Role of Oxidative Stress in Stem, Cancer, and Cancer Stem Cells




Abstract
:
1. Introduction
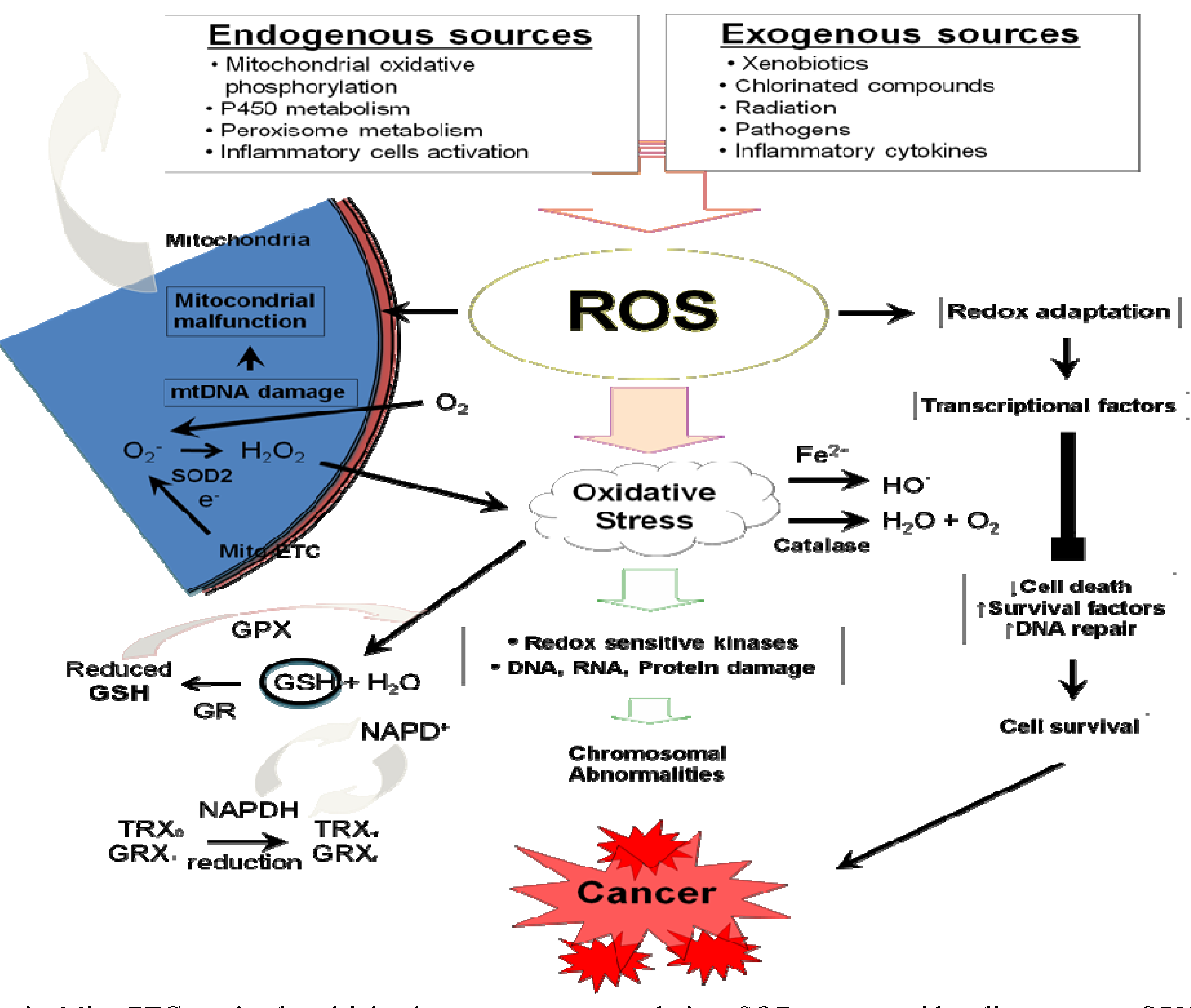
2. Stem Cells
2.1. Embryonic Stem Cells (ESCs)
2.2. Adult Stem Cells (ASCs)
3. Cancer Cells
4. Cancer Stem Cells (CSCs)
- (1)
- Self-renewal ability (Asymmetric divisions): This property contributes toward developing a critical mass of cells. Moreover, it generates a quiescent stem cell and a committed progenitor [21];
- (2)
- Self-renewal regulation: Control of the self-renewal ability occurs by similar signaling pathways such as, Wnt, Sonic Hedgehog, Notch, and Polycomb genes (BMI-1 and EZH2);
- (3)
- Telomeres and telomerase activity: This telomerase activity increases the cellular life span. Both have extended telomeres and telomerase activity;
- (4)
- ATP-binding cassette (ABC) transporters: Both express the ABC transporters, which are implicated in the cellular resistance against specific growth-inhibitory drugs;
- (5)
- Surface receptor expression: Both express similar surface receptors such as, c-kit, c-met, LIF-R, CD133, and CXCR4. These surface receptors were identified as stem cell markers or associated with metastasis;
- (6)
- Longevity (Long life span): Both are long-lived;
- (7)
- Resistance to deleterious agents: Both are resistant to deleterious agents;
- (8)
- Metastasis: Both have the metastatic property;
- (9)

{kind=link}
{kind=link}
{kind=link}
| Signal pathway | Normal stem cells | Cancer and cancer stem cells |
|---|---|---|
| Polycomb-group protein family (Bmi-1) |
|
|
| Notch |
|
|
| Wnt/β-catenin | ||
| PTEN |
|
|
| Sonic hedgehog (Shh) | ||
| Hox family |
|
| Cancer stem cells | Normal stem cells | |
|---|---|---|
| Surfacemarkers | AML (CD123+/CD117–), Prostate (CD133+/–), Breast (CD44+/CD24–) | Absent |
| Self-renewalcapacity | Extensive and indefinite | Limited |
| Nature | Tumorigenic | Organogenic |
| Karyotype | Abnormal | Normal |
| Tumorsuppressorgenes | Present (Interferon factor-1, Death associated protein kinase-1) | Absent |
4.1. Breast CSCs
4.2. Prostate CSCs
4.3. Neuronal CSCs
4.4. ROS and CSCs
5. ROS and Apoptosis
6. ROS and Autophagy
7. Signaling Pathways, Transcription Factors, and Their Roles in Oxidative Stress
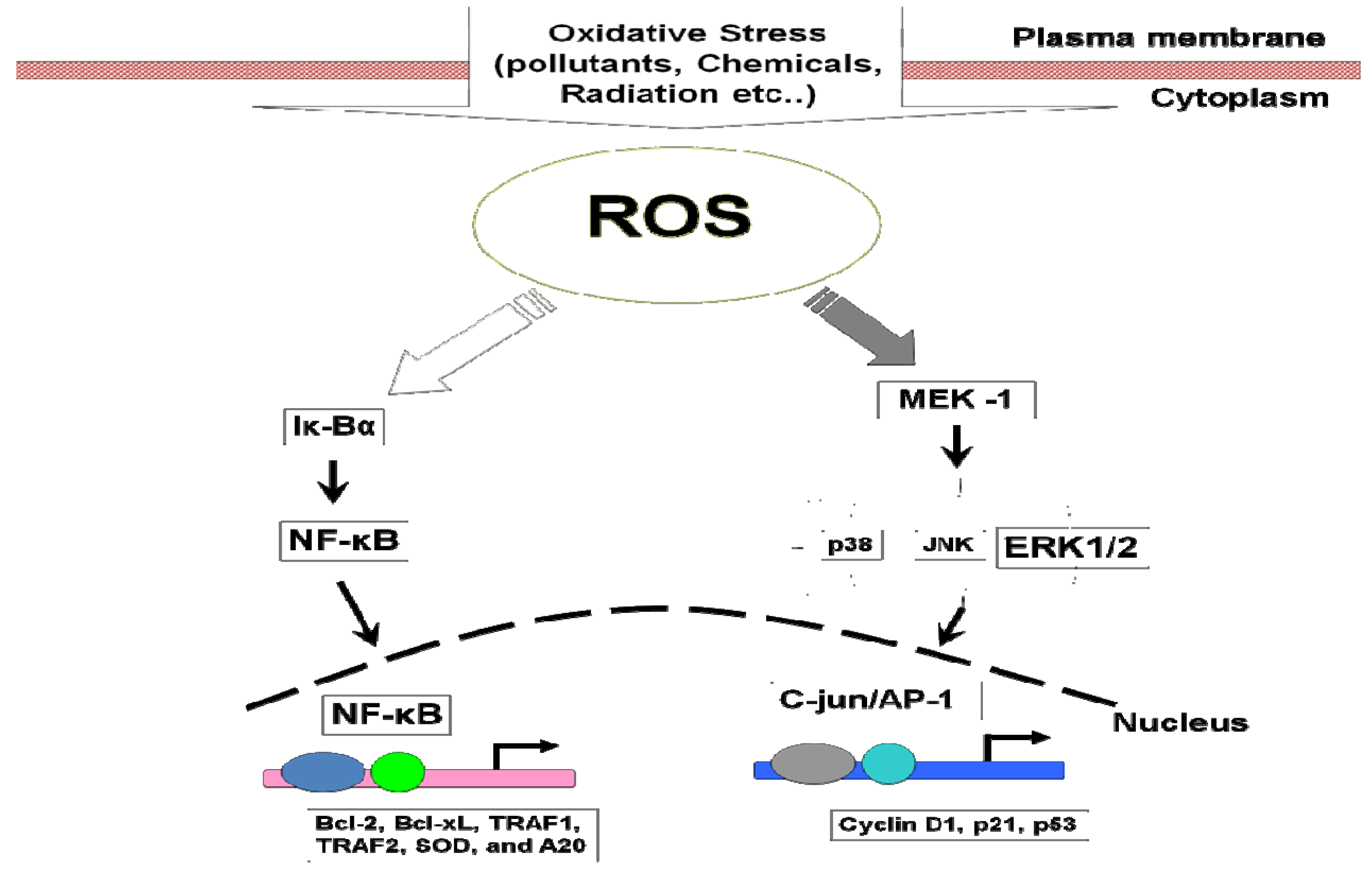
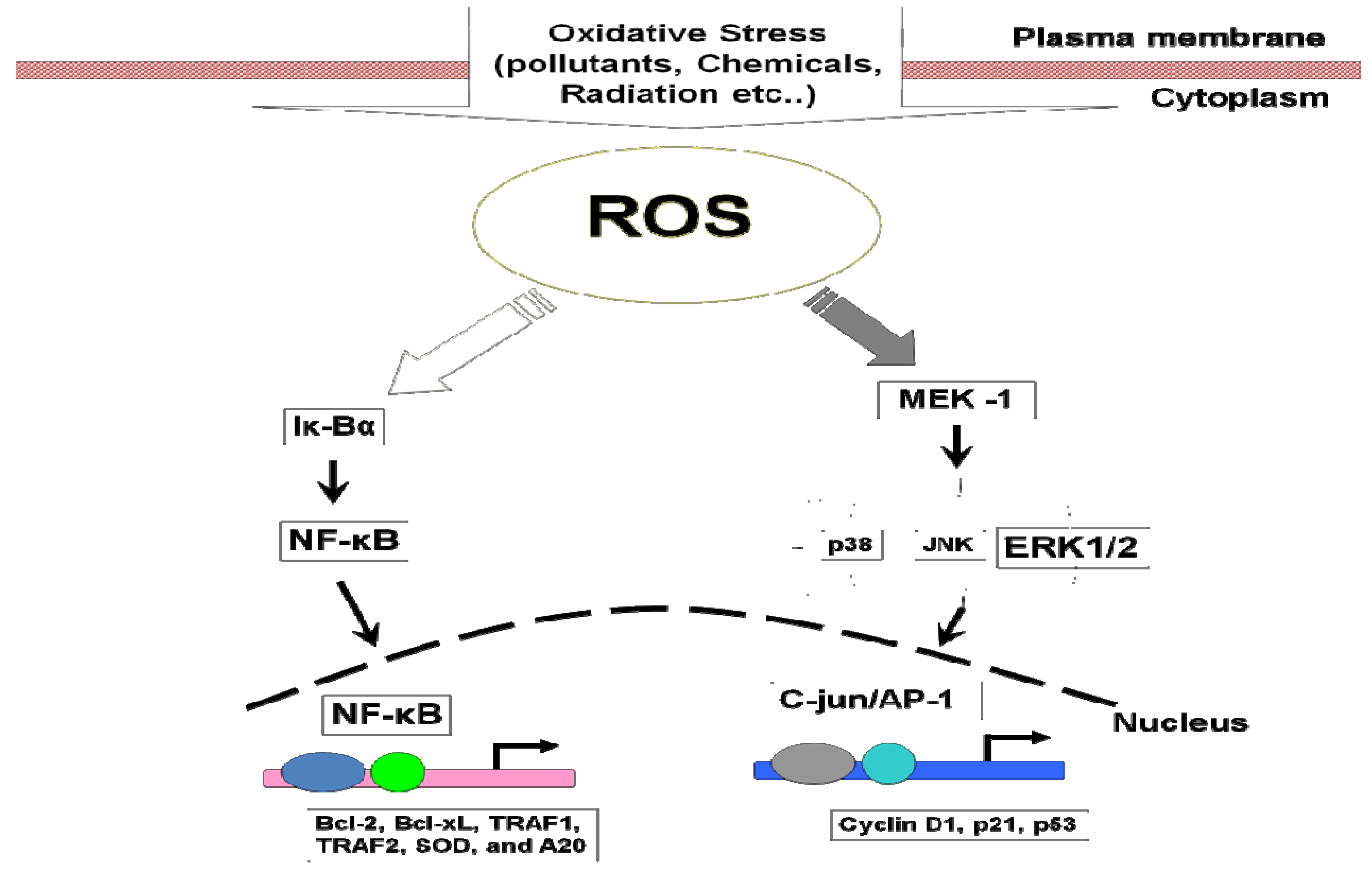
7.1. Mitogen-Activated Protein Kinases (MAPKs)
7.1.1. MAPKs and Cancer
7.1.2. MAPKs and Stem Cells
7.1.3. MAPKs and CSCs
7.2. NF-κB
7.2.1. NF-κB and Cancer
7.2.2. NF-κB and Stem Cells
7.2.3. NF-κB and CSCs
8. Conclusions
Acknowledgements
References
- Vishal, R.T.; Sharma, S.; Mahajan, A.; Bardi, G.H. Oxidative Stress: A Novel Strategy in Cancer Treatment. JK Sci. 2005, 7, 1–3. [Google Scholar]
- Chandra, J.; Samali, A.; Orrenius, S. Triggering and modulation of apoptosis by oxidative stress. Free Rad. Med. Biol. 2000, 29, 323–333. [Google Scholar] [CrossRef]
- Poli, G.; Biasi, F.; Chiarpotto, E. Oxidative stress and cell signaling. Curr. Med. Chem. 2004, 11, 1163–1182. [Google Scholar] [CrossRef]
- Pillai, C.K.; Pillai, K.S. Antioxidants in health. Int. J. Physiol. Pharmacol. 2002, 46, 1–5. [Google Scholar]
- Sohal, R.S. Oxidative stress, caloric restriction, and aging. Science 1996, 273, 59–63. [Google Scholar]
- Valko, M.; Rhodes, C.J.; Moncol, J.; Izakovic, M.; Mazur, M. Free radicals, metals and antioxidants in oxidative stress-induced cancer. Chem. Biol. Interact. 2006, 160, 1–40. [Google Scholar] [CrossRef]
- Inoue, M.; Nishikawa, M.; Park, A.M.; Kira, Y.; Imada, I.; Utsumi, K. Mitochondrial generation of reactive oxygen species and its role in aerobic life. Curr. Med. Chem. 2003, 10, 2495–2505. [Google Scholar] [CrossRef]
- Parke, D.V. Chemical toxicity and reactive oxygen species. Int. J. Occup. Med. Environ. Health 1996, 9, 331–340. [Google Scholar]
- Waris, G.; Ahsan, H. Reactive oxygen species: Role in the development of cancer and various chronic conditions. J. Carcinog. 2006, 5, 1–8. [Google Scholar] [CrossRef]
- Ogasawara, M.A.; Zhang, H. Redox regulation and its emerging roles in stem cells and stem-like cancer cells. Antioxid. Redox Signal. 2009, 5, 1107–1122. [Google Scholar] [CrossRef]
- Nesti, C.; Pasquali, L.; Mancuso, M.; Siciliano, G. The Role of Mitochondria in StemCell Biology. In Stem Cell Biology and Regenerative Medicine Regulatory Networks in Stem Cells; Vinagolu, K.R., Mohan, C.V., Eds.; Memorial Sloan-Kettering Cancer Center: New York, NY, USA, 2009; pp. 135–143. [Google Scholar]
- Thomson, J.A.; Odorico, J.S. Human embryonic stem cell and embryonic germ cell lines. Trends Biotechnol. 2000, 18, 53–57. [Google Scholar] [CrossRef]
- Serakinci, N; Keith, N.W. Therapeutic potential of adult stem cells. Eur. J. Cancer 2006, 42, 1243–1246. [Google Scholar] [CrossRef]
- Jacks, T.; Weinberg, R.A. Cell-cycle control and its watchman. Nature 1996, 381, 643–644. [Google Scholar] [CrossRef]
- Sugimura, T. A new concept of co-mutagenicity from a phenomenon forgotten for the past two decades: Is it more important than previously expected? Environ. Health Perspect. 1998, 106, A522–A523. [Google Scholar] [CrossRef]
- Visvader, J.E.; Lindeman, G.J. Cancer stem cells in solid tumours: accumulating evidence and unresolved questions. Nat. Rev. Cancer 2008, 8, 755–768. [Google Scholar] [CrossRef]
- Eyler, C.E; Rich, J.N. Survival of the fittest: cancer stem cells in therapeutic resistance and angiogenesis. J. Clin. Oncol. 2008, 26, 2839–2845. [Google Scholar] [CrossRef]
- Li, X.; Lewi, M.T.; Huang, J.; Gutierrez, C.; Osborne, C.K.; Wu, M.; Hilsenbeck, S.G.; Pavlick, A.; Xiaomei Zhang, X.; Chamness, G.C.; Wong, H.; Rosen, J.; Chang, J.C. Intrinsic resistance of tumorigenic breast cancer cells to chemotherapy. J. Natl. Cancer Inst. 2008, 100, 672–679. [Google Scholar] [CrossRef]
- Bao, S.; Wu, Q.; McLendon, R.E.; Hao, Y.; Shi, Q.; Hjelmeland, A.B.; Dewhirst, M.W.; Bigner, D.D.; Rich, J.N. Glioma stem cells promote radioresistance by preferential activation of the DNA damage response. Nature 2006, 444, 756–760. [Google Scholar] [CrossRef]
- Dalerba, P.; Cho, R.W.; Clarke, M.F. Cancer stem cells: models and concepts. Annu. Rev. Med. 2007, 58, 267–284. [Google Scholar] [CrossRef]
- Wicha, M.S.; Liu, S.; Dontu, G. Cancer stem cells: an old idea—a paradigm shifts. Cancer Res. 2006, 66, 1883–1890. [Google Scholar] [CrossRef]
- Al-Hajj, M.; Wicha, M.S.; Benito-Hernandez, A.; Morrison, S.J.; Clarke, M.F. Prospective identification of tumorigenic breast cancer cells. Proc. Natl. Acad. Sci. USA 2003, 100, 3983–3988. [Google Scholar] [CrossRef]
- Collins, A.T.; Berry, P.A.; Hyde, C.; Michael, J.; Stower, M.J.; Maitland, N.J. Prospective identification of tumorigenic prostate cancer stem cells. Cancer Res. 2005, 65, 10946–10951. [Google Scholar] [CrossRef]
- Soltysova, A.; Altanerova, V.; Altaner, C. Cancer stem cells. Neoplasma 2005, 52, 435–440. [Google Scholar]
- Jones, R.J.; Matsui, W.H.; Smith, B.D. Cancer stem cells: are we missing the target? J. Natl. Cancer Inst. 2004, 96, 583–585. [Google Scholar] [CrossRef]
- Dontu, G.; Jackson, K.W.; McNicholas, E.; Kawamura, M.J.; Abdallah, W.M.; Wicha, M.S. Role of Notch signaling in cell-fate determination of human mammary stem/progenitor cells. BreastCancer Res. 2004, 6, R605–R615. [Google Scholar]
- Lowe, S.W.; Sherr, C.J. Tumor suppression by Ink4a-Arf: Progress and puzzles. Curr. Opin. Genet. Dev. 2003, 13, 77–83. [Google Scholar] [CrossRef]
- Hatsell, S.; Frost, A.R. Hedgehog signaling in mammary gland development and breast cancer. J. Mammary Gland Biol. Neoplasia 2007, 12, 163–173. [Google Scholar] [CrossRef]
- Lindvall, C.; Bu, W.; Williams, B.O. Wnt signaling, stem cells, and the cellular origin of breast cancer. Stem Cell Rev. 2007, 3, 157–168. [Google Scholar] [CrossRef]
- Zhou, J.; Wulfkuhle, J.; Zhang, H.; Gu, P.; Yang, Y.; Deng, J.; Margolick, J.B.; Liotta, L.A.; Petricoin , E.; Zhang, Y. Activation of the PTEN/mTOR/STAT3 pathway in breast cancer stem-like cells is required for viability and maintenance. Proc. Natl. Acad. Sci. USA 2007, 104, 16158–16163. [Google Scholar] [CrossRef]
- Prince, M.E.; Sivanandan, R.; Kaczorowski, A.; Wolf, G.T.; Kaplan, M.J.; Dalerba, P.; Weissman, I.L.; Clarke, M.F.; Ailles, L.E. Identification of a subpopulation of cells with cancer stem cell properties in head and neck squamous cell carcinoma. Proc. Natl. Acad. Sci. USA 2007, 104, 973–978. [Google Scholar] [CrossRef]
- Shmelkov, S.V.; St Clair, R.; Lyden, D.; Rafii, S. AC133/CD133/Prominin-1. Int. J. Biochem. Cell Biol. 2005, 37, 715–719. [Google Scholar] [CrossRef]
- Mimeault, M.; Batra, S.K. Recent advances in cancer stem/progenitor cell research: therapeutic implications for overcoming resistance to the most aggressive cancers. J. Cell. Mol. Med. 2007, 11, 981–1011. [Google Scholar] [CrossRef]
- Kai, K.; Arima, Y.; Kamiya, T.; Saya, H. Breast cancer stem cells. Breast Cancer 2009, 17, 80–85. [Google Scholar]
- Zhang, M.; Rosen, J.M. Stem cells in the etiology and treatment of cancer. Curr. Opin. Genet. Dev. 2006, 16, 60–64. [Google Scholar] [CrossRef]
- Sun, W.; Kang, K.S.; Morita, I.; Trosko, J.E.; Chang, C.C. High susceptibility of a human breast epithelial cell type with stem cell characteristics to telomerase activation and immortalization. Cancer Res. 1999, 59, 6118–6123. [Google Scholar]
- Benny, K.; Abraham, B.K.; Fritz, P.; McClellan, M.; Hauptvogel, P.; Athelogou, M.; Brauch, H. Prevalence of CD44+/CD24−/low cells in breast cancer may not be associated with clinical outcome but may favor distant metastasis. Clin. Cancer Res. 2005, 11, 1154–1159. [Google Scholar]
- Kritikou, E.A.; Sharkey, A.; Abell, K.; Came, P.J.; Anderson, E.; Clarkson, R.W.E.; Watson, C.J. A dual, non-redundant, role for LIF as a regulator of development and STAT3-mediated cell death in mammary gland. Development 2003, 130, 3459–3468. [Google Scholar] [CrossRef]
- Boulanger, C.A.; Wagner, K.U.; Smith, G.H. Parity-induced mouse mammary epithelial cells are pluripotent, self-renewing and sensitive to TGFb1 expression. Oncogene 2005, 24, 552–56. [Google Scholar] [CrossRef]
- Shipitsin, M.; Campbell, L.L.; Argani, P.; Weremowicz, S.; Bloushtain-Qimron, N.; Yao, J.; Nikolskaya, T.; Serebryiskaya, T.; Beroukhim, R.; Hu, M.; Halushka, M.K.; Saraswati, S.; Parker, L.M.; Anderson, K.S.; Lyndsay, N.; Harris, L.N.; Garber, J.E.; Richardson, A.L.; Schnitt, S.J.; Nikolsky, Y.; Gelman, R.S.; Polyak, K. Molecular definition of breast tumor heterogeneity. Cancer Cell 2007, 11, 259–273. [Google Scholar] [CrossRef]
- Azad, N.; Rojanasakul, Y.; Vallyathan, V. Inflammation and lung cancer: Roles of reactive oxygen/nitrogen species. J. Toxicol. Environ. Health B 2008, 11, 1–15. [Google Scholar] [CrossRef]
- Vercauteren, S.M.; Sutherland, H.J. CD133 (AC133) expression on AML cells and progenitors. Cytotherapy 2001, 3, 449–459. [Google Scholar] [CrossRef]
- Kurata, S. Selective activation of p38 MAPK cascade and mitotic arrest caused by low level oxidative stress. J. Biol. Chem. 2000, 275, 23413–23416. [Google Scholar] [CrossRef]
- Marshall, H.E.; Hess, D.T.; Stamler, J.S. S-Nitrosylation: Physiological regulation of NF-kappaB. Proc. Natl. Acad. Sci. USA 2004, 101, 8841–8842. [Google Scholar] [CrossRef]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38MAPkinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar]
- Demicco, E.G.; Kavanagh, K.T.; Romieu-Mourez, R.; Xiaobo Wang, X.; Shin, S.R.; Esther Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. RelB/p52 NF-kappaB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IkappaB-alpha expression and promote carcinogenesis of the mammary gland. Mol. Cell Biol. 2005, 25, 10136–10147. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, J.L.; Karin, M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15852–15857. [Google Scholar] [CrossRef]
- Kasper, M.; Jaks, V.; Marie Fiaschi, M.; Rune Toftgård, R. Hedgehog signalling in breast cancer. Carcinogenesis 2009, 30, 903–911. [Google Scholar] [CrossRef]
- Lam, J.S.; Reiter, R.E. Stem cells in prostate and prostate cancer development. Urol. Oncol. 2006, 24, 131–140. [Google Scholar] [CrossRef]
- Tang, D.G.; Patrawala, L.; Calhoun, T.; Bhatia, B.; Schneider-Broussard, R.; Choy, G.; Jeter, C. Prostate cancer stem/progenitor cells: identification, characterization and implications. Mol. Carcinogen 2007, 46, 1–14. [Google Scholar] [CrossRef]
- Liu, A.Y.; True, L.D.; LaTray, L.; Nelson, P.S.; Ellis, W.J.; Vessella, R.L.; Lange, P.H.; Hood, L.; Van Den Engh, G. Cell-cell interaction in prostate gene regulation and cytodifferentiation. Proc. Natl. Acad. Sci. USA 1997, 94, 10705–10710. [Google Scholar] [CrossRef]
- Patrawala, L.; Tang, D.G. CD44 as a functional cancer stem cell marker and therapeutic target. In Progress in Gene Therapy: Autologous and Cancer Stem Cell Gene Therapy; Bertolotti, R., Ozawka, K., Eds.; World Scientific: Hacxkensack, NJ, USA, 2008; Volume 3, pp. 317–334. [Google Scholar]
- Ricci-Vitiani, L.; Lombardi, D.G.; Pilozzi, E.; Biffoni, M.; Todaro, M.; Peschle, C.; De Maria, R. Identification and expansion of human colon-cancer-initiating cells. Nature 2007, 445, 111–115. [Google Scholar] [CrossRef]
- Reynolds, B.A.; Weiss, S. Generation of neurons and astrocytes from isolated cells of the adult mammalian central nervous system. Science 1992, 255, 1707–1710. [Google Scholar]
- Honeth, G.; Bendahl, P.; Ringnér, M.; Saal, L.H.; Gruvberger-Saal, S.K.; Lövgren, K.; Grabau, D.; Fernö, M.; Borg, A.; Hegardt, C. The CD44+/CD24− phenotype is enriched in basal-like breast tumors. Breast Cancer Res. 2008, 10, R53. [Google Scholar] [CrossRef]
- Bertucci, F.; Finetti, P.; Cervera, N.; Charafe-Jauffret, E.; Mamessier, E.; Adélaïde, J.; Debono, S.; Houvenaeghel, G.; Maraninchi, D.; Viens, P.; Charpin, C.; Jacquemier, J.; Birnbaum, D. Gene expression profiling of breast cell lines identifies potential new basal markers. Oncogene 2006, 6, 2273–2284. [Google Scholar]
- Mark, A.; La Barge, M.A.; Bissell, M.J. Is CD133 a marker of metastatic colon cancer stem cells? J. Clin. Invest. 2008, 118, 2021–2024. [Google Scholar]
- Singh, S.K.; Clarke, I.D.; Hide, T.; Dirks, P.B. Cancer stem cells in nervous system tumors. Oncogene 2004, 23, 7267–7273. [Google Scholar] [CrossRef]
- Vercauteren, S.M.; Sutherland, H.J. CD133 (AC133) expression on AML cells and progenitors. Cytotherapy 2001, 3, 449–459. [Google Scholar] [CrossRef]
- Yin, S.; Li, J.; Hu, C.; Chen, X.; Yao, M.; Yan, M.; Jiang, G.; Ge, C.; Xie, H.; Wan, D.; Yang, S.; Zheng, S.; Gu, J. CD133 positive hepatocellular carcinoma cells possess high capacity for tumorigenicity. Int. J. Cancer 2007, 120, 1444–1450. [Google Scholar] [CrossRef]
- Wright, M.H.; Calcagno, A.M.; Salcido, C.D.; Carlson, M.D.; Ambudkar, S.V.; Varticovski, L. Brca1 breast tumors contain distinct CD44+/CD24− and CD133+ cells with cancer stem cell characteristics. Breast Cancer Res. 2008, 10, R10. [Google Scholar] [CrossRef]
- Brantley, D.M.; Chen, C.L.; Muraoka, R.S.; Bushdid, P.B.; Bradberry, J.L.; Kittrell, F.; Medina, D.; Matrisian, L.M.; Kerr, L.D.; Yull, F.E. Nuclear factor-kappa B (NF-kappa B) regulates proliferation and branching in mouse mammary epithelium. Mol. Biol. Cell 2011, 12, 1445–1455. [Google Scholar]
- Demicco, E.G.; Kavanagh, K.T.; Romieu-Mourez, R.; Wang, X.; Shin, S.R.; Landesman-Bollag, E.; Seldin, D.C.; Sonenshein, G.E. RelB/p52 NF-kappaB complexes rescue an early delay in mammary gland development in transgenic mice with targeted superrepressor IkappaB-alpha expression and promote carcinogenesis of the mammary gland. Mol. Cell Biol. 2005, 25, 10136–10147. [Google Scholar] [CrossRef]
- Cao, Y.; Luo, J.L.; Karin, M. IkappaB kinase alpha kinase activity is required for self-renewal of ErbB2/Her2-transformed mammary tumor-initiating cells. Proc. Natl. Acad. Sci. USA 2007, 104, 15852–15857. [Google Scholar] [CrossRef]
- Liu, R.; Wang, X.; Chen, G.Y.; Dalerba, P.; Gurney, A.; Hoey, T.; Sherlock, G.; Lewicki, J.; Shedden, K.; Clarke, M.F. The prognostic role of a gene signature from tumorigenic breast-cancer cells. N. Engl. J. Med. 2007, 18, 217–226. [Google Scholar]
- Lam, J.S.; Reiter, R.E. Stem cells in prostate and prostate cancer development. Urol. Oncol. 2006, 24, 131–140. [Google Scholar]
- Klonisch, T.; Wiechec, E.; Hombach-Klonisch, S.; Ande, S.R.; Wesselborg, S.; Schulze-Osthoff, K.; Los, M. Cancer Stem cell markers in common cancers-therapeutic implications. Trends Mol. Med. 2008, 14, 450–460. [Google Scholar] [CrossRef]
- Takaishi, S.; Okumura, T.; Tu, S.; Wang, S.W.; Shibata, W.; Vigneshwaran, R.; Gordon, S.A.K.; Shimada, Y.; Wang, T.C. Identification of gastric cancer stem cells using the cell surface marker CD44. Stem Cells 2009, 27, 1006–1020. [Google Scholar] [CrossRef]
- Jang, Y.Y.; Sharkis, S.J. A low level of reactive oxygen species selects for primitive hematopoietic stem cells that may reside in the low-oxygenic niche. Blood 2007, 110, 3056–3063. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Matsuoka, S.; Takubo, K.; Hamaguchi, I.; Nomiyama, K.; Hosokawa, K.; Sakurada, K.; Nakagata, N.; Ikeda, Y.; Mak, T.W.; Suda, T. Regulation of oxidative stress by ATM is required for self-renewal of haematopoietic stem cells. Nature 2004, 431, 997–1002. [Google Scholar] [CrossRef]
- Diehn, M.; Cho, R.W.; Lobo, N.A.; Kalisky, T.; Dorie, M.J.; Kulp, A.N.; Qian, D.; Jessica S. Lam, J.S.; Ailles, L.E.; Wong, M.; Joshua, B.; Kaplan, M.J.; Wapnir, I.; Dirbas, F.M.; Somlo, G.; Garberoglio, C.; Paz, B.; Shen, J.; Lau, S.K.; Stephen R. Quake, S.R.; Brown, J.M.; Weissman, I.L.; Clarke, M.F. Association of reactive oxygen species levels and radioresistance in cancer stem cells. Nature 2009, 458, 780–783. [Google Scholar] [CrossRef]
- Szatrowski, T.P.; Nathan, C.F. Production of large amounts of hydrogen peroxide by human tumor cells. Cancer Res. 1991, 51, 794–798. [Google Scholar]
- Kawanishi, S.; Hiraku, Y.; Pinlaor, S.; Ma, N. Oxidative and nitrative DNA damage in animals and patients with inflammatory diseases in relation to inflammation-related carcinogenesis. Biol. Chem. 2006, 387, 365–372. [Google Scholar]
- Zhou, Y.; Hileman, E.O.; Plunkett, W.; Keating, M.J.; Huang, P. Free radical stress in chronic lymphocytic leukemia cells and its role in cellular sensitivity to ROS generating anticancer agents. Blood 2003, 101, 4098–4104. [Google Scholar] [CrossRef]
- Kamiguti, A.S; Serrander, L.; Lin, K.; Harris, R.J.; Cawley, J.C.; Allsup, D.J.; Slupsky, J.R.; Kraus, K.H.; Zuzel, M. Expression and activity of NOX5 in the circulating malignant B cells of hairy cell leukemia. J. Immunol. 2005, 175, 8424–8430. [Google Scholar]
- Oberley, T.D.; Oberley, L.W. Antioxidant enzyme levels in cancer. Histol. Histopathol. 1997, 12, 525–535. [Google Scholar]
- Hu, Y.; Rosen, D.G.; Zhou, Y.; Feng, L.; Yang, G.; Liu, J. L.; Huang, P. Mitochondrial manganese-superoxide dismutase expression in ovarian cancer: role in cell proliferation and response to oxidative stress. J. Biol. Chem. 2005, 280, 39485–39492. [Google Scholar]
- Saydama, N.; Kirb, A.; Demirb, Ö.; Hazanc, E.; Otoc, Ö; Saydama, O.; Güner, G. Determination of glutathione, glutathione reductase, glutathione peroxidase and glutathione S-transferase levels in human lung cancer tissues. Cancer 1997, 119, 13–19. [Google Scholar]
- Townsend, D.M.; Tew, K.D. The role of glutathione-S-transferase in anti-cancer drug resistance. Oncogene 2003, 22, 7369–7375. [Google Scholar] [CrossRef]
- Yadav, S.; Zajac, E.; Singhal, S.S.; Awasthi, S. Linking stress-signaling, glutathione metabolism, signaling pathways and xenobiotic transporter. Cancer Metastasis Rev. 2007, 26, 59–69. [Google Scholar] [CrossRef]
- Trachootham, D.; Zhang, H.; Zhang, W.; Feng, L.; Du, M.; Zhou, Y.; Chen, Z.; Pelicano, H.; Plunkett, W.; Wierda, W.G.; Keating, M.J.; Huang, P. Effective elimination of fludarabine-resistant CLL cells by PEITC through a redox-mediated mechanism. Blood 2008, 112, 1912–1922. [Google Scholar] [CrossRef]
- Zhang, H.; Trachootham, D.; Lu, W.; Carew, J.; Giles, F.J.; Keating, M.J.; Arlinghaus, R.B.; Huang, P. Effective killing of Gleevec-resistant CML cells with T315I mutation by a natural compound PEITC through redox-mediated mechanism. Leukemia 2008, 22, 1191–1199. [Google Scholar] [CrossRef]
- Naka, K.; Muraguchi, T.; Hoshii, T.; Hirao, A. Regulation of reactive oxygen species and genomic stability in hematopoietic stem cells. Antioxid.Redox Signal. 2008, 10, 1883–1894. [Google Scholar] [CrossRef]
- Toyokuni, S. Novel aspects of oxidative stress associated carcinogenesis. Antioxid. Redox Signal. 2006, 8, 1373–1377. [Google Scholar] [CrossRef]
- Ghaffari, S. Oxidative stress in the regulation of normal and neoplastic hematopoiesis. Antioxid. RedoxSignal. 2008, 10, 1923–1940. [Google Scholar] [CrossRef]
- Lockshin, R.A.; Zakeri, Z. Apoptosis, autophagy, and more. Inter. J. Biochem. Cell Biol. 2004, 36, 2405–2419. [Google Scholar] [CrossRef]
- Schwartzman, R.A.; Cidlowski, L.A. Apoptosis: the biochemistry and molecular biology of programmed cell death. Endocrine Rev. 1993, 14, 133–151. [Google Scholar]
- Ashkenazi, A.; Dixit, V.M. Death receptors: signaling and modulation. Science 1998, 281, 1305–1308. [Google Scholar] [CrossRef]
- Gorman, A.; McGowan, A.; Cotter, T.G. Role of peroxide and superoxide anion during tumour cell apoptosis. FEBSLett. 1997, 404, 27–33. [Google Scholar] [CrossRef]
- Meyer, M.; Schreck, R.; Baeuerle, P.A. H2O2 and antioxidants have opposite effects on activation of NF-kappa B and AP-1 in intact cells: AP-1 as secondary anti-oxidant responsive factor. EMBO J. 2005, 12, 2005–2015. [Google Scholar]
- Gottlieb, E.; Vander Heiden, M.G.; Thompson, C.B. Bcl-x(L) prevents the initial decrease in mitochondrial membrane potential and subsequent reactive oxygen species production during tumor necrosis factor alpha-induced apoptosis. Mol. Cell Biol. 2000, 20, 5680–5689. [Google Scholar] [CrossRef]
- Price, R.; Vugt, M.V.; Nosten, F.; Luxemburger, C.; Brockman, A.; Phaipun, L.; Chongsuphajaisiddhi, T.; White, N. Artesunate versus artemether for the treatment of recrudescent multidrugresistant Plasmodium falciparum malaria. Am. J. Trop. Med. 1998, 59, 883–888. [Google Scholar]
- Ribeiro, I.R.; Ollario, P. Safety of artemisinin and its derivatives. A review of published and unpublished clinical trials. Med. Trop. (Mars) 1998, 58, 50–53. [Google Scholar]
- Adjuik, M.; Babiker, A.; Garner, P.; Olliato, P.; Taylor, W.; White, N. International Artemisinin Study Group. Artesunate combinations for treatment of malaria: meta-analysis. Lancet 2004, 363, 9–17. [Google Scholar] [CrossRef]
- Efferth, T. Molecular pharmacology and pharmacogenomics of artemisinin and its derivatives in cancer cells. Curr. Drug Targets 2006, 7, 407–421. [Google Scholar] [CrossRef]
- Schulze-Bergkamen, H.; Krar, P.H. Apoptosis in cancer–implications for therapy. Semin. Oncol. 2004, 31, 90–119. [Google Scholar] [CrossRef]
- Debatin, K.M.; Krammer, P.H. Death receptors in chemotherapy and cancer. Oncogene 2004, 23, 2950–2966. [Google Scholar] [CrossRef]
- Gordi, T.; Lepist, E.I. Artemisinin derivatives: toxic for laboratory animals, safe for humans? Toxicol. Lett. 2004, 147, 539–545. [Google Scholar]
- Dell’Eva, R.; Pfeffer, U.; Vene, R.; Anfosso, L.; Forlani, A.; Albini, A.; Efferth, T. Inhibition of angiogenesis in vivo and growth of Kaposi’s sarcoma xenograft tumors by the anti-malarial artesunate. Biochem. Pharmacol. 2004, 68, 2359–2366. [Google Scholar] [CrossRef]
- Orrenius, S. Mitochondrial regulation of apoptotic cell death. Toxicol. Lett. 2004, 149, 19–23. [Google Scholar] [CrossRef]
- Muller, I.; Niethammer, D.; Bruchelt, G. Anthracycline-derived chemotherapeutics in apoptosis and free radical Cytotoxicity. Int. J. Mol. Med. 1998, 1, 491–494. [Google Scholar]
- Efferth, T.; Giaisi, M.; Merling, A.; Krammer, P.H.; Li-Weber, M. Artesunate Induces ROS-Mediated Apoptosis in Doxorubicin-Resistant T Leukemia Cells. PLoS ONE 2007, 2, e693. [Google Scholar]
- Eskelinen, E.L. New insights into the mechanisms of macroautophagy in Mammalian cells. Inter. Rev.Cell Mol. Biol. 2008, 266, 207–247. [Google Scholar] [CrossRef]
- Galluzzi, L.; Maiuri, M.C.; Vitale, I.; Zischka, H.; Castedo, M.; Zitvogel, L.; Kroemer, G. Cell death modalities: classification and pathophysiological implications. Cell Death Differ. 2007, 14, 1237–1243. [Google Scholar] [CrossRef]
- Kihara, A.; Noda, T.; Ishihara, N.; Ohsumi, Y. Two distinct vps34 phosphatidylinositol-3-kinase complexes function in autophagy and carboxypeptidase Y sorting in Saccharomyces cerevisiae. J. Cell Biol. 2001, 152, 519–530. [Google Scholar] [CrossRef]
- Ohsumi, Y. Molecular dissection of autophagy: two ubiquitin-like systems. Nat. Rev. Mol. Cell Biol. 2001, 2, 211–216. [Google Scholar] [CrossRef]
- Shintani, T.; Klionsky, D.J. Autophagy in Health and Disease: A Double-Edged Sword. Science 2004, 306, 990–995. [Google Scholar] [CrossRef]
- Cuervo, A.M. Autophagy: In sickness and in health. Trends Cell Biol. 2004, 14, 70–77. [Google Scholar] [CrossRef]
- Levine, B. Eating Oneself and Uninvited Guests: Minireview Autophagy-Related Pathways in Cellular Defense. Cell 2005, 120, 159–162. [Google Scholar]
- Tu, B.P.; Weissman, J.S. Oxidative protein folding in eukaryotes: mechanisms and consequences. J. Cell Biol. 2004, 164, 341–346. [Google Scholar] [CrossRef]
- Fariss, M.W.; Chan, C.B.; Patel, M.; Van Houten, B.; Orrenius, S. Role of mitochondria in toxic oxidative stress. Mol. Inter. 2005, 5, 94–111. [Google Scholar]
- Wang, S.H.; Shih, Y.L.; Ko, W.C.; Wei, Y.H.; Shih, C.M. Cadmium-induced autophagy and apoptosis are mediated by a calcium signaling pathway. Cell. Mol. Life Sci. 2008, 65, 3640–3652. [Google Scholar] [CrossRef]
- Wang, S.H.; Shih, Y.L.; Kuo, T.C; Ko, W.C.; Shih, C.M. Cadmium Toxicity toward Autophagy through ROS-Activated GSK-3b in Mesangial Cells. Toxicol. Sci. 2009, 108, 124–131. [Google Scholar] [CrossRef]
- Gozuacik, D.; Kimchi, A. Autophagy as a cell death and tumor suppressor mechanism. Oncogene 2004, 23, 2891–2906. [Google Scholar] [CrossRef]
- Levine, B.; Yuan, J. Autophagy in cell death: an innocent convict? J. Clin. Invest. 2005, 115, 2679–2688. [Google Scholar] [CrossRef]
- Li, N.; Ragheb, K.; Lawler, G.; Sturgis, J.; Rajwa, B.; Melendez, J.A.; Robinson, J.P. Mitochondrial complex I inhibitor rotenone induces apoptosis through enhancing mitochondrial reactive oxygen species production. J. Biol. Chem. 2003, 278, 8516–8525. [Google Scholar]
- Pelicano, H.; Carney, D.; Huang, P. ROS stress in cancer cells and therapeutic implications. Drug Resist. Updat. 2004, 7, 97–110. [Google Scholar] [CrossRef]
- Scherz-Shouval, R.; Shvets, E.; Fass, E.; Shorer, H.; Gil, L.; Elazar, Z. Reactive oxygen species are essential for autophagy and specifically regulate the activity of Atg4. EMBO J. 2007, 26, 1749–1760. [Google Scholar] [CrossRef]
- Yu, L.; Wan, F.; Dutta, S.; Welsh, S.; Liu, Z.; Freundt, E.; Baehrecke, E.H.; Lenardo, M. Autophagic programmed cell death by selective catalase degradation. Proc. Natl. Acad. Sci. USA 2006, 103, 4952–4957. [Google Scholar]
- Reynolds, B.A.; Tetzlaff, W.; Weiss, S. A multipotent EGF-responsive striatal embryonic progenitor cell produces neurons and astrocytes. J. Neurosci. 1992, 12, 4565–4574. [Google Scholar]
- Jacks, T.; Weinberg, R.A. Cell-cycle control and its watchman. Nature 1996, 381, 643–644. [Google Scholar] [CrossRef]
- Sugimura, T. A new concept of co-mutagenicity from a phenomenon forgotten for the past two decades: Is it more important than previously expected? Environ. Health Perspect. 1998, 106, A522–A523. [Google Scholar] [CrossRef]
- Norman, J.; Cristina Cellurale, K.; Davis, R.J. A Radical Role for p38 MAPK in Tumor Initiation. Cancer Cell 2007, 11, 101–103. [Google Scholar] [CrossRef]
- Martindale, J.L.; Holbrook, N.J. Cellular response to oxidative stress: signaling for suicide and survival. J. Cell. Physiol. 2002, 192, 1–15. [Google Scholar] [CrossRef]
- Chatterjee, S.; Fisher, A.B. ROS to the rescue. Am. J. Physiol. Lung Cell Mol. Physiol. 2004, 287, L704–L705. [Google Scholar] [CrossRef]
- Kurata, S. Selective activation of p38 MAPK cascade and mitotic arrest caused by low level oxidative stress. J. Biol. Chem. 2000, 275, 23413–23416. [Google Scholar] [CrossRef]
- Marshall, H.E.; Hess, D.T.; Stamler, J.S. S-Nitrosylation: Physiological regulation of NF-kappaB. Proc. Natl. Acad. Sci. USA 2004, 101, 8841–8842. [Google Scholar] [CrossRef]
- Xia, Z.; Dickens, M.; Raingeaud, J.; Davis, R.J.; Greenberg, M.E. Opposing effects of ERK and JNK-p38MAPkinases on apoptosis. Science 1995, 270, 1326–1331. [Google Scholar]
- Wang, D.; Kreutzer, D.A.; Esigmann, J.M. Mutagenicity and repair of oxidative DNA damage: insights from studies using defined lesions. Mutat. Res. 1998, 400, 99–115. [Google Scholar] [CrossRef]
- Bours, V.; Bentires-Alj, M.; Hellin, A-C.; Viatour, P.; Robe, P.; Delhalle, S.; Benoit, V.; Merville, M-P. Nuclear factor-κB, cancer, and apoptosis. Biochem. Pharmacol. 1999, 60, 1085–1090. [Google Scholar]
- Kennedy, N.J.; Davis, R.J. Role of JNK in tumor development. Cell Cycle 2003, 2, 199–201. [Google Scholar]
- Bulavin, D.V.; Phillips, C.; Nannenga, B.; Timofeev, O.; Donehower, L.A.; Anderson, C.W.; Appella, E.; Fornace, A.J., Jr. Inactivation of the Wip1 phosphatase inhibits mammary tumorigenesis through p38 MAPK-mediated activation of the p16(Ink4a)-p19(Arf) pathway. Nat. Genet. 2004, 36, 343–350. [Google Scholar] [CrossRef]
- Ivanova, N.B.; Dimos, J.T.; Schaniel, C.; Hackney, J.A.; Moore, K.A.; Lemischka, I.R. A stem cell molecular signature. Science 2002, 298, 601–604. [Google Scholar] [CrossRef]
- Kunduzova, O.R.; Bianchi, P.; Pizzinat, N.; Escourrou, G.; Seguelas, M.H.; Parini, A.; Cambon, C. Regulation of JNK/ERK activation, cell apoptosis, and tissue regeneration by monoamine oxidases after renal ischemia-reperfusion. FASEB J. 2002, 16, 1129–1131. [Google Scholar]
- Morel, A.P.; Marjory Lievre, M.; Thomas, C.; Hinkal, G.; Ansieau, S.; Alain Puisieux, A. Generation of Breast Cancer Stem Cells through Epithelial-Mesenchymal Transition. PLoS ONE 2008, 3, 1–7. [Google Scholar]
- Das, B.; Tsuchida, R.; Baruchel, S.; Malkin, D.; Yeger, H. The role of VEGF/Flt1 signaling in the maintenance of neuroblastoma side-population celll “stemness” during hypoxia. In Advances in Neuroblastoma Research meeting, Los Angeles, CA, USA, 17–20 May 2006.
- Das, B. The role of VEGF autocrine signaling in hypoxia and oxidative stress driven “stemness switch”: implications in solid tumor progression and metastasis. PhD thesis, Institute of Medical Sciences, University of Toronto, Canada, 2007. [Google Scholar]
- Tsuchida, R.; Das, B.; Yeger, H.; Koren, G.; Shibuya, M.; Thorner, P.S.; Baruchel, S.; Malkin, D. Cisplatin treatment increases survival and expansion of a highly tumorigenic side-population fraction by upregulating VEGF/Flt1 autocrine signaling. Oncogene 2008, 27, 3923–3934. [Google Scholar] [CrossRef]
- Kunath, T.; Saba-El-Leil, M.; Almousailleakh, M.; Wray, J.; Meloche, S.; Smith, A. FGF stimulation of the Erk1/2 signalling cascade triggers transition of pluripotent embryonic stem cells from self-renewal to lineage commitment. Development 2007, 134, 2895–2902. [Google Scholar] [CrossRef]
- Ito, K.; Hirao, A.; Arai, F.; Takubo, K.; Matsuoka, S.; Miyamoto, K.; Ohmura, M.; Naka, K.; Hosokawa, K.; Ikeda, Y.; Suda, T. Reactive oxygen species act through p38 MAPK to limit the lifespan of hematopoietic stem cells. Nat. Med. 2006, 12, 446–451. [Google Scholar] [CrossRef]
- Bonnet, D.; Dick, J.E. Human acute myeloid leukemia is organized as a hierarchy that originates from a primitive hematopoietic cell. Nat. Med. 1997, 3, 730–737. [Google Scholar] [CrossRef]
- Cook, J.A.; Giu, D.; Wink, D.A.; Krishna, M.C.; Russo, A.; Mitchell, J.B. Oxidative stress, redox, and the tumor microenvironment. Semin. Radiat. Oncol. 2004, 14, 259–266. [Google Scholar] [CrossRef]
- Azad, N.; Rojanasakul, Y.; Vallyathan, V. Inflammation and lung cancer: Roles of reactive oxygen/nitrogen species. J. Toxicol. Environ. Health B 2008, 11, 1–15. [Google Scholar]
- Dobrovolskaia, M.A.; Kozlov, S.V. Inflammation and cancer: when NF-kappaB amalgamates the perilous partnership. Curr. Cancer Drug Targets 2005, 5, 5325–5344. [Google Scholar]
- Karin, M. Nuclear factor-κB in cancer development and progression. Nature 2006, 441, 431–436. [Google Scholar] [CrossRef]
- Drost, J.; Agami, R. Transformation Locked in a Loop. Cell 2009, 139, 654–656. [Google Scholar] [CrossRef]
- Klaunig, J.E.; Kamendulis, L.M. The Role of Oxidative Stress in Carcinogenesis. Ann. Rev. Pharmacol. Toxicol. 2004, 44, 239–267. [Google Scholar] [CrossRef]
- Kaltschmidt, B.; Kaltschmidt, C. NF-kB in the nervous system. Beyaert, R., Ed.; Kluwer Academic Publishers: Norwell, MA, USA, 2003; pp. 375–394. [Google Scholar]
- Denk, A.; Wirth, T.; Baumann, B. NF-kappaB transcription factors: critical regulators of hematopoiesis and neuronal survival. Cytokine Growth Factor Rev. 2000, 11, 303–320. [Google Scholar] [CrossRef]
- Qiu, P.; Pan, P.C.; Govind, S. A role for the Drosophila Toll/Cactus pathway in larval hematopoiesis. Development 1998, 125, 1909–1920. [Google Scholar]
- Ghosh, S.; May, M.J.; Kopp, E.B. NF-kappa B and Rel proteins: evolutionarily conserved mediators of immune responses. Annu. Rev. Immunol. 1998, 16, 225–260. [Google Scholar] [CrossRef]
- Liou, H.C.; Baltimore, D. Regulation of the NF-kappa B/rel transcription factor and I kappa B inhibitor system. Curr. Opin. Cell Biol. 1993, 5, 477–487. [Google Scholar] [CrossRef]
- Kang, H.B.; Kim, Y.E.; Kwon, H.J.; Sok, D.E.; Lee, Y. Enhancement of NF kappaB expression and activity upon differentiation of human embryonic stem cell line SNUhES3. Stem Cells Dev. 2007, 16, 615–623. [Google Scholar] [CrossRef]
- Molofsky, A.V.; Pardal, R.; Iwashita, T.; Park, I.K.; Clarke, M.F.; Morrison, S. J. Bmi-1 dependence distinguishes neural stem cell self-renewal from progenitor proliferation. Nature 2003, 425, 962–967. [Google Scholar] [CrossRef]
- Lessard, J.; Sauvageau, G. Bmi-1 determines the proliferative capacity of normal and leukaemic stem cells. Nature 2003a, 423, 255–260. [Google Scholar] [CrossRef]
- Park, I.K.; Qian, D.; Kiel, M.; Becker, M.W.; Pihalja, M.; Weissman, I.L.; Morrison, S.J.; Clarke, M.F. Bmi-1 is required for maintenance of adult self-renewing haematopoietic stem cells. Nature 2003, 423, 302–305. [Google Scholar] [CrossRef]
- Sawa, M.; Yamamoto, K.; Yokozawa, T.; Kiyoi, H.; Hishida, A.; Kajiguchi, T.; Seto, M.; Kohno, A.; Kitamura, K.; Itoh, Y.; Asou, N.; Hamajima, N.; Emi, N.; Naoe, T. BMI-1 is highly expressed in M0-subtype acute myeloid leukemia. Int. J. Hematol. 2005, 82, 42–47. [Google Scholar] [CrossRef]
- Androutsellis-Theotokis, A.; Leker, R.R.; Soldner, F.; Hoeppner, D.J.; Ravin, R.; Poser, S.W.; Rueger, M.A.; Bae, S.K.; Kittappa, R.; McKay, R.D.G. Notch signalling regulates stem cell numbers in vitro and in vivo. Nature 2006, 442, 823–826. [Google Scholar] [CrossRef]
- Lobo, N.A.; Shimono, Y.; Qian, D.; Clarke, M. F. The Biology of Cancer Stem Cells. Annu. Rev. Cell Dev. Biol. 2007, 23, 675–699. [Google Scholar] [CrossRef]
- Taipale, J.; Beachy, P.A. The Hedgehog and Wnt signaling pathways in cancer. Nature 2001, 411, 349–354. [Google Scholar] [CrossRef]
- Reya, T.; Duncan, A.W.; Ailles, L.; Domen, J.; Scherer, D.C.; Willert, K.; Hintz, L.; Nusse, R.; Weissman, I.L. A role for Wnt signaling in self-renewal of hematopoietic stem cells. Nature 2003, 423, 409–414. [Google Scholar] [CrossRef]
- Reguart, N.; He, B.; Taron, M.; You, L.; Jablons, D.M.; Rosell, R. The role of Wnt signaling in cancer and stem cells. Fut. Oncol. 2005, 1, 787–797. [Google Scholar] [CrossRef]
- Zhang, J.; Grindley, J.C.; Yin, T.; Jayasinghe, S.; He, X.C.; Ross, J.T.; Haug, J.S.; Rupp, D.; Porter-Westpfahl, K.S.; Wiedemann, L.M.; Wu, H.; Li, L. PTEN maintains haematopoietic stem cells and acts in lineage choice and leukaemia prevention. Nature 2006, 441, 518–522. [Google Scholar] [CrossRef]
- Chow, L.M.; Baker, S.J. PTEN function in normal and neoplastic growth. Cancer Lett. 2006, 241, 184–196. [Google Scholar] [CrossRef]
- Leung, C.; Lingbeek, M.; Shakhova, O.; Liu, J.; Tanger, E.; Saremaslani, P.; Van Lohuizen, M.; Marino, S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Nature 2004, 428, 337–341. [Google Scholar] [CrossRef]
- Athar, M.; Tang, X.; Lee, J.L.; Kopelovich, L.; Kim, A.L. Hedgehog signalling in skin development and cancer. Exp. Dermatol. 2006, 15, 667–677. [Google Scholar] [CrossRef]
- Palma, V.; Lim, D.A.; Dahmane, N.; Sanchez, P.; Brionne, T.C.; Herzberg, C.D.; Gitton, Y.; Carleton, A.; Alvarez-Buylla, A.; Ruiz i Altaba, A. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development 2005, 132, 335–344. [Google Scholar] [CrossRef]
- Ruizi Altaba, A.; Sanchez, P.; Dahmane, N. Gli and hedgehog in cancer: tumours, embryos and stem cells. Nat. Rev. Cancer 2002, 2, 361–372. [Google Scholar] [CrossRef]
- Roche, J.; Zeng, C.; Barón, A; Gadgil, S.; Gemmill, R.M.; Tigaud, I.; Thomas, X.; Drabkin, H.A. Hox expression in AML identifies a distinct subset of patients with intermediate cytogenetics. Leukemia 2004, 18, 1059–1063. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Dayem, A.A.; Choi, H.-Y.; Kim, J.-H.; Cho, S.-G. Role of Oxidative Stress in Stem, Cancer, and Cancer Stem Cells. Cancers 2010, 2, 859-884. https://doi.org/10.3390/cancers2020859
Dayem AA, Choi H-Y, Kim J-H, Cho S-G. Role of Oxidative Stress in Stem, Cancer, and Cancer Stem Cells. Cancers. 2010; 2(2):859-884. https://doi.org/10.3390/cancers2020859
Chicago/Turabian StyleDayem, Ahmed Abdal, Hye-Yeon Choi, Jung-Hyun Kim, and Ssang-Goo Cho. 2010. "Role of Oxidative Stress in Stem, Cancer, and Cancer Stem Cells" Cancers 2, no. 2: 859-884. https://doi.org/10.3390/cancers2020859