E.; Dike, A.; Drozd, A.; Edwards, K.; Forbes, S.; Foster, R.; Gray, K.; Greenman, C.; Halliday, K.; Hills, K.; Kosmidou, V.; Lugg, R.; Menzies, A.; Perry, J.; Petty, R.; Raine, K.; Ratford, L.; Shepherd, R.; Small, A.; Stephens, Y.; Tofts, C.; Varian, J.; West, S.; Widaa, S.; Yates, A.; Brasseur, F.; Cooper, C.S.; Flanagan, A.M.; Knowles, M.; Leung, S.Y.; Louis, D.N.; Looijenga, L.H.; Malkowicz, B.; Pierotti, M.A.; Teh, B.; Chenevix-Trench, G.; Weber, B.L.; Yuen, S.T.; Harris, G.; Goldstraw, P.; Nicholson, A.G.; Futreal, P.A.; Wooster, R.; Stratton, M.R. Lung cancer: Intragenic ERBB2 kinase mutations in tumours. Nature 2004, 431, 525–526. [Google Scholar]
- Shigematsu, H.; Takahashi, T.; Nomura, M.; Majmudar, K.; Suzuki, M.; Lee, H.; Wistuba, I.I.; Fong, K.M.; Toyooka, S.; Shimizu, N.; Fujisawa, T.; Minna, J.D.; Gazdar, A.F. Somatic Mutations of the HER2 Kinase Domain in Lung Adenocarcinomas. Cancer Res. 2005, 65, 1642–1646. [Google Scholar]
- Buttitta, F.; Barassi, F.; Fresu, G.; Felicioni, L.; Chella, A.; Paolizzi, D.; Lattanzio, G.; Salvatore, S.; Camplese, P.P.; Rosini, S.; Iarussi, T.; Mucilli, F.; Sacco, R.; Mezzetti, A.; Marchetti, A. Mutational analysis of the HER2 gene in lung tumors from Caucasian patients: Mutations are mainly present in adenocarcinomas with bronchioloalveolar features. Int. J. Cancer 2006, 119, 2586–2591. [Google Scholar]
- Shah, S.P.; Morin, R.D.; Khattra, J.; Prentice, L.; Pugh, T.; Burleigh, A.; Delaney, A.; Gelmon, K.; Guliany, R.; Senz, J.; Steidl, C.; Holt, R.A.; Jones, S.; Sun, M.; Leung, G.; Moore, R.; Severson, T.; Taylor, G.A.; Teschendorff, A.E.; Tse, K.; Turashvili, G.; Varhol, R.; Warren, R.L.; Watson, P.; Zhao, Y.; Caldas, C.; Huntsman, D.; Hirst, M.; Marra, M.A.; Aparicio, S. Mutational evolution in a lobular breast tumour profiled at single nucleotide resolution. Nature 2009, 461, 809–813. [Google Scholar]
- Soung, Y.H.; Lee, J.W.; Kim, S.Y.; Wang, Y.P.; Jo, K.H.; Moon, S.W.; Park, W.S.; Nam, S.W.; Lee, J.Y.; Yoo, N.J.; Lee, S.H. Somatic mutations of the ERBB4 kinase domain in human cancers. Int. J. Cancer 2006, 118, 1426–1429. [Google Scholar]
- Prickett, T.D.; Agrawal, N.S.; Wei, X.; Yates, K.E.; Lin, J.C.; Wunderlich, J.R.; Cronin, J.C.; Cruz, P.; Rosenberg, S.A.; Samuels, Y. Analysis of the tyrosine kinome in melanoma reveals recurrent mutations in ERBB4. Nat. Genet. 2009, 41, 1127–1132. [Google Scholar]
- Ding, L.; Getz, G.; Wheeler, D.A.; Mardis, E.R.; McLellan, M.D.; Cibulskis, K.; Sougnez, C.; Greulich, H.; Muzny, D.M.; Morgan, M.B.; Fulton, L.; Fulton, R.S.; Zhang, Q.; Wendl, M.C.; Lawrence, M.S.; Larson, D.E.; Chen, K.; Dooling, D.J.; Sabo, A.; Hawes, A.C.; Shen, H.; Jhangiani, S.N.; Lewis, L.R.; Hall, O.; Zhu, Y.; Mathew, T.; Ren, Y.; Yao, J.; Scherer, S.E.; Clerc, K.; Metcalf, G.A.; Ng, B.; Milosavljevic, A.; Gonzalez-Garay, M.L.; Osborne, J.R.; Meyer, R.; Shi, X.; Tang, Y.; Koboldt, D.C.; Lin, L.; Abbott, R.; Miner, T.L.; Pohl, C.; Fewell, G.; Haipek, C.; Schmidt, H.; Dunford-Shore, B.H.; Kraja, A.; Crosby, S.D.; Sawyer, C.S.; Vickery, T.; Sander, S.; Robinson, J.; Winckler, W.; Baldwin, J.; Chirieac, L.R.; Dutt, A.; Fennell, T.; Hanna, M.; Johnson, B.E.; Onofrio, R.C.; Thomas, R.K.; Tonon, G.; Weir, B.A.; Zhao, X.; Ziaugra, L.; Zody, M.C.; Giordano, T.; Orringer, M.B.; Roth, J.A.; Spitz, M.R.; Wistuba, I.I.; Ozenberger, B.; Good, P.J.; Chang, A.C.; Beer, D.G.; Watson, M.A.; Ladanyi, M.; Broderick, S.; Yoshizawa, A.; Travis, W.D.; Pao, W.; Province, M.A.; Weinstock, G.M.; Varmus, H.E.; Gabriel, S.B.; Lander, E.S.; Gibbs, R.A.; Meyerson, M.; Wilson, R.K. Somatic mutations affect key pathways in lung adenocarcinoma. Nature 2008, 455, 1069–1075. [Google Scholar]
- Davies, H.; Hunter, C.; Smith, R.; Stephens, P.; Greenman, C.; Bignell, G.; Teague, J.; Butler, A.; Edkins, S.; Stevens, C.; Parker, A.; O'Meara, S.; Avis, T.; Barthorpe, S.; Brackenbury, L.; Buck, G.; Clements, J.; Cole, J.; Dicks, E.; Edwards, K.; Forbes, S.; Gorton, M.; Gray, K.; Halliday, K.; Harrison, R.; Hills, K.; Hinton, J.; Jones, D.; Kosmidou, V.; Laman, R.; Lugg, R.; Menzies, A.; Perry, J.; Petty, R.; Raine, K.; Shepherd, R.; Small, A.; Solomon, H.; Stephens, Y.; Tofts, C.; Varian, J.; Webb, A.; West, S.; Widaa, S.; Yates, A.; Brasseur, F.; Cooper, C.S.; Flanagan, A.M.; Green, A.; Knowles, M.; Leung, S.Y.; Looijenga, L.H.J.; Malkowicz, B.; Pierotti, M.A.; Teh, B.T.; Yuen, S.T.; Lakhani, S.R.; Easton, D.F.; Weber, B.L.; Goldstraw, P.; Nicholson, A.G.; Wooster, R.; Stratton, M.R.; Futreal, P.A. Somatic Mutations of the Protein Kinase Gene Family in Human Lung Cancer. Cancer Res. 2005, 65, 7591–7595. [Google Scholar]
- Lierman, E.; Michaux, L.; Beullens, E.; Pierre, P.; Marynen, P.; Cools, J.; Vandenberghe, P. FIP1L1-PDGFR[alpha] D842V, a novel panresistant mutant, emerging after treatment of FIP1L1-PDGFR[alpha] T674I eosinophilic leukemia with single agent sorafenib. Leukemia 2009, 23, 845–851. [Google Scholar]
- Ridge, S.A.; Worwood, M.; Oscier, D.; Jacobs, A.; Padua, R.A. FMS mutations in myelodysplastic, leukemic, and normal subjects. Proc. Natl. Acad. Sci. USA 1990, 87, 1377–1380. [Google Scholar]
- Tobal, K.; Pagliuca, A.; Bhatt, B.; Bailey, N.; Layton, D.; Mufti, G. Mutation of the human FMS gene (M-CSF receptor) in myelodysplastic syndromes and acute myeloid leukemia. Leukemia 1990, 4, 486–489. [Google Scholar]
- Boldrini, L.; Ursino, S.; Gisfredi, S.; Faviana, P.; Donati, V.; Camacci, T.; Lucchi, M.; Mussi, A.; Basolo, F.; Pingitore, R.; Fontanini, G. Expression and Mutational Status of c-kit in Small-Cell Lung Cancer. Clin. Cancer Res. 2004, 10, 4101–4108. [Google Scholar]
- Willmore-Payne, C.; Holden, J.A.; Tripp, S.; Layfield, L.J. Human malignant melanoma: detection of BRAF- and c-kit-activating mutations by high-resolution amplicon melting analysis. Hum. Pathol. 2005, 36, 486–493. [Google Scholar]
- Curtin, J.A.; Busam, K.; Pinkel, D.; Bastian, B.C. Somatic Activation of KIT in Distinct Subtypes of Melanoma. J. Clin. Oncol. 2006, 24, 4340–4346. [Google Scholar]
- Beadling, C.; Jacobson-Dunlop, E.; Hodi, F.S.; Le, C.; Warrick, A.; Patterson, J.; Town, A.; Harlow, A.; Cruz, F.; Azar, S.; Rubin, B.P.; Muller, S.; West, R.; Heinrich, M.C.; Corless, C.L. KIT Gene Mutations and Copy Number in Melanoma Subtypes. Clin. Cancer Res. 2008, 14, 6821–6828. [Google Scholar]
- Torres-Cabala, C.A.; Wang, W.-L.; Trent, J.; Yang, D.; Chen, S.; Galbincea, J.; Kim, K.B.; Woodman, S.; Davies, M.; Plaza, J.A.; Nash, J.W.; Prieto, V.G.; Lazar, A.J.; Ivan, D. Correlation between KIT expression and KIT mutation in melanoma: A study of 173 cases with emphasis on the acral-lentiginous/mucosal type. Mod. Pathol. 2009, 22, 1446–1456. [Google Scholar]
- Furitsu, T.; Tsujimura, T.; Tono, T.; Ikeda, H.; Kitayama, H.; Koshimizu, U.; Sugahara, H.; Butterfield, J.H.; Ashman, L.K.; Kanayama, Y. Identification of mutations in the coding sequence of the proto-oncogene c-kit in a human mast cell leukemia cell line causing ligand-independent activation of c-kit product. J. Clin. Invest. 1993, 92, 1736–1744. [Google Scholar]
- Kitayama, H.; Kanakura, Y.; Furitsu, T.; Tsujimura, T.; Oritani, K.; Ikeda, H.; Sugahara, H.; Mitsui, H.; Kanayama, Y.; Kitamura, Y. Constitutively activating mutations of c-kit receptor tyrosine kinase confer factor-independent growth and tumorigenicity of factor-dependent hematopoietic cell lines. Blood 1995, 85, 790–798. [Google Scholar]
- Nagata, H.; Worobec, A.S.; Oh, C.K.; Chowdhury, B.A.; Tannenbaum, S.; Suzuki, Y.; Metcalfe, D.D. Identification of a point mutation in the catalytic domain of the protooncogene c-kit in peripheral blood mononuclear cells of patients who have mastocytosis with an associated hematologic disorder. Proc. Natl. Acad. Sci. USA 1995, 92, 10560–10564. [Google Scholar]
- Longley, B.J.; Tyrrell, L.; Lu, S.-Z.; Ma, Y.-S.; Langley, K.; Ding, T.-g.; Duffy, T.; Jacobs, P.; Tang, L.H.; Modlin, I. Somatic c-KIT activating mutation in urticaria pigmentosa and aggressive mastocytosis: establishment of clonality in a human mast cell neoplasm. Nat. Genet. 1996, 12, 312–314. [Google Scholar]
- Nakao, M.; Yokota, S.; Iwai, T.; Kaneko, H.; Horiike, S.; Kashima, K.; Sonoda, Y.; Fujimoto, T.; Misawa, S. Internal tandem duplication of the flt3 gene found in acute myeloid leukemia. Leukemia 1996, 10, 1911–1918. [Google Scholar]
- Yokota, S.; Kiyoi, H.; Nakao, M.; Iwai, T.; Misawa, S.; Okuda, T.; Sonoda, Y.; Abe, T.; Kahsima, K.; Matsuo, Y.; Naoe, T. Internal tandem duplication of the FLT3 gene is preferentially seen in acute myeloid leukemia and myelodysplastic syndrome among various hematological malignancies. A study on a large series of patients and cell lines. Leukemia 1997, 11, 1605–1609. [Google Scholar]
- Kiyoi, H.; Naoe, T.; Yokota, S.; Nakao, M.; Minami, S.; Kuriyama, K.; Takeshita, A.; Saito, K.; Hasegawa, S.; Shimodaira, S.; Tamura, J.; Shimazaki, C.; Matsue, K.; Kobayashi, H.; Arima, N.; Suzuki, R.; Morishita, H.; Saito, H.; Ueda, R.; Ohno, R. Internal tandem duplication of FLT3 associated with leukocytosis in acute promyelocytic leukemia. Leukemia Study Group of the Ministry of Health and Welfare (Kohseisho). Leukemia 1997, 11, 1447–1452. [Google Scholar]
- Yamamoto, Y.; Kiyoi, H.; Nakano, Y.; Suzuki, R.; Kodera, Y.; Miyawaki, S.; Asou, N.; Kuriyama, K.; Yagasaki, F.; Shimazaki, C.; Akiyama, H.; Saito, K.; Nishimura, M.; Motoji, T.; Shinagawa, K.; Takeshita, A.; Saito, H.; Ueda, R.; Ohno, R.; Naoe, T. Activating mutation of D835 within the activation loop of FLT3 in human hematologic malignancies. Blood 2001, 97, 2434–2439. [Google Scholar]
- Abu-Duhier, F.M.; Goodeve, A.C.; Wilson, G.A.; Care, R.S.; Peake, I.R.; Reilly, J.T. Identification of novel FLT-3 Asp835 mutations in adult acute myeloid leukaemia. Br. J. Haematol. 2001, 113, 983–988. [Google Scholar]
- Antonescu, C.R.; Yoshida, A.; Guo, T.; Chang, N.-E.; Zhang, L.; Agaram, N.P.; Qin, L.-X.; Brennan, M.F.; Singer, S.; Maki, R.G. KDR Activating Mutations in Human Angiosarcomas Are Sensitive to Specific Kinase Inhibitors. Cancer Res. 2009, 69, 7175–7179. [Google Scholar]
- Gartside, M.G.; Chen, H.; Ibrahimi, O.A.; Byron, S.A.; Curtis, A.V.; Wellens, C.L.; Bengston, A.; Yudt, L.M.; Eliseenkova, A.V.; Ma, J.; Curtin, J.A.; Hyder, P.; Harper, U.L.; Riedesel, E.; Mann, G.J.; Trent, J.M.; Bastian, B.C.; Meltzer, P.S.; Mohammadi, M.; Pollock, P.M. Loss-of-Function Fibroblast Growth Factor Receptor-2 Mutations in Melanoma. Mol. Cancer Res. 2009, 7, 41–54. [Google Scholar]
- Logie, A.; Dunois-Larde, C.; Rosty, C.; Levrel, O.; Blanche, M.; Ribeiro, A.; Gasc, J.-M.; Jorcano, J.; Werner, S.; Sastre-Garau, X.; Thiery, J.P.; Radvanyi, F. Activating mutations of the tyrosine kinase receptor FGFR3 are associated with benign skin tumors in mice and humans. Hum. Mol. Genet. 2005, 14, 1153–1160. [Google Scholar]
- Hafner, C.; van Oers, J.M.M.; Hartmann, A.; Landthaler, M.; Stoehr, R.; Blaszyk, H.; Hofstaedter, F.; Zwarthoff, E.C.; Vogt, T. High Frequency of FGFR3 Mutations in Adenoid Seborrheic Keratoses. J. Invest. Dermatol. 2006, 126, 2404–2407. [Google Scholar]
- Hafner, C.; Hartmann, A.; Real, F.X.; Hofstaedter, F.; Landthaler, M.; Vogt, T. Spectrum of FGFR3 Mutations in Multiple Intraindividual Seborrheic Keratoses. J. Invest. Dermatol. 2007, 127, 1883–1885. [Google Scholar]
- Chesi, M.; Nardini, E.; Brents, L.A.; Schröck, E.; Ried, T.; Kuehl, W.M.; Bergsagel, P.L. Frequent translocation t(4;14)(p16.3;q32.3) in multiple myeloma is associated with increased expression and activating mutations of fibroblast growth factor receptor 3. Oncogene 1997, 16, 260–264. [Google Scholar]
- Intini, D.; Baldini, L.; Fabris, S.; Lombardi, L.; Ciceri, G.; Maiolo, A.T.; Neri, A. Analysis of FGFR3 gene mutations in multiple myeloma patients with t(4;14). Br. J. Haematol. 2001, 114, 362–364. [Google Scholar]
- Ronchetti, D.; Greco, A.; Compasso, S.; Colombo, G.; Dell'Era, P.; Otsuki, T.; Lombardi, L.; Neri, A. Deregulated FGFR3 mutants in multiple myeloma cell lines with t(4;14): comparative analysis of Y373C, K650E and the novel G384D mutations. Oncogene 2001, 20, 3553–3562. [Google Scholar]
- Kong-Beltran, M.; Seshagiri, S.; Zha, J.; Zhu, W.; Bhawe, K.; Mendoza, N.; Holcomb, T.; Pujara, K.; Stinson, J.; Fu, L.; Severin, C.; Rangell, L.; Schwall, R.; Amler, L.; Wickramasinghe, D.; Yauch, R. Somatic Mutations Lead to an Oncogenic Deletion of Met in Lung Cancer. Cancer Res. 2006, 66, 283–289. [Google Scholar]
- Lorenzato, A.; Olivero, M.; Patanè, S.; Rosso, E.; Oliaro, A.; Comoglio, P.M.; Di Renzo, M.F. Novel Somatic Mutations of the MET Oncogene in Human Carcinoma Metastases Activating Cell Motility and Invasion. Cancer Res. 2002, 62, 7025–7030. [Google Scholar]
- Forbes, S.A.; Tang, G.; Bindal, N.; Bamford, S.; Dawson, E.; Cole, C.; Kok, C.Y.; Jia, M.; Ewing, R.; Menzies, A.; Teague, J.W.; Stratton, M.R.; Futreal, P.A. COSMIC (the Catalogue of Somatic Mutations in Cancer): a resource to investigate acquired mutations in human cancer. Nucleic Acids Res. 2010, 38, D652–D657. [Google Scholar]
- Citri, A.; Gan, J.; Mosesson, Y.; Vereb, G.; Szollosi, J.; Yarden, Y. Hsp90 restrains ErbB-2/HER2 signalling by limiting heterodimer formation. EMBO Rep. 2004, 5, 1165–1170. [Google Scholar]
- Xu, W.; Yuan, X.; Xiang, Z.; Mimnaugh, E.; Marcu, M.; Neckers, L. Surface charge and hydrophobicity determine ErbB2 binding to the Hsp90 chaperone complex. Nat. Struct. Mol. Biol. 2005, 12, 120–126. [Google Scholar]
- Tvorogov, D.; Sundvall, M.; Kurppa, K.; Hollmen, M.; Repo, S.; Johnson, M.S.; Elenius, K. Somatic Mutations of ErbB4. J. Biol. Chem. 2009, 284, 5582–5591. [Google Scholar]
- Franco-Hernandez, C.; Martinez-Glez, V.; Arjona, D.; de Campos, J.M.; Isla, A.; Gutierrez, M.; Vaquero, J.; Rey, J.A. EGFR sequence variations and real-time quantitative polymerase chain reaction analysis of gene dosage in brain metastases of solid tumors. Cancer Genet. Cytogenet. 2007, 173, 63–67. [Google Scholar]
- Iyevleva, A.G.; Novik, A.V.; Moiseyenko, V.M.; Imyanitov, E.N. EGFR mutation in kidney carcinoma confers sensitivity to gefitinib treatment: A case report. Urol. Oncol. 2009, 27, 548–550. [Google Scholar]
- Masago, K.; Asato, R.; Fujita, S.; Hirano, S.; Tamura, Y.; Kanda, T.; Mio, T.; Katakami, N.; Mishima, M.; Ito, J. Epidermal growth factor receptor gene mutations in papillary thyroid carcinoma. Int. J. Cancer 2009, 124, 2744–2749. [Google Scholar]
- Mitsiades, C.S.; Kotoula, V.; Poulaki, V.; Sozopoulos, E.; Negri, J.; Charalambous, E.; Fanourakis, G.; Voutsinas, G.; Tseleni-Balafouta, S.; Mitsiades, N. Epidermal Growth Factor Receptor as a Therapeutic Target in Human Thyroid Carcinoma: Mutational and Functional Analysis. J. Clin. Endocrinol. Metab. 2006, 91, 3662–3666. [Google Scholar]
- Anglesio, M.S.; Arnold, J.M.; George, J.; Tinker, A.V.; Tothill, R.; Waddell, N.; Simms, L.; Locandro, B.; Fereday, S.; Traficante, N.; Russell, P.; Sharma, R.; Birrer, M.J.; Group, A.S.; deFazio, A.; Chenevix-Trench, G.; Bowtell, D.D.L. Mutation of ERBB2 Provides a Novel Alternative Mechanism for the Ubiquitous Activation of RAS-MAPK in Ovarian Serous Low Malignant Potential Tumors. Mol. Cancer Res. 2008, 6, 1678–1690. [Google Scholar]
- Lassus, H.; Sihto, H.; Leminen, A.; Joensuu, H.; Isola, J.; Nupponen, N.; Butzow, R. Gene amplification, mutation, and protein expression of EGFR and mutations of ERBB2 in serous ovarian carcinoma. J. Mol. Med. 2006, 84, 671–681. [Google Scholar]
- Nakayama, K.; Nakayama, N.; Kurman, R.; Cope, L.; Pohl, G.; Samuels, Y.; Velculescu, V.; Wang, T.; Shih, I. Sequence mutations and amplification of PIK3CA and AKT2 genes in purified ovarian serous neoplasms. Cancer Biol. Ther. 2006, 5, 779–785. [Google Scholar]
- Heinrich, M.C.; Corless, C.L.; Duensing, A.; McGreevey, L.; Chen, C.-J.; Joseph, N.; Singer, S.; Griffith, D.J.; Haley, A.; Town, A.; Demetri, G.D.; Fletcher, C.D.M.; Fletcher, J.A. PDGFRA Activating Mutations in Gastrointestinal Stromal Tumors. Science 2003, 299, 708–710. [Google Scholar]
- Heinrich, M.C.; Corless, C.L.; Demetri, G.D.; Blanke, C.D.; von Mehren, M.; Joensuu, H.; McGreevey, L.S.; Chen, C.-J.; Van den Abbeele, A.D.; Druker, B.J.; Kiese, B.; Eisenberg, B.; Roberts, P.J.; Singer, S.; Fletcher, C.D.M.; Silberman, S.; Dimitrijevic, S.; Fletcher, J.A. Kinase Mutations and Imatinib Response in Patients With Metastatic Gastrointestinal Stromal Tumor. J. Clin. Oncol. 2003, 21, 4342–4349. [Google Scholar]
- Hirota, S.; Ohashi, A.; Nishida, T.; Isozaki, K.; Kinoshita, K.; Shinomura, Y.; Kitamura, Y. Gain-of-function mutations of platelet-derived growth factor receptor [alpha] gene in gastrointestinal stromal tumors. Gastroenterology 2003, 125, 660–667. [Google Scholar]
- Hoei-Hansen, C.; Kraggerud, S.; Abeler, V.; Kaern, J.; Rajpert-De Meyts, E.; Lothe, R. Ovarian dysgerminomas are characterised by frequent KIT mutations and abundant expression of pluripotency markers. Mol. Cancer 2007, 6, 12. [Google Scholar]
- Tian, Q.; Frierson, H.F., Jr.; Krystal, G.W.; Moskaluk, C.A. Activating c-kit Gene Mutations in Human Germ Cell Tumors. Am. J. Pathol. 1999, 154, 1643–1647. [Google Scholar]
- Hirota, S.; Isozaki, K.; Moriyama, Y.; Hashimoto, K.; Nishida, T.; Ishiguro, S.; Kawano, K.; Hanada, M.; Kurata, A.; Takeda, M.; Muhammad Tunio, G.; Matsuzawa, Y.; Kanakura, Y.; Shinomura, Y.; Kitamura, Y. Gain-of-Function Mutations of c-kit in Human Gastrointestinal Stromal Tumors. Science 1998, 279, 577–580. [Google Scholar]
- Lasota, J.; Jasinski, M.; Sarlomo-Rikala, M.; Miettinen, M. Mutations in Exon 11 of c-Kit Occur Preferentially in Malignant versus Benign Gastrointestinal Stromal Tumors and Do Not Occur in Leiomyomas or Leiomyosarcomas. Am. J. Pathol. 1999, 154, 53–60. [Google Scholar]
- Moskaluk, C.; Tian, Q.; Marshall, C.; Rumpel, C.; Franquemont, D.; Frierson, H. Mutations of c-kit JM domain are found in a minority of human gastrointestinal stromal tumors. Oncogene 1999, 18, 1897–1902. [Google Scholar]
- Taniguchi, M.; Nishida, T.; Hirota, S.; Isozaki, K.; Ito, T.; Nomura, T.; Matsuda, H.; Kitamura, Y. Effect of c-kit Mutation on Prognosis of Gastrointestinal Stromal Tumors. Cancer Res. 1999, 59, 4297–4300. [Google Scholar]
- Wasenius, V.-M.; Hemmer, S.; Karjalainen-Lindsberg, M.-L.; Nupponen, N.N.; Franssila, K.; Joensuu, H. MET Receptor Tyrosine Kinase Sequence Alterations in Differentiated Thyroid Carcinoma. Am. J. Surg. Pathol. 2005, 29, 544–549. [Google Scholar]
- Ricarte-Filho, J.C.; Ryder, M.; Chitale, D.A.; Rivera, M.; Heguy, A.; Ladanyi, M.; Janakiraman, M.; Solit, D.; Knauf, J.A.; Tuttle, R.M.; Ghossein, R.A.; Fagin, J.A. Mutational Profile of Advanced Primary and Metastatic Radioactive Iodine-Refractory Thyroid Cancers Reveals Distinct Pathogenetic Roles for BRAF, PIK3CA, and AKT1. Cancer Res. 2009, 69, 4885–4893. [Google Scholar]
- Hofstra, R.M.W.; Landsvater, R.M.; Ceccherini, I.; Stulp, R.P.; Stelwagen, T.; Luo, Y.; Pasini, B.; Hoppener, J.W.M.; van Amstel, H.K.P.; Romeo, G.; Lips, C.J.M.; Buys, C.H.C.M. A mutation in the RET proto-oncogene associated with multiple endocrine neoplasia type 2B and sporadic medullary thyroid carcinoma. Nature 1994, 367, 375–376. [Google Scholar]
- Zedenlus, J.; Wallin, G.; Hamberger, B.; Nordenskjōld, M.; Weber, G.n.; Larsson, C. Somatic and MEN 2A de novo mutations identified in the RET proto-oncogene by screening of sporadic MTC: s. Hum. Mol. Genet. 1994, 3, 1259–1262. [Google Scholar]
- Eng, C.; Mulligan, L.M.; Smith, D.P.; Healey, C.S.; Frilling, A.; Raue, F.; Neumann, H.P.H.; Pfragner, R.; Behmel, A.; Lorenzo, M.J.; Stonehouse, T.J.; Ponder, M.A.; Ponder, B.A.J. Mutation of the RET protooncogene in sporadic medullary thyroid carcinoma. Genes. Chromosomes Cancer 1995, 12, 209–212. [Google Scholar]
- Romei, C.; Elisei, R.; Pinchera, A.; Ceccherini, I.; Molinaro, E.; Mancusi, F.; Martino, E.; Romeo, G.; Pacini, F. Somatic mutations of the ret protooncogene in sporadic medullary thyroid carcinoma are not restricted to exon 16 and are associated with tumor recurrence. J. Clin. Endocrinol. Metab. 1996, 81, 1619–1622. [Google Scholar]
- Alemi, M.; Lucas, S.; Sällström, J.; Bergholm, U.; Akerström, G.; Wilander, E. A complex nine base pair deletion in RET exon 11 common in sporadic medullary thyroid carcinoma. Oncogene 1997, 14, 2041–2045. [Google Scholar]
- Uchino, S.; Noguchi, S.; Yamashita, H.; Sato, M.; Adachi, M.; Yamashita, H.; Watanabe, S.; Ohshima, A.; Mitsuyama, S.; Iwashita, T.; Takahashi, M. Somatic Mutations in RET Exons 12 and 15 in Sporadic Medullary Thyroid Carcinomas: Different Spectrum of Mutations in Sporadic Type from Hereditary Type. Cancer Sci. 1999, 90, 1231–1237. [Google Scholar]
- Chen, Y.; Takita, J.; Choi, Y.L.; Kato, M.; Ohira, M.; Sanada, M.; Wang, L.; Soda, M.; Kikuchi, A.; Igarashi, T.; Nakagawara, A.; Hayashi, Y.; Mano, H.; Ogawa, S. Oncogenic mutations of ALK kinase in neuroblastoma. Nature 2008, 455, 971–974. [Google Scholar]
- George, R.E.; Sanda, T.; Hanna, M.; Frohling, S.; Ii, W.L.; Zhang, J.; Ahn, Y.; Zhou, W.; London, W.B.; McGrady, P.; Xue, L.; Zozulya, S.; Gregor, V.E.; Webb, T.R.; Gray, N.S.; Gilliland, D.G.; Diller, L.; Greulich, H.; Morris, S.W.; Meyerson, M.; Look, A.T. Activating mutations in ALK provide a therapeutic target in neuroblastoma. Nature 2008, 455, 975–978. [Google Scholar]
- Janoueix-Lerosey, I.; Lequin, D.; Brugieres, L.; Ribeiro, A.; de Pontual, L.; Combaret, V.; Raynal, V.; Puisieux, A.; Schleiermacher, G.; Pierron, G.; Valteau-Couanet, D.; Frebourg, T.; Michon, J.; Lyonnet, S.; Amiel, J.; Delattre, O. Somatic and germline activating mutations of the ALK kinase receptor in neuroblastoma. Nature 2008, 455, 967–970. [Google Scholar]
- Mosse, Y.P.; Laudenslager, M.; Longo, L.; Cole, K.A.; Wood, A.; Attiyeh, E.F.; Laquaglia, M.J.; Sennett, R.; Lynch, J.E.; Perri, P.; Laureys, G.; Speleman, F.; Kim, C.; Hou, C.; Hakonarson, H.; Torkamani, A.; Schork, N.J.; Brodeur, G.M.; Tonini, G.P.; Rappaport, E.; Devoto, M.; Maris, J.M. Identification of ALK as a major familial neuroblastoma predisposition gene. Nature 2008, 455, 930–935. [Google Scholar]
- Weinstein, I.B. Cancer. Addiction to oncogenes--the Achilles heel of cancer. Science 2002, 297, 63–64. [Google Scholar]
- Kris, M.G.; Natale, R.B.; Herbst, R.S.; Lynch, T.J.; Prager, D.; Belani, C.P.; Schiller, J.H.; Kelly, K.; Spiridonidis, H.; Sandler, A.; Albain, K.S.; Cella, D.; Wolf, M.K.; Averbuch, S.D.; Ochs, J.J.; Kay, A.C. Efficacy of Gefitinib, an Inhibitor of the Epidermal Growth Factor Receptor Tyrosine Kinase, in Symptomatic Patients With Non-Small Cell Lung Cancer. JAMA 2003, 290, 2149–2158. [Google Scholar]
- Fukuoka, M.; Yano, S.; Giaccone, G.; Tamura, T.; Nakagawa, K.; Douillard, J.-Y.; Nishiwaki, Y.; Vansteenkiste, J.; Kudoh, S.; Rischin, D.; Eek, R.; Horai, T.; Noda, K.; Takata, I.; Smit, E.; Averbuch, S.; Macleod, A.; Feyereislova, A.; Dong, R.-P.; Baselga, J. Multi-Institutional Randomized Phase II Trial of Gefitinib for Previously Treated Patients With Advanced Non-Small-Cell Lung Cancer. J. Clin. Oncol. 2003, 21, 2237–2246. [Google Scholar]
- Thatcher, N.; Chang, A.; Parikh, P.; Rodrigues Pereira, J.; Ciuleanu, T.; von Pawel, J.; Thongprasert, S.; Tan, E.H.; Pemberton, K.; Archer, V.; Carroll, K. Gefitinib plus best supportive care in previously treated patients with refractory advanced non-small-cell lung cancer: results from a randomised, placebo-controlled, multicentre study (Iressa Survival Evaluation in Lung Cancer). Lancet 2005, 366, 1527–1537. [Google Scholar]
- Pao, W.; Miller, V.A.; Politi, K.A.; Riely, G.J.; Somwar, R.; Zakowski, M.F.; Kris, M.G.; Varmus, H. Acquired Resistance of Lung Adenocarcinomas to Gefitinib or Erlotinib Is Associated with a Second Mutation in the EGFR Kinase Domain. PLoS Med. 2005, 2, e73. 225–235. [Google Scholar]
- Blencke, S.; Ullrich, A.; Daub, H. Mutation of Threonine 766 in the Epidermal Growth Factor Receptor Reveals a Hotspot for Resistance Formation against Selective Tyrosine Kinase Inhibitors. J. Biol. Chem. 2003, 278, 15435–15440. [Google Scholar]
- Balak, M.N.; Gong, Y.; Riely, G.J.; Somwar, R.; Li, A.R.; Zakowski, M.F.; Chiang, A.; Yang, G.; Ouerfelli, O.; Kris, M.G.; Ladanyi, M.; Miller, V.A.; Pao, W. Novel D761Y and Common Secondary T790M Mutations in Epidermal Growth Factor Receptor–Mutant Lung Adenocarcinomas with Acquired Resistance to Kinase Inhibitors. Clin. Cancer Res. 2006, 12, 6494–6501. [Google Scholar]
- Kosaka, T.; Yatabe, Y.; Endoh, H.; Yoshida, K.; Hida, T.; Tsuboi, M.; Tada, H.; Kuwano, H.; Mitsudomi, T. Analysis of Epidermal Growth Factor Receptor Gene Mutation in Patients with Non-Small Cell Lung Cancer and Acquired Resistance to Gefitinib. Clin. Cancer Res. 2006, 12, 5764–5769. [Google Scholar]
- Engelman, J.A.; Mukohara, T.; Zejnullahu, K.; Lifshits, E.; Borrás, A.M.; Gale, C.-M.; Naumov, G.N.; Yeap, B.Y.; Jarrell, E.; Sun, J.; Tracy, S.; Zhao, X.; Heymach, J.V.; Johnson, B.E.; Cantley, L.C.; Jänne, P.A. Allelic dilution obscures detection of a biologically significant resistance mutation in EGFR-amplified lung cancer. J. Clin. Invest. 2006, 116, 2695–2706. [Google Scholar]
- Frey, K.M.; Georgiev, I.; Donald, B.R.; Anderson, A.C. Predicting resistance mutations using protein design algorithms. Proc. Natl. Acad. Sci. USA 2010, 107, 13707–13712. [Google Scholar]
- Telesco, S.E.; Shih, A.J.; Jia, F.; Radhakrishnan, R. A multiscale modeling approach to investigate molecular mechanisms of pseudokinase activation and drug resistance in the HER3/ErbB3 receptor tyrosine kinase signaling network. Mol. Biosyst. 2011. In Review. [Google Scholar]
- Telesco, S.E.; Radhakrishnan, R. Atomistic Insights into Regulatory Mechanisms of the HER2 Tyrosine Kinase Domain: A Molecular Dynamics Study. Biophys. J. 2009, 96, 2321–2334. [Google Scholar]
- Shih, A.J.; Telesco, S.E.; Choi, S.H.; Lemmon, M.A.; Radhakrishnan, R. Molecular Dynamics Analysis of Conserved Hydrophobic and Hydrophilic Bond Interaction Networks in ErbB Family Kinases. Biochem. J. 2011. In Review. [Google Scholar]
- Ozkirimli, E.; Post, C.B. Src kinase activation: A switched electrostatic network. Protein Sci. 2006, 15, 1051–1062. [Google Scholar]
- Ozkirimli, E.; Yadav, S.S.; Miller, W.T.; Post, C.B. An electrostatic network and long-range regulation of Src kinases. Protein Sci. 2008, 17, 1871–1880. [Google Scholar]
- Citri, A.; Kochupurakkal, B.S.; Yarden, Y. The Achilles heel of ErbB-2/HER2: regulation by the Hsp90 chaperone machine and potential for pharmacological intervention. Cell Cycle 2003, 3, 51–60. [Google Scholar]
- Godawat, R.; Jamadagni, S.N.; Garde, S. Characterizing hydrophobicity of interfaces by using cavity formation, solute binding, and water correlations. Proc. Natl. Acad. Sci. USA 2009, 106, 15119–15124. [Google Scholar]
- Acharya, H.; Vembanur, S.; Jamadagni, S.N.; Garde, S. Mapping hydrophobicity at the nanoscale: Applications to heterogeneous surfaces and proteins. Faraday Discuss. 2010. [Google Scholar] [CrossRef]
- Shi, F.; Telesco, S.E.; Liu, Y.; Radhakrishnan, R.; Lemmon, M.A. ErbB3/HER3 intracellular domain is competent to bind ATP and catalyze autophosphorylation. Proc. Natl. Acad. Sci. USA 2010, 107, 7692–7697. [Google Scholar]
- Tice, D.A.; Biscardi, J.S.; Nickles, A.L.; Parsons, S.J. Mechanism of biological synergy between cellular Src and epidermal growth factor receptor. Proc. Natl. Acad. Sci. USA 1999, 96, 1415–1420. [Google Scholar]
- Bose, R.; Molina, H.; Patterson, A.S.; Bitok, J.K.; Periaswamy, B.; Bader, J.S.; Pandey, A.; Cole, P.A. Phosphoproteomic analysis of Her2/neu signaling and inhibition. Proc. Natl. Acad. Sci. USA 2006, 103, 9773–9778. [Google Scholar]
- Zhang, H.T.; O'Rourke, D.M.; Zhao, H.; Murali, R.; Mikami, Y.; Davis, J.G.; Greene, M.I.; Qian, X. Absence of autophosphorylation site Y882 in the p185neu oncogene product correlates with a reduction of transforming potential. Oncogene 1998, 16, 2835–2842. [Google Scholar]
- Wang, S.E.; Narasanna, A.; Perez-Torres, M.; Xiang, B.; Wu, F.Y.; Yang, S.; Carpenter, G.; Gazdar, A.F.; Muthuswamy, S.K.; Arteaga, C.L. HER2 kinase domain mutation results in constitutive phosphorylation and activation of HER2 and EGFR and resistance to EGFR tyrosine kinase inhibitors. Cancer Cell 2006, 10, 25–38. [Google Scholar]
- Liu, Y.; Radhakrishnan, R. Computational Delineation of the Tyrosyl-Substrate Binding, Pre-Catalytic, and Catalytic Landscapes in the Epidermal Growth Factor Receptor (EGFR) Tyrosine Kinase Domain. 2011. in preparation. [Google Scholar]
- Sharma, S.V.; Gajowniczek, P.; Way, I.P.; Lee, D.Y.; Jiang, J.; Yuza, Y.; Classon, M.; Haber, D.A.; Settleman, J. A common signaling cascade may underlie “addiction” to the Src, BCR-ABL, and EGF receptor oncogenes. Cancer Cell 2006, 10, 425–435. [Google Scholar]
- Shih, A.; Purvis, J.; Radhakrishnan, R. Molecular systems biology of ErbB1 signaling: bridging the gap through multiscale modeling and high-performance computing. Mol. Biosyst. 2008, 4, 1151–1159. [Google Scholar]
- Purvis, J.; Ilango, V.; Radhakrishnan, R. Role of Network Branching in Eliciting Differential Short-Term Signaling Responses in the Hyper-Sensitive Epidermal Growth Factor Receptor Mutants Implicated in Lung Cancer. Biotechnol. Prog. 2008, 24, 540–553. [Google Scholar]
- Purvis, J.; Liu, Y.; Ilango, V.; Radhakrishnan, R. Efficacy of tyrosine kinase inhibitors in the mutants of the epidermal growth factor receptor: A multiscale molecular/systems model for phosphorylation and inhibition. In Proc. Found. Syst. Biol. II; 2007; IRB Verlag Stuttgart; pp. 289–294. [Google Scholar]
- Duesbery, N.; Vande Woude, G. BRAF and MEK mutations make a late entrance. Sci. STKE 2006, 2006, pe15. [Google Scholar]
- Bentires-Alj, M.; Gil, S.G.; Chan, R.; Wang, Z.C.; Wang, Y.; Imanaka, N.; Harris, L.N.; Richardson, A.; Neel, B.G.; Gu, H. A role for the scaffolding adapter GAB2 in breast cancer. Nat. Med. 2006, 12, 114–121. [Google Scholar]
- Mascaux, C.; Iannino, N.; Martin, B.; Paesmans, M.; Berghmans, T.; Dusart, M.; Haller, A.; Lothaire, P.; Meert, A.P.; Noel, S.; Lafitte, J.J.; Sculier, J.P. The role of RAS oncogene in survival of patients with lung cancer: a systematic review of the literature with meta-analysis. Br. J. Cancer 2005, 92, 131–139. [Google Scholar]
- Samuels, Y.; Wang, Z.; Bardelli, A.; Silliman, N.; Ptak, J.; Szabo, S.; Yan, H.; Gazdar, A.; Powell, S.M.; Riggins, G.J.; Willson, J.K.V.; Markowitz, S.; Kinzler, K.W.; Vogelstein, B.; Velculescu, V.E. High Frequency of Mutations of the PIK3CA Gene in Human Cancers. Science 2004, 304, 554. [Google Scholar]
- Sjoblom, T.; Jones, S.; Wood, L.D.; Parsons, D.W.; Lin, J.; Barber, T.D.; Mandelker, D.; Leary, R.J.; Ptak, J.; Silliman, N.; Szabo, S.; Buckhaults, P.; Farrell, C.; Meeh, P.; Markowitz, S.D.; Willis, J.; Dawson, D.; Willson, J.K.V.; Gazdar, A.F.; Hartigan, J.; Wu, L.; Liu, C.; Parmigiani, G.; Park, B.H.; Bachman, K.E.; Papadopoulos, N.; Vogelstein, B.; Kinzler, K.W.; Velculescu, V.E. The Consensus Coding Sequences of Human Breast and Colorectal Cancers. Science 2006, 314, 268–274. [Google Scholar]
- Schulze, W.X.; Deng, L.; Mann, M. Phosphotyrosine interactome of the ErbB-receptor kinase family. Mol. Syst. Biol. 2005. [Google Scholar] [CrossRef]
- Bing Liu, B.B.J.H.W. Impact of EGFR point mutations on the sensitivity to gefitinib: Insights from comparative structural analyses and molecular dynamics simulations. Proteins: Struct. Funct. Bioinf. 2006, 65, 331–346. [Google Scholar]
- Mulloy, R.; Ferrand, A.; Kim, Y.; Sordella, R.; Bell, D.W.; Haber, D.A.; Anderson, K.S.; Settleman, J. Epidermal growth factor receptor mutants from human lung cancers exhibit enhanced catalytic activity and increased sensitivity to gefitinib. Cancer Res. 2007, 67, 2325–2330. [Google Scholar]
- Sergina, N.V.; Rausch, M.; Wang, D.; Blair, J.; Hann, B.; Shokat, K.M.; Moasser, M.M. Escape from HER-family tyrosine kinase inhibitor therapy by the kinase-inactive HER3. Nature 2007, 445, 437–441. [Google Scholar]
- Engelman, J.A.; Zejnullahu, K.; Mitsudomi, T.; Song, Y.; Hyland, C.; Park, J.O.; Lindeman, N.; Gale, C.M.; Zhao, X.; Christensen, J.; Kosaka, T.; Holmes, A.J.; Rogers, A.M.; Cappuzzo, F.; Mok, T.; Lee, C.; Johnson, B.E.; Cantley, L.C.; Janne, P.A. MET amplification leads to gefitinib resistance in lung cancer by activating ERBB3 signaling. Science 2007, 316, 1039–1043. [Google Scholar]
- Baselga, J.; Swain, S.M. Novel anticancer targets: revisiting ERBB2 and discovering ERBB3. Nat. Rev. Cancer 2009, 9, 463–475. [Google Scholar]
- Hutcheson, I.R.; Knowlden, J.M.; Hiscox, S.E.; Barrow, D.; Gee, J.M.; Robertson, J.F.; Ellis, I.O.; Nicholson, R.I. Heregulin beta1 drives gefitinib-resistant growth and invasion in tamoxifen-resistant MCF-7 breast cancer cells. Breast Cancer Res. 2007, 9, R50. [Google Scholar]
- Menendez, J.A.; Lupu, R. Transphosphorylation of kinase-dead HER3 and breast cancer progression: a new standpoint or an old concept revisited? Breast Cancer Res. 2007, 9, 111. [Google Scholar]
- Hsieh, M.H.; Fang, Y.F.; Chang, W.C.; Kuo, H.P.; Lin, S.Y.; Liu, H.P.; Liu, C.L.; Chen, H.C.; Ku, Y.C.; Chen, Y.T.; Chang, Y.H.; Chen, Y.T.; Hsi, B.L.; Tsai, S.F.; Huang, S.F. Complex mutation patterns of epidermal growth factor receptor gene associated with variable responses to gefitinib treatment in patients with non-small cell lung cancer. Lung Cancer 2006, 53, 311–322. [Google Scholar]
- Hamburger, A.W. The role of ErbB3 and its binding partners in breast cancer progression and resistance to hormone and tyrosine kinase directed therapies. J. Mammary Gland Biol. Neoplasia 2008, 13, 225–233. [Google Scholar]
- Offterdinger, M.; Schofer, C.; Weipoltshammer, K.; Grunt, T.W. c-erbB-3: a nuclear protein in mammary epithelial cells. J. Cell. Biol. 2002, 157, 929–939. [Google Scholar]
- Schoeberl, B.; Pace, E.A.; Fitzgerald, J.B.; Harms, B.D.; Xu, L.; Nie, L.; Linggi, B.; Kalra, A.; Paragas, V.; Bukhalid, R.; Grantcharova, V.; Kohli, N.; West, K.A.; Leszczyniecka, M.; Feldhaus, M.J.; Kudla, A.J.; Nielsen, U.B. Therapeutically Targeting ErbB3: A Key Node in Ligand-Induced Activation of the ErbB Receptor-PI3K Axis. Sci. Signal. 2009, 2, ra31. [Google Scholar]
- Ferrer-Soler, L.; Vazquez-Martin, A.; Brunet, J.; Menendez, J.A.; De Llorens, R.; Colomer, R. An update of the mechanisms of resistance to EGFR-tyrosine kinase inhibitors in breast cancer: Gefitinib (Iressa) -induced changes in the expression and nucleo-cytoplasmic trafficking of HER-ligands (Review). Int. J. Mol. Med. 2007, 20, 3–10. [Google Scholar]
- Motoyama, A.B.; Hynes, N.E. {BAD}: a good therapeutic target? Breast Cancer Res. 2003, 5, 27–30. [Google Scholar]
- Shih, A.; Telesco, S.T.; Purvis, J.; Liu, Y.; Radhakrishnan, R. The role for molecular modeling in multiscale cancer models; Multiscale Cancer Modeling of Cancer; Mathematical and Computational Biology Series; Deisboeck, T.S., Stamatakos, G., Eds.; Chapman & Hall-CRC: Boca Rotan, FL, USA, 2010; in press. [Google Scholar]
- Ramanathan, A.; Agarwal, P.K.; Kurnikova, M.; Langmead, C.J. An Online Approach for Mining Collective Behaviors from Molecular Dynamics Simulations. J. Comput. Biol. 2010, 17, 309–324. [Google Scholar]
- Kholodenko, B.N.; Demin, O.V.; Moehren, G.; Hoek, J.B. Quantification of short term signaling by the epidermal growth factor receptor. J. Biol. Chem. 1999, 274, 30169–30181. [Google Scholar]
- Schoeberl, B.; Jonsson, C.E.; Gilles, E.D.; Muller, G. Computational modeling of the dynamics of MAPKinase cascade activated by surface and internalized receptors. Nature Biotech. 2002, 20, 370–375. [Google Scholar]
- Brown, K.S.; Hill, C.C.; Calero, G.A.; Myers, C.R.; Lee, K.H.; Sethna, J.P.; Cerione, R.A. The statistical mechanics of complex signaling networks: Nerve growth factor signaling. Phys. Biol. 2004, 1, 184–195. [Google Scholar]
© 2011 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Shih, A.J.; Telesco, S.E.; Radhakrishnan, R. Analysis of Somatic Mutations in Cancer: Molecular Mechanisms of Activation in the ErbB Family of Receptor Tyrosine Kinases. Cancers 2011, 3, 1195-1231. https://doi.org/10.3390/cancers3011195
Shih AJ, Telesco SE, Radhakrishnan R. Analysis of Somatic Mutations in Cancer: Molecular Mechanisms of Activation in the ErbB Family of Receptor Tyrosine Kinases. Cancers. 2011; 3(1):1195-1231. https://doi.org/10.3390/cancers3011195
Chicago/Turabian StyleShih, Andrew J., Shannon E. Telesco, and Ravi Radhakrishnan. 2011. "Analysis of Somatic Mutations in Cancer: Molecular Mechanisms of Activation in the ErbB Family of Receptor Tyrosine Kinases" Cancers 3, no. 1: 1195-1231. https://doi.org/10.3390/cancers3011195
APA StyleShih, A. J., Telesco, S. E., & Radhakrishnan, R. (2011). Analysis of Somatic Mutations in Cancer: Molecular Mechanisms of Activation in the ErbB Family of Receptor Tyrosine Kinases. Cancers, 3(1), 1195-1231. https://doi.org/10.3390/cancers3011195