Molecular Pathogenesis of Pancreatic Neuroendocrine Tumors

Abstract
:1. Introduction
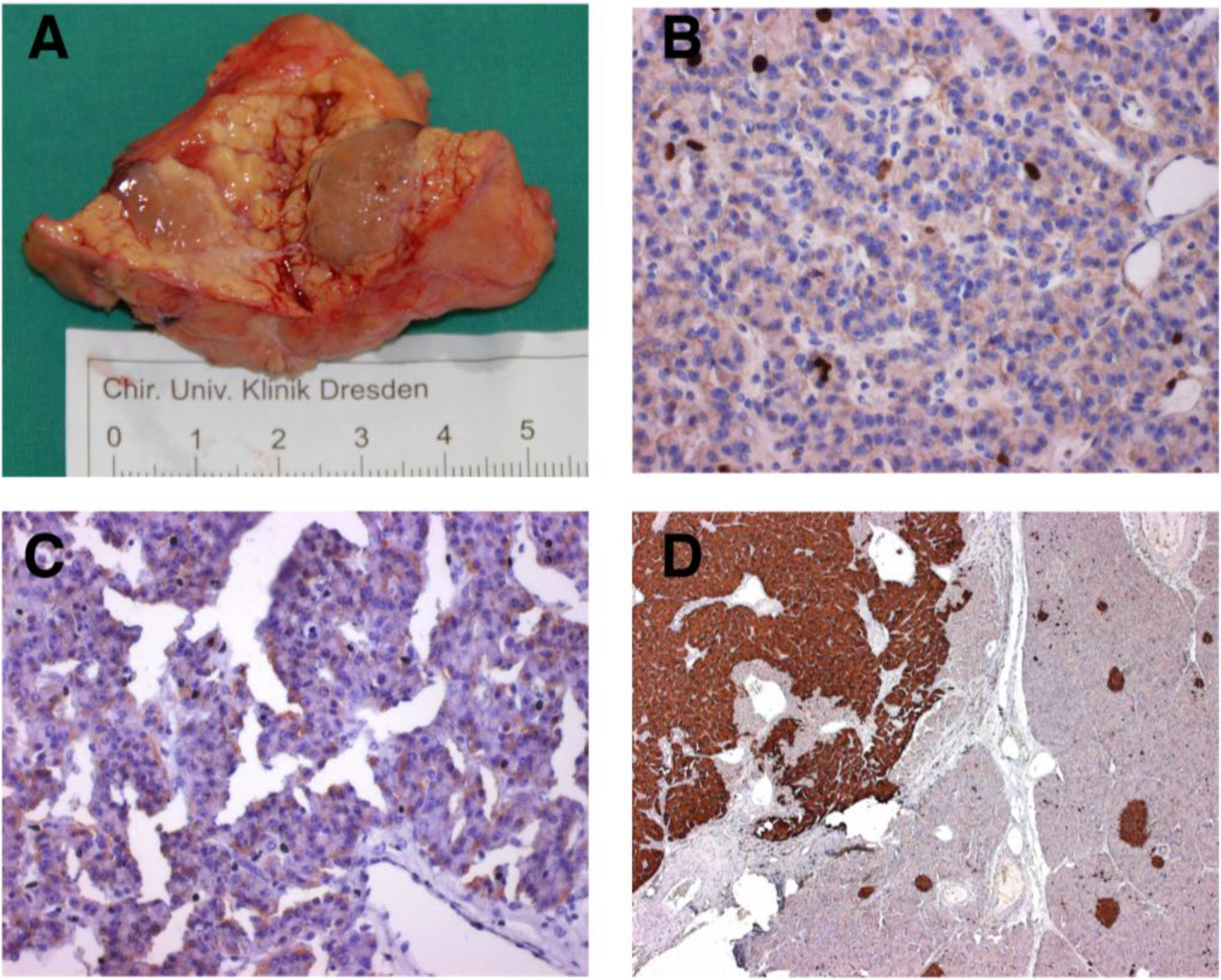
2. Methods
3. Review of Literature
3.1. MEN-1 Syndrome and the Role of Menin in PNET Initiation

{kind=link}
{kind=link}
| Mechanism | Co-factor | Regulated Factor | Consequence |
|---|---|---|---|
| Transcription activation | HMT | p27kip1 | Cell growth inhibition |
| p18ink4c | |||
| Hoxc8 | Cell differentiation | ||
| Transcription repression | HDAC | IGFBP-2 | Decreased cell proliferation |
| Inhibition | ? | Cyclin D/CDK4 | Inhibition of G1/S transition |
| cdc7/ASK | Inhibition of DNA synthesis | ||
| Transcription activation | ? | Caspase 8 | TNFα- sensitizing/apoptosis |
| Protein-protein interactions | FancD2/RPA2/ cdc7/ASK | hTERT | Genome stabilization |
3.2. Role of Angiogensis in PNET Progression
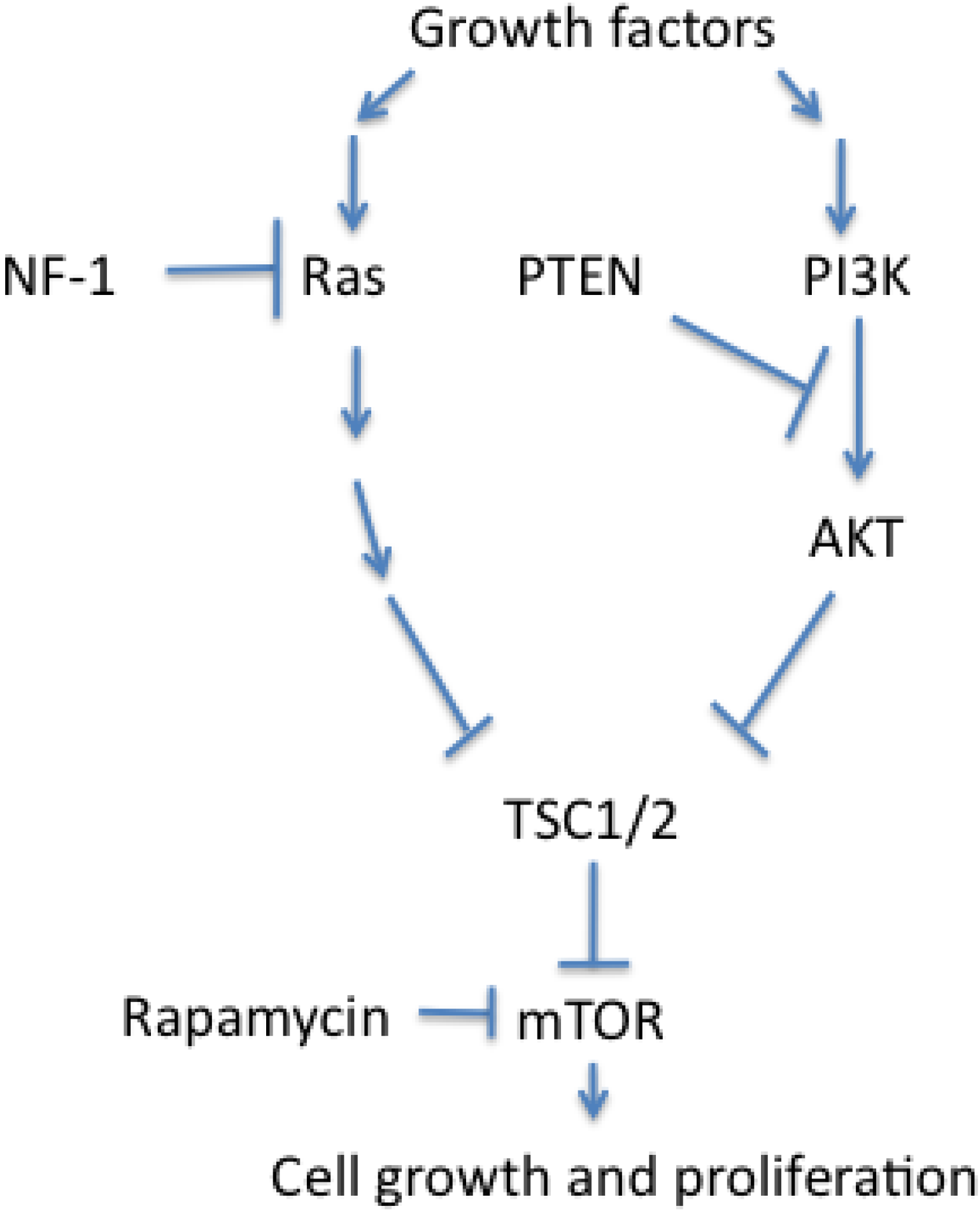
3.3. Dysregulated Cell Growth and Proliferation in PNETs
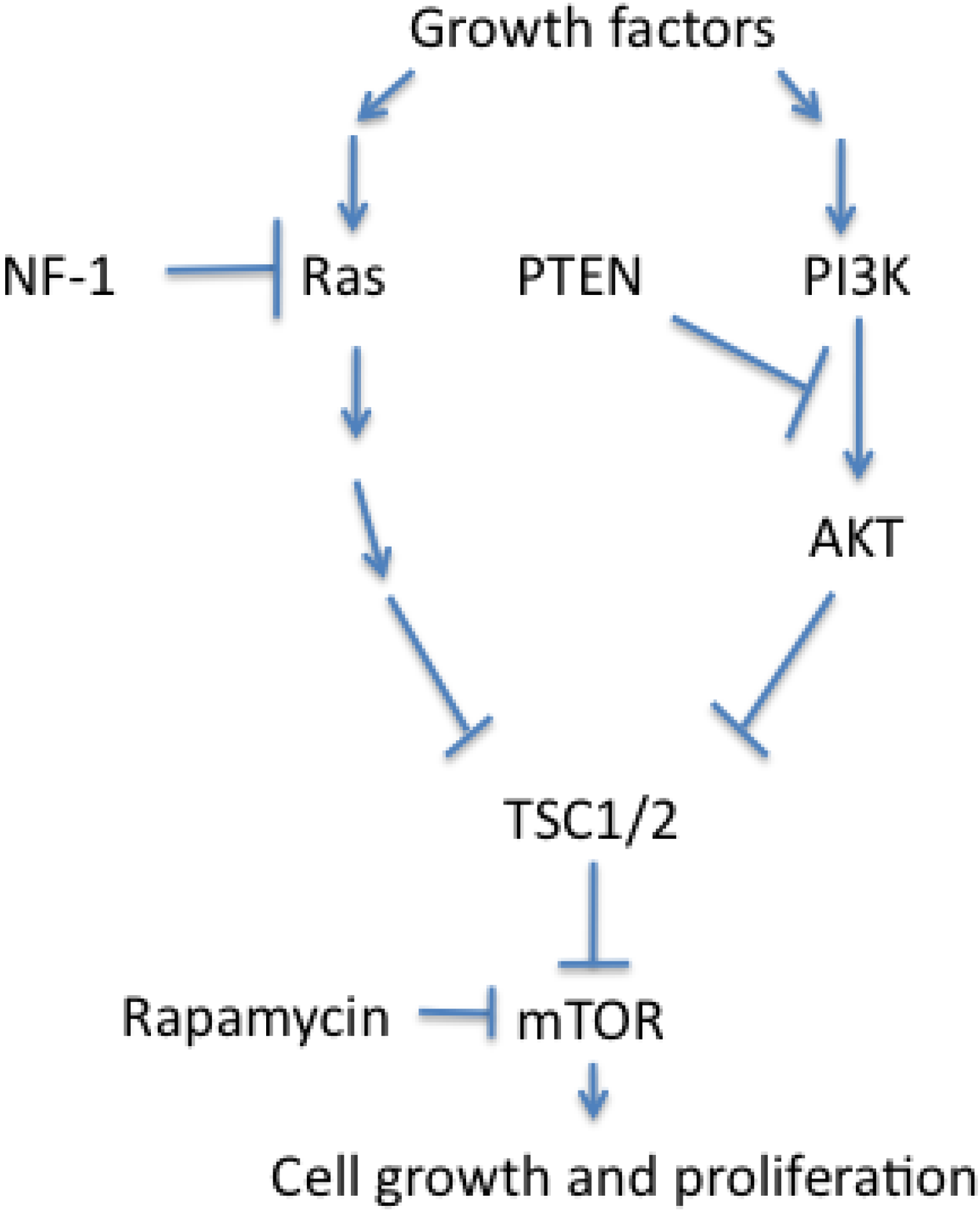
4. Conclusions
References
- Yao, J.C.; Eisner, M.P.; Leary, C.; Dagohoy, C.; Phan, A.; Rashid, A.; Hassan, M.; Evans, D.B. Population-based study of islet cell carcinoma. Ann. Surg. Oncol. 2007, 14, 3492–3500. [Google Scholar] [CrossRef]
- Halfdanarson, T.R.; Rabe, K.G.; Rubin, J.; Petersen, G.M. Pancreatic neuroendocrine tumors (pnets): Incidence, prognosis and recent trend toward improved survival. Ann. Oncol. 2008, 19, 1727–1733. [Google Scholar] [CrossRef]
- Mansour, J.C.; Chen, H. Pancreatic endocrine tumors. J. Surg. Res. 2004, 120, 139–161. [Google Scholar] [CrossRef]
- Ehehalt, F.; Saeger, H.D.; Schmidt, C.M.; Grützmann, R. Neuroendocrine tumors of the pancreas. Oncologist 2009, 14, 456–467. [Google Scholar] [CrossRef]
- Modlin, I.M.; Moss, S.F.; Chung, D.C.; Jensen, R.T.; Snyderwine, E. Priorities for improving the management of gastroenteropancreatic neuroendocrine tumors. J. Natl. Cancer. Inst. 2008, 100, 1282–1289. [Google Scholar] [CrossRef]
- Leotlela, P.D.; Jauch, A.; Holtgreve-Grez, H.; Thakker, R.V. Genetics of neuroendocrine and carcinoid tumours. Endocr. Relat. Cancer 2003, 10, 437–450. [Google Scholar] [CrossRef]
- Yang, Y.; Hua, X. In search of tumor suppressing functions of menin. Mol. Cell. Endocrinol. 2007, 265-266, 34–41. [Google Scholar] [CrossRef]
- Akerstrom, G.; Hessman, O.; Hellman, P.; Skogseid, B. Pancreatic tumours as part of the men-1 syndrome. Best Pract. Res. Clin. Gastroenterol. 2005, 19, 819–830. [Google Scholar] [CrossRef]
- Anlauf, M.; Garbrecht, N.; Henopp, T.; Schmitt, A.; Schlenger, R.; Raffel, A.; Krausch, M.; Gimm, O.; Eisenberger, C.F.; Knoefel, W.T.; Dralle, H.; Komminoth, P.; Heitz, P.U.; Perren, A.; Kloppel, G. Sporadic versus hereditary gastrinomas of the duodenum and pancreas: Distinct clinico-pathological and epidemiological features. World J. Gastroenterol. 2006, 12, 5440–5446. [Google Scholar]
- Shan, L.; Nakamura, Y.; Nakamura, M.; Yokoi, T.; Tsujimoto, M.; Arima, R.; Kameya, T.; Kakudo, K. Somatic mutations of multiple endocrine neoplasia type 1 gene in the sporadic endocrine tumors. Lab. Invest. 1998, 78, 471–475. [Google Scholar]
- Wang, E.H.; Ebrahimi, S.A.; Wu, A.Y.; Kashefi, C.; Passaro, E., Jr.; Sawicki, M.P. Mutation of the menin gene in sporadic pancreatic endocrine tumors. Cancer Res. 1998, 58, 4417–4420. [Google Scholar]
- Oberg, K. Genetics and molecular pathology of neuroendocrine gastrointestinal and pancreatic tumors (gastroenteropancreatic neuroendocrine tumors). Curr. Opin. Endocrinol. Diabetes Obes. 2009, 16, 72–78. [Google Scholar] [CrossRef]
- Corbo, V.; Dalai, I.; Scardoni, M.; Barbi, S.; Beghelli, S.; Bersani, S.; Albarello, L.; Doglioni, C.; Schott, C.; Capelli, P.; Chilosi, M.; Boninsegna, L.; Becker, K.F.; Falconi, M.; Scarpa, A. Men1 in pancreatic endocrine tumors: Analysis of gene and protein status in 169 sporadic neoplasms reveals alterations in the vast majority of cases. Endocr. Relat. Cancer 17, 771–783.
- Bertolino, P.; Tong, W.M.; Galendo, D.; Wang, Z.Q.; Zhang, C.X. Heterozygous men1 mutant mice develop a range of endocrine tumors mimicking multiple endocrine neoplasia type 1. Mol. Endocrinol. 2003, 17, 1880–1892. [Google Scholar] [CrossRef]
- Lu, J.; Herrera, P.L.; Carreira, C.; Bonnavion, R.; Seigne, C.; Calender, A.; Bertolino, P.; Zhang, C.X. Alpha cell-specific men1 ablation triggers the transdifferentiation of glucagon-expressing cells and insulinoma development. Gastroenterology 138, 1954–1965.
- Vortmeyer, A.O.; Huang, S.; Lubensky, I.; Zhuang, Z. Non-islet origin of pancreatic islet cell tumors. J. Clin. Endocrinol. Metab. 2004, 89, 1934–1938. [Google Scholar] [CrossRef]
- Corcos, O.; Couvelard, A.; Giraud, S.; Vullierme, M.P.; Dermot, O.T.; Rebours, V.; Stievenart, J.L.; Penfornis, A.; Niccoli-Sire, P.; Baudin, E.; Sauvanet, A.; Levy, P.; Ruszniewski, P.; Richard, S.; Hammel, P. Endocrine pancreatic tumors in von hippel-lindau disease: Clinical, histological, and genetic features. Pancreas 2008, 37, 85–93. [Google Scholar]
- Lubensky, I.A.; Pack, S.; Ault, D.; Vortmeyer, A.O.; Libutti, S.K.; Choyke, P.L.; Walther, M.M.; Linehan, W.M.; Zhuang, Z. Multiple neuroendocrine tumors of the pancreas in von hippel-lindau disease patients: Histopathological and molecular genetic analysis. Am. J. Pathol. 1998, 153, 223–231. [Google Scholar] [CrossRef]
- Latif, F.; Tory, K.; Gnarra, J.; Yao, M.; Duh, F.M.; Orcutt, M.L.; Stackhouse, T.; Kuzmin, I.; Modi, W.; Geil, L.; et al. Identification of the von hippel-lindau disease tumor suppressor gene. Science 1993, 260, 1317–1320. [Google Scholar]
- Lonser, R.R.; Glenn, G.M.; Walther, M.; Chew, E.Y.; Libutti, S.K.; Linehan, W.M.; Oldfield, E.H. Von hippel-lindau disease. Lancet 2003, 361, 2059–2067. [Google Scholar] [CrossRef]
- Kaelin, W.G., Jr. Molecular basis of the vhl hereditary cancer syndrome. Nat. Rev. Cancer 2002, 2, 673–682. [Google Scholar] [CrossRef]
- Kersting, S.; Konopke, R.; Kersting, F.; Volk, A.; Distler, M.; Bergert, H.; Saeger, H.D.; Grützmann, R.; Bunk, A. Quantitative perfusion analysis of transabdominal contrast-enhanced ultrasonography of pancreatic masses and carcinomas. Gastroenterology 2009, 137, 1903–1911. [Google Scholar] [CrossRef]
- Lott, S.T.; Chandler, D.S.; Curley, S.A.; Foster, C.J.; El-Naggar, A.; Frazier, M.; Strong, L.C.; Lovell, M.; Killary, A.M. High frequency loss of heterozygosity in von hippel-lindau (vhl)-associated and sporadic pancreatic islet cell tumors: Evidence for a stepwise mechanism for malignant conversion in vhl tumorigenesis. Cancer Res. 2002, 62, 1952–1955. [Google Scholar]
- Starker, L.F.; Carling, T. Molecular genetics of gastroenteropancreatic neuroendocrine tumors. Curr. Opin. Oncol. 2009, 21, 29–33. [Google Scholar] [CrossRef]
- Takahashi, Y.; Akishima-Fukasawa, Y.; Kobayashi, N.; Sano, T.; Kosuge, T.; Nimura, Y.; Kanai, Y.; Hiraoka, N. Prognostic value of tumor architecture, tumor-associated vascular characteristics, and expression of angiogenic molecules in pancreatic endocrine tumors. Clin. Cancer Res. 2007, 13, 187–196. [Google Scholar] [CrossRef]
- Papouchado, B.; Erickson, L.A.; Rohlinger, A.L.; Hobday, T.J.; Erlichman, C.; Ames, M.M.; Lloyd, R.V. Epidermal growth factor receptor and activated epidermal growth factor receptor expression in gastrointestinal carcinoids and pancreatic endocrine carcinomas. Mod. Pathol. 2005, 18, 1329–1335. [Google Scholar] [CrossRef]
- Silva, S.R.; Bowen, K.A.; Rychahou, P.G.; Jackson, L.N.; Weiss, H.L.; Lee, E.Y.; Townsend, C.M., Jr.; Evers, B.M. Vegfr-2 expression in carcinoid cancer cells and its role in tumor growth and metastasis. Int. J. Cancer 2010, in press. [Google Scholar]
- Detjen, K.M.; Rieke, S.; Deters, A.; Schulz, P.; Rexin, A.; Vollmer, S.; Hauff, P.; Wiedenmann, B.; Pavel, M.; Scholz, A. Angiopoietin-2 promotes disease progression of neuroendocrine tumors. Clin. Cancer Res. 2010, 16, 420–429. [Google Scholar] [CrossRef]
- Ulanet, D.B.; Hanahan, D. Loss of p19(arf) facilitates the angiogenic switch and tumor initiation in a multi-stage cancer model via p53-dependent and independent mechanisms. PLoS One 2010, 5, e12454. [Google Scholar]
- Eriksson, B. New drugs in neuroendocrine tumors: Rising of new therapeutic philosophies? Curr. Opin. Oncol. 2010, 22, 381–386. [Google Scholar] [CrossRef]
- Francalanci, P.; Diomedi-Camassei, F.; Purificato, C.; Santorelli, F.M.; Giannotti, A.; Dominici, C.; Inserra, A.; Boldrini, R. Malignant pancreatic endocrine tumor in a child with tuberous sclerosis. Am. J. Surg. Pathol. 2003, 27, 1386–1389. [Google Scholar] [CrossRef]
- Eledrisi, M.S.; Stuart, C.A.; Alshanti, M. Insulinoma in a patient with tuberous sclerosis: Is there an association? Endocr. Pract. 2002, 8, 109–112. [Google Scholar] [CrossRef]
- Verhoef, S.; van Diemen-Steenvoorde, R.; Akkersdijk, W.L.; Bax, N.M.; Ariyurek, Y.; Hermans, C.J.; van Nieuwenhuizen, O.; Nikkels, P.G.; Lindhout, D.; Halley, D.J.; Lips, K.; van den Ouweland, A.M. Malignant pancreatic tumour within the spectrum of tuberous sclerosis complex in childhood. Eur. J. Pediatr. 1999, 158, 284–287. [Google Scholar] [CrossRef] [Green Version]
- Cappelli, C.; Agosti, B.; Braga, M.; Cumetti, D.; Gandossi, E.; Rizzoni, D.; Agabiti Rosei, E. Von recklinghausen's neurofibromatosis associated with duodenal somatostatinoma. A case report and review of the literature. Minerva Endocrinol. 2004, 29, 19–24. [Google Scholar]
- Tan, C.C.; Hall, R.I.; Semeraro, D.; Irons, R.P.; Freeman, J.G. Ampullary somatostatinoma associated with von recklinghausen's neurofibromatosis presenting as obstructive jaundice. Eur. J. Surg. Oncol. 1996, 22, 298–301. [Google Scholar] [CrossRef]
- Inoki, K.; Corradetti, M.N.; Guan, K.L. Dysregulation of the tsc-mtor pathway in human disease. Nat. Genet. 2005, 37, 19–24. [Google Scholar] [CrossRef]
- Johannessen, C.M.; Reczek, E.E.; James, M.F.; Brems, H.; Legius, E.; Cichowski, K. The nf1 tumor suppressor critically regulates tsc2 and mtor. Proc. Natl. Acad. Sci. USA 2005, 102, 8573–8578. [Google Scholar]
- Brugarolas, J.; Lei, K.; Hurley, R.L.; Manning, B.D.; Reiling, J.H.; Hafen, E.; Witters, L.A.; Ellisen, L.W.; Kaelin, W.G., Jr. Regulation of mtor function in response to hypoxia by redd1 and the tsc1/tsc2 tumor suppressor complex. Genes Dev. 2004, 18, 2893–2904. [Google Scholar] [CrossRef] [Green Version]
- Ulanet, D.B.; Ludwig, D.L.; Kahn, C.R.; Hanahan, D. Insulin receptor functionally enhances multistage tumor progression and conveys intrinsic resistance to igf-1r targeted therapy. Proc. Natl. Acad. Sci. USA 2010, 107, 10791–10798. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sabatini, D.M. Growing roles for the mtor pathway. Curr. Opin. Cell. Biol. 2005, 17, 596–603. [Google Scholar] [CrossRef]
- Maitra, A.; Hansel, D.E.; Argani, P.; Ashfaq, R.; Rahman, A.; Naji, A.; Deng, S.; Geradts, J.; Hawthorne, L.; House, M.G.; Yeo, C.J. Global expression analysis of well-differentiated pancreatic endocrine neoplasms using oligonucleotide microarrays. Clin. Cancer Res. 2003, 9, 5988–5995. [Google Scholar]
- Hansel, D.E.; Rahman, A.; House, M.; Ashfaq, R.; Berg, K.; Yeo, C.J.; Maitra, A. Met proto-oncogene and insulin-like growth factor binding protein 3 overexpression correlates with metastatic ability in well-differentiated pancreatic endocrine neoplasms. Clin. Cancer Res. 2004, 10, 6152–6158. [Google Scholar] [CrossRef]
- Capurso, G.; Lattimore, S.; Crnogorac-Jurcevic, T.; Panzuto, F.; Milione, M.; Bhakta, V.; Campanini, N.; Swift, S.M.; Bordi, C.; Delle Fave, G.; Lemoine, N.R. Gene expression profiles of progressive pancreatic endocrine tumours and their liver metastases reveal potential novel markers and therapeutic targets. Endocr. Relat. Cancer 2006, 13, 541–558. [Google Scholar] [CrossRef]
- Couvelard, A.; Hu, J.; Steers, G.; O'Toole, D.; Sauvanet, A.; Belghiti, J.; Bedossa, P.; Gatter, K.; Ruszniewski, P.; Pezzella, F. Identification of potential therapeutic targets by gene-expression profiling in pancreatic endocrine tumors. Gastroenterology 2006, 131, 1597–1610. [Google Scholar] [CrossRef]
- Bloomston, M.; Durkin, A.; Yang, I.; Rojiani, M.; Rosemurgy, A.S.; Enkmann, S.; Yeatman, T.J.; Zervos, E.E. Identification of molecular markers specific for pancreatic neuroendocrine tumors by genetic profiling of core biopsies. Ann. Surg. Oncol. 2004, 11, 413–419. [Google Scholar] [CrossRef]
- Dilley, W.G.; Kalyanaraman, S.; Verma, S.; Cobb, J.P.; Laramie, J.M.; Lairmore, T.C. Global gene expression in neuroendocrine tumors from patients with the men1 syndrome. Mol. Cancer 2005, 4, 9. [Google Scholar] [CrossRef] [Green Version]
- Duerr, E.M.; Mizukami, Y.; Ng, A.; Xavier, R.J.; Kikuchi, H.; Deshpande, V.; Warshaw, A.L.; Glickman, J.; Kulke, M.H.; Chung, D.C. Defining molecular classifications and targets in gastroenteropancreatic neuroendocrine tumors through DNA microarray analysis. Endocr. Relat. Cancer 2008, 15, 243–256. [Google Scholar] [CrossRef]
- Missiaglia, E.; Dalai, I.; Barbi, S.; Beghelli, S.; Falconi, M.; della Peruta, M.; Piemonti, L.; Capurso, G.; Di Florio, A.; delle Fave, G.; Pederzoli, P.; Croce, C.M.; Scarpa, A. Pancreatic endocrine tumors: Expression profiling evidences a role for akt-mtor pathway. J. Clin. Oncol. 2010, 28, 245–255. [Google Scholar] [CrossRef]
- Duran, I.; Kortmansky, J.; Singh, D.; Hirte, H.; Kocha, W.; Goss, G.; Le, L.; Oza, A.; Nicklee, T.; Ho, J.; Birle, D.; Pond, G.R.; Arboine, D.; Dancey, J.; Aviel-Ronen, S.; Tsao, M.S.; Hedley, D.; Siu, L.L. A phase ii clinical and pharmacodynamic study of temsirolimus in advanced neuroendocrine carcinomas. Br. J. Cancer 2006, 95, 1148–1154. [Google Scholar] [CrossRef]
- O'Donnell, P.H.; Ratain, M.J. Evaluating the activity of temsirolimus in neuroendocrine cancer. Br. J. Cancer 2007, 96, 177, author reply 178-179. [Google Scholar] [CrossRef]
- Yao, J.C.; Phan, A.T.; Chang, D.Z.; Wolff, R.A.; Hess, K.; Gupta, S.; Jacobs, C.; Mares, J.E.; Landgraf, A.N.; Rashid, A.; Meric-Bernstam, F. Efficacy of rad001 (everolimus) and octreotide lar in advanced low- to intermediate-grade neuroendocrine tumors: Results of a phase ii study. J. Clin. Oncol. 2008, 26, 4311–4318. [Google Scholar] [CrossRef]
- Zitzmann, K.; Ruden, J.; Brand, S.; Goke, B.; Lichtl, J.; Spottl, G.; Auernhammer, C.J. Compensatory activation of akt in response to mtor and raf inhibitors - a rationale for dual-targeted therapy approaches in neuroendocrine tumor disease. Cancer Lett. 2010, 295, 100–109. [Google Scholar] [CrossRef]
- House, M.G.; Herman, J.G.; Guo, M.Z.; Hooker, C.M.; Schulick, R.D.; Lillemoe, K.D.; Cameron, J.L.; Hruban, R.H.; Maitra, A.; Yeo, C.J. Aberrant hypermethylation of tumor suppressor genes in pancreatic endocrine neoplasms. Ann. Surg. 2003, 238, 423-431; discussion 431-422. [Google Scholar]
- Roldo, C.; Missiaglia, E.; Hagan, J.P.; Falconi, M.; Capelli, P.; Bersani, S.; Calin, G.A.; Volinia, S.; Liu, C.G.; Scarpa, A.; Croce, C.M. Microrna expression abnormalities in pancreatic endocrine and acinar tumors are associated with distinctive pathologic features and clinical behavior. J. Clin. Oncol. 2006, 24, 4677–4684. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Ehehalt, F.; Franke, E.; Pilarsky, C.; Grützmann, R. Molecular Pathogenesis of Pancreatic Neuroendocrine Tumors. Cancers 2010, 2, 1901-1910. https://doi.org/10.3390/cancers2041901
Ehehalt F, Franke E, Pilarsky C, Grützmann R. Molecular Pathogenesis of Pancreatic Neuroendocrine Tumors. Cancers. 2010; 2(4):1901-1910. https://doi.org/10.3390/cancers2041901
Chicago/Turabian StyleEhehalt, Florian, Ellen Franke, Christian Pilarsky, and Robert Grützmann. 2010. "Molecular Pathogenesis of Pancreatic Neuroendocrine Tumors" Cancers 2, no. 4: 1901-1910. https://doi.org/10.3390/cancers2041901