Platelet Proteome and Tumor Dormancy: Can Platelets Content Serve as Predictive Biomarkers for Exit of Tumors from Dormancy?

Abstract
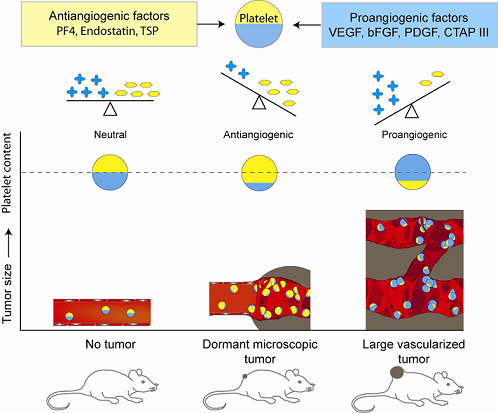
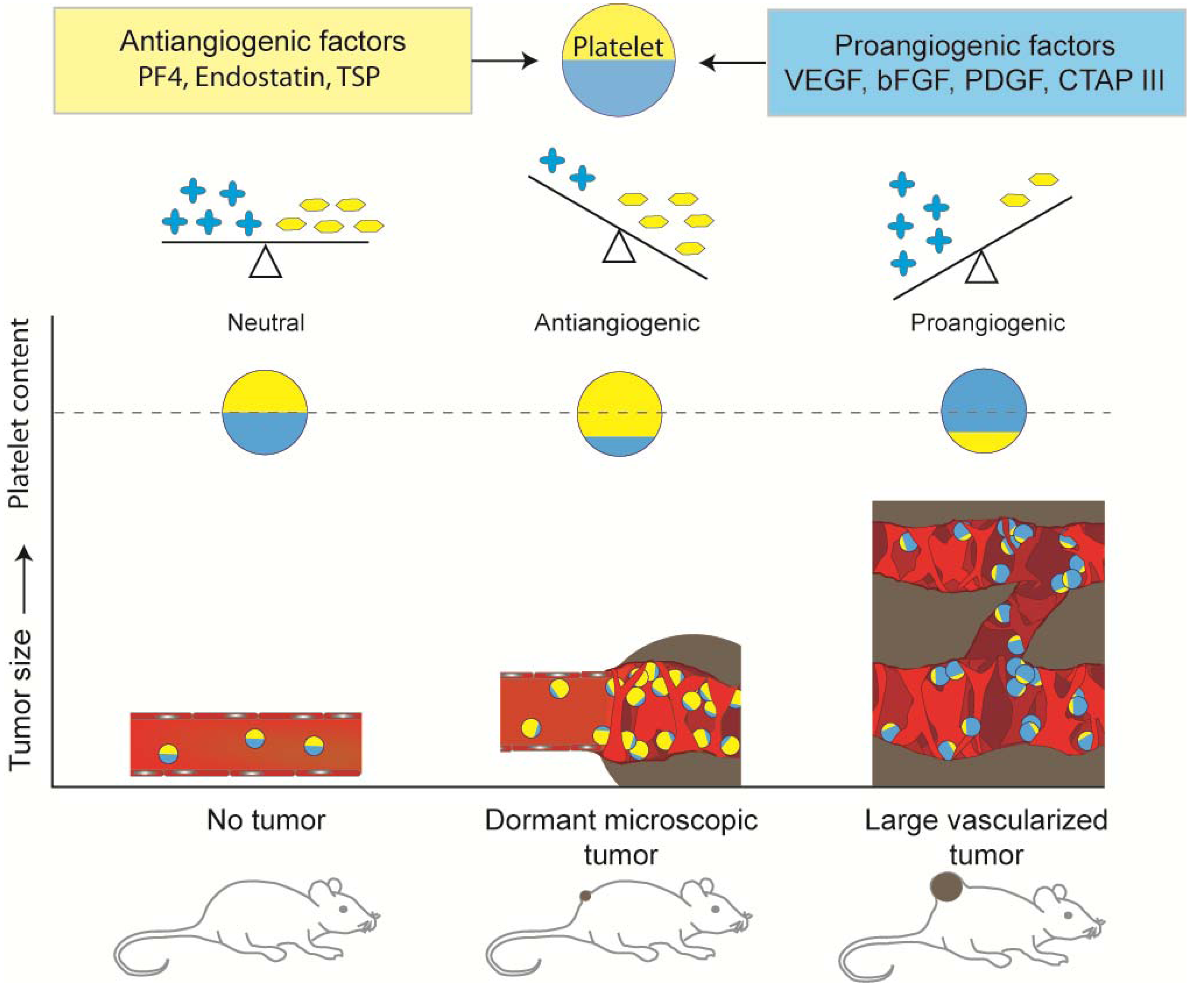
:
{kind=link}
{kind=link}
1. Introduction
2. Tumor Dormancy
3. Induction of Angiogenesis as a Decision Point
4. Platelets and Their Role in Tumor Biology
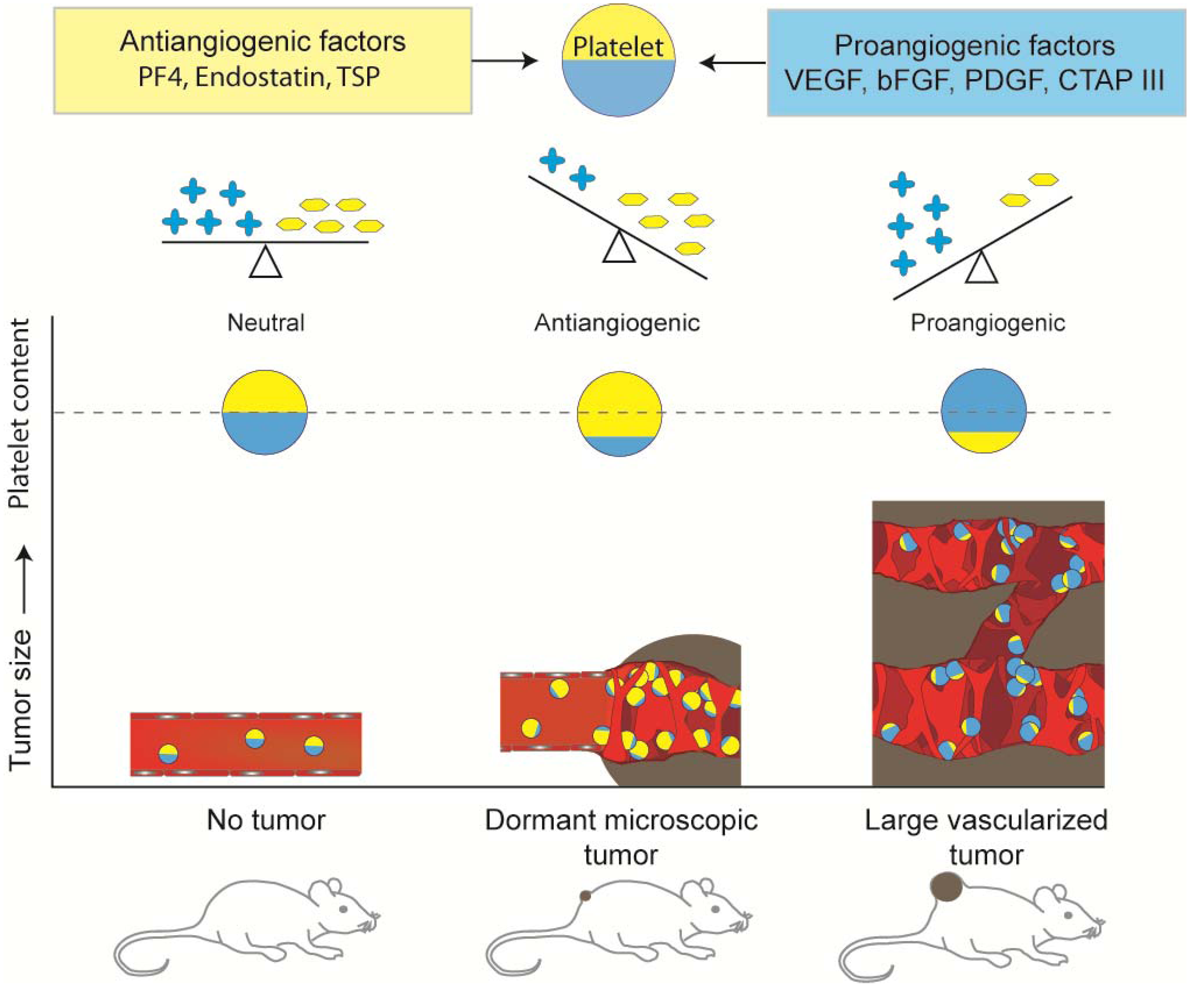
5. Platelet Protein Content as a Biomarker of Early Tumor Presence
6. Conclusions
Acknowledgments
References
- Almog, N.; Henke, V.; Flores, L.; Hlatky, L.; Kung, A.L.; Wright, R.D.; Berger, R.; Hutchinson, L.; Naumov, G.N.; Bender, E.; Akslen, L.A.; Achilles, E.G.; Folkman, J. Prolonged dormancy of human liposarcoma is associated with impaired tumor angiogenesis. FASEB J. 2006, 20, 947–949. [Google Scholar] [CrossRef]
- Naumov, G.N.; Bender, E.; Zurakowski, D.; Kang, S.Y.; Sampson, D.; Flynn, E.; Watnick, R.S.; Straume, O.; Akslen, L.A.; Folkman, J.; Almog, N. A model of human tumor dormancy: an angiogenic switch from the nonangiogenic phenotype. J. Natl. Cancer Inst. 2006, 98, 316–325. [Google Scholar] [CrossRef]
- Almog, N.; Ma, L.; Raychowdhury, R.; Schwager, C.; Erber, R.; Short, S.; Hlatky, L.; Vajkoczy, P.; Huber, P.E.; Folkman, J.; Abdollahi, A. Transcriptional switch of dormant tumors to fast-growing angiogenic phenotype. Cancer Res. 2009, 69, 836–844. [Google Scholar] [CrossRef]
- Cervi, D.; Yip, T.T.; Bhattacharya, N.; Podust, V.N.; Peterson, J.; Abou-Slaybi, A.; Naumov, G.N.; Bender, E.; Almog, N.; Italiano, J.E., Jr.; Folkman, J.; Klement, G.L. Platelet-associated PF-4 as a biomarker of early tumor growth. Blood 2008, 111, 1201–1207. [Google Scholar]
- Klement, G.L.; Yip, T.T.; Cassiola, F.; Kikuchi, L.; Cervi, D.; Podust, V.; Italiano, J.E.; Wheatley, E.; Abou-Slaybi, A.; Bender, E.; Almog, N.; Kieran, M.W.; Folkman, J. Platelets actively sequester angiogenesis regulators. Blood 2009, 113, 2835–2842. [Google Scholar] [CrossRef]
- Black, W.C.; Welch, H.G. Advances in diagnostic imaging and overestimations of disease prevalence and the benefits of therapy. N. Engl. J. Med. 1993, 328, 1237–1243. [Google Scholar] [CrossRef]
- Brackstone, M.; Townson, J.L.; Chambers, A.F. Tumour dormancy in breast cancer: an update. Breast Cancer Res. 2007, 9, 208. [Google Scholar] [CrossRef]
- Folkman, J.; Kalluri, R. Cancer without disease. Nature 2004, 427, 787. [Google Scholar] [CrossRef]
- Nielsen, M.; Thomsen, J.L.; Primdahl, S.; Dyreborg, U.; Andersen, J.A. Breast cancer and atypia among young and middle-aged women: a study of 110 medicolegal autopsies. Br. J. Cancer 1987, 56, 814–819. [Google Scholar] [CrossRef]
- Hart, I.R. Perspective: tumour spread--the problems of latency. J. Pathol. 1999, 187, 91–94. [Google Scholar] [CrossRef]
- Harach, H.R.; Franssila, K.O.; Wasenius, V.M. Occult papillary carcinoma of the thyroid. A "normal" finding in Finland. A systematic autopsy study. Cancer 1985, 56, 531–538. [Google Scholar] [CrossRef]
- Hedley, B.D.; Chambers, A.F. Tumor dormancy and metastasis. Adv. Cancer Res. 2009, 102, 67–101. [Google Scholar] [CrossRef]
- Wikman, H.; Vessella, R.; Pantel, K. Cancer micrometastasis and tumour dormancy. APMIS 2008, 116, 754–770. [Google Scholar] [CrossRef]
- Retsky, M.; Demicheli, R.; Hrushesky, W. Breast cancer screening for women aged 40–49 years: screening may not be the benign process usually thought. J. Natl. Cancer Inst. 2001, 93, 1572. [Google Scholar]
- Baum, M.; Demicheli, R.; Hrushesky, W.; Retsky, M. Does surgery unfavourably perturb the "natural history" of early breast cancer by accelerating the appearance of distant metastases? Eur. J. Cancer 2005, 41, 508–515. [Google Scholar] [CrossRef]
- Demicheli, R.; Retsky, M.W.; Hrushesky, W.J.; Baum, M. Tumor dormancy and surgery-driven interruption of dormancy in breast cancer: learning from failures. Nat. Clin. Pract. Oncol. 2007, 4, 699–710. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. Models, mechanisms and clinical evidence for cancer dormancy. Nat. Rev. Cancer 2007, 7, 834–846. [Google Scholar] [CrossRef]
- Demicheli, R.; Terenziani, M.; Valagussa, P.; Moliterni, A.; Zambetti, M.; Bonadonna, G. Local recurrences following mastectomy: support for the concept of tumor dormancy. J. Natl. Cancer Inst. 1994, 86, 45–48. [Google Scholar] [CrossRef]
- Achilles, E.G.; Fernandez, A.; Allred, E.N.; Kisker, O.; Udagawa, T.; Beecken, W.D.; Flynn, E.; Folkman, J. Heterogeneity of angiogenic activity in a human liposarcoma: a proposed mechanism for "no take" of human tumors in mice. J. Natl. Cancer Inst. 2001, 93, 1075–1081. [Google Scholar] [CrossRef]
- Folkman, J. Endogenous angiogenesis inhibitors. APMIS 2004, 112, 496–507. [Google Scholar] [CrossRef]
- Gimbrone, M.A., Jr.; Leapman, S.B.; Cotran, R.S.; Folkman, J. Tumor dormancy in vivo by prevention of neovascularization. J. Exp. Med. 1972, 136, 261–276. [Google Scholar]
- Hahnfeldt, P.; Panigrahy, D.; Folkman, J.; Hlatky, L. Tumor development under angiogenic signaling: a dynamical theory of tumor growth, treatment response, and postvascular dormancy. Cancer Res. 1999, 59, 4770–4775. [Google Scholar]
- Holmgren, L.; O'Reilly, M.S.; Folkman, J. Dormancy of micrometastases: balanced proliferation and apoptosis in the presence of angiogenesis suppression. Nat. Med. 1995, 1, 149–153. [Google Scholar] [CrossRef]
- Naumov, G.N.; Folkman, J.; Straume, O. Tumor dormancy due to failure of angiogenesis: role of the microenvironment. Clin. Exp. Metastasis 2009, 26, 51–60. [Google Scholar] [CrossRef]
- O'Reilly, M.S.; Holmgren, L.; Chen, C.; Folkman, J. Angiostatin induces and sustains dormancy of human primary tumors in mice. Nat. Med. 1996, 2, 689–692. [Google Scholar] [CrossRef]
- Udagawa, T.; Fernandez, A.; Achilles, E.G.; Folkman, J.; D'Amato, R.J. Persistence of microscopic human cancers in mice: alterations in the angiogenic balance accompanies loss of tumor dormancy. FASEB J. 2002, 16, 1361–1370. [Google Scholar] [CrossRef]
- Indraccolo, S.; Favaro, E.; Amadori, A. Dormant tumors awaken by a short-term angiogenic burst: the spike hypothesis. Cell Cycle 2006, 5, 1751–1755. [Google Scholar] [CrossRef]
- Aguirre-Ghiso, J.A. The problem of cancer dormancy: understanding the basic mechanisms and identifying therapeutic opportunities. Cell Cycle 2006, 5, 1740–1743. [Google Scholar] [CrossRef]
- Barkan, D.; Kleinman, H.; Simmons, J.L.; Asmussen, H.; Kamaraju, A.K.; Hoenorhoff, M.J.; Liu, Z.Y.; Costes, S.V.; Cho, E.H.; Lockett, S.; Khanna, C.; Chambers, A.F.; Green, J.E. Inhibition of metastatic outgrowth from single dormant tumor cells by targeting the cytoskeleton. Cancer Res. 2008, 68, 6241–6250. [Google Scholar] [CrossRef]
- Uhr, J.W.; Marches, R. Dormancy in a model of murine B cell lymphoma. Semin. Cancer Biol. 2001, 11, 277–283. [Google Scholar] [CrossRef]
- Koebel, C.M.; Vermi, W.; Swann, J.B.; Zerafa, N.; Rodig, S.J.; Old, L.J.; Smyth, M.J.; Schreiber, R.D. Adaptive immunity maintains occult cancer in an equilibrium state. Nature 2007, 450, 903–907. [Google Scholar] [CrossRef]
- Soucek, L.; Lawlor, E.R.; Soto, D.; Shchors, K.; Swigart, L.B.; Evan, G.I. Mast cells are required for angiogenesis and macroscopic expansion of Myc-induced pancreatic islet tumors. Nat. Med. 2007, 13, 1211–1218. [Google Scholar] [CrossRef]
- Quesnel, B. Tumor dormancy and immunoescape. APMIS 2008, 116, 685–694. [Google Scholar] [CrossRef]
- Horak, C.E.; Lee, J.H.; Marshall, J.C.; Shreeve, S.M.; Steeg, P.S. The role of metastasis suppressor genes in metastatic dormancy. APMIS 2008, 116, 586–601. [Google Scholar] [CrossRef]
- Chambers, A.F. Influence of diet on metastasis and tumor dormancy. Clin. Exp. Metastasis 2009, 26, 61–66. [Google Scholar] [CrossRef]
- Gewirtz, D.A. Autophagy, senescence and tumor dormancy in cancer therapy. Autophagy 2009, 5, 1232–1234. [Google Scholar] [CrossRef]
- Lu, Z.; Luo, R.Z.; Lu, Y.; Zhang, X.; Yu, Q.; Khare, S.; Kondo, S.; Kondo, Y.; Yu, Y.; Mills, G.B.; Liao, W.S.; Bast, R.C., Jr. The tumor suppressor gene ARHI regulates autophagy and tumor dormancy in human ovarian cancer cells. J. Clin. Invest. 2008, 118, 3917–3929. [Google Scholar]
- Folkman, J. Angiogenesis in cancer, vascular, rheumatoid and other disease. Nat. Med. 1995, 1, 27–31. [Google Scholar] [CrossRef]
- Folkman, J. The role of angiogenesis in tumor growth. Semin. Cancer Biol. 1992, 3, 65–71. [Google Scholar]
- Folkman, J. Tumor angiogenesis: therapeutic implications. N. Engl. J. Med. 1971, 285, 1182–1186. [Google Scholar] [CrossRef]
- Hanahan, D.; Folkman, J. Patterns and emerging mechanisms of the angiogenic switch during tumorigenesis. Cell 1996, 86, 353–364. [Google Scholar] [CrossRef]
- Moserle, L.; Amadori, A.; Indraccolo, S. The angiogenic switch: implications in the regulation of tumor dormancy. Curr. Mol. Med. 2009, 9, 935–941. [Google Scholar] [CrossRef]
- Bayko, L.; Rak, J.; Man, S.; Bicknell, R.; Ferrara, N.; Kerbel, R.S. The dormant in vivo phenotype of early stage primary human melanoma: termination by overexpression of vascular endothelial growth factor. Angiogenesis 1998, 2, 203–217. [Google Scholar] [CrossRef]
- Arbiser, J.L.; Moses, M.A.; Fernandez, C.A.; Ghiso, N.; Cao, Y.; Klauber, N.; Frank, D.; Brownlee, M.; Flynn, E.; Parangi, S.; Byers, H.R.; Folkman, J. Oncogenic H-ras stimulates tumor angiogenesis by two distinct pathways. Proc. Natl. Acad. Sci. USA 1997, 94, 861–866. [Google Scholar] [CrossRef]
- Cao, Y.; O'Reilly, M.S.; Marshall, B.; Flynn, E.; Ji, R.W.; Folkman, J. Expression of angiostatin cDNA in a murine fibrosarcoma suppresses primary tumor growth and produces long-term dormancy of metastases. J. Clin. Invest. 1998, 101, 1055–1063. [Google Scholar]
- Gilead, A.; Meir, G.; Neeman, M. The role of angiogenesis, vascular maturation, regression and stroma infiltration in dormancy and growth of implanted MLS ovarian carcinoma spheroids. Int. J. Cancer 2004, 108, 524–531. [Google Scholar] [CrossRef]
- Tang, Y.; Wang, M.T.; Chen, Y.; Yang, D.; Che, M.; Honn, K.V.; Akers, G.D.; Johnson, S.R.; Nie, D. Downregulation of vascular endothelial growth factor and induction of tumor dormancy by 15-lipoxygenase-2 in prostate cancer. Int. J. Cancer 2009, 124, 1545–1551. [Google Scholar] [CrossRef]
- Udagawa, T. Tumor dormancy of primary and secondary cancers. APMIS 2008, 116, 615–628. [Google Scholar] [CrossRef]
- Watnick, R.S.; Cheng, Y.N.; Rangarajan, A.; Ince, T.A.; Weinberg, R.A. Ras modulates Myc activity to repress thrombospondin-1 expression and increase tumor angiogenesis. Cancer Cell 2003, 3, 219–231. [Google Scholar] [CrossRef]
- Kang, S.Y.; Watnick, R.S. Regulation of tumor dormancy as a function of tumor-mediated paracrine regulation of stromal Tsp-1 and VEGF expression. APMIS 2008, 116, 638–647. [Google Scholar] [CrossRef]
- Pietramaggiori, G.; Scherer, S.S.; Cervi, D.; Klement, G.; Orgill, D.P. Tumors stimulate platelet delivery of angiogenic factors in vivo: an unexpected benefit. Am. J. Pathol. 2008, 173, 1609–1616. [Google Scholar] [CrossRef]
- Handagama, P.; Rappolee, D.A.; Werb, Z.; Levin, J.; Bainton, D.F. Platelet alpha-granule fibrinogen, albumin, and immunoglobulin G are not synthesized by rat and mouse megakaryocytes. J. Clin. Invest. 1990, 86, 1364–1368. [Google Scholar] [CrossRef]
- Italiano, J.E., Jr.; Richardson, J.L.; Patel-Hett, S.; Battinelli, E.; Zaslavsky, A.; Short, S.; Ryeom, S.; Folkman, J.; Klement, G.L. Angiogenesis is regulated by a novel mechanism: pro- and antiangiogenic proteins are organized into separate platelet alpha granules and differentially released. Blood 2008, 111, 1227–1233. [Google Scholar]
- Pinedo, H.M.; Verheul, H.M.; D'Amato, R.J.; Folkman, J. Involvement of platelets in tumour angiogenesis? Lancet 1998, 352, 1775–1777. [Google Scholar] [CrossRef]
- Gasic, G.J. Role of plasma, platelets, and endothelial cells in tumor metastasis. Cancer Metastasis Rev. 1984, 3, 99–114. [Google Scholar] [CrossRef]
- Gasic, G.J.; Gasic, T.B.; Galanti, N.; Johnson, T.; Murphy, S. Platelet-tumor-cell interactions in mice. The role of platelets in the spread of malignant disease. Int. J. Cancer 1973, 11, 704–718. [Google Scholar] [CrossRef]
- Nierodzik, M.L.; Karpatkin, S. Thrombin induces tumor growth, metastasis, and angiogenesis: Evidence for a thrombin-regulated dormant tumor phenotype. Cancer Cell 2006, 10, 355–362. [Google Scholar] [CrossRef]
- Tang, N.; Tornatore, P.; Weinberger, S.R. Current developments in SELDI affinity technology. Mass Spectrom. Rev. 2004, 23, 34–44. [Google Scholar] [CrossRef]
- Seibert, V.; Wiesner, A.; Buschmann, T.; Meuer, J. Surface-enhanced laser desorption ionization time-of-flight mass spectrometry (SELDI TOF-MS) and ProteinChip technology in proteomics research. Pathol. Res. Pract. 2004, 200, 83–94. [Google Scholar] [CrossRef]
- Dvorak, H.F. Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N. Engl. J. Med. 1986, 315, 1650–1659. [Google Scholar] [CrossRef]
- Ma, L.; Perini, R.; McKnight, W.; Dicay, M.; Klein, A.; Hollenberg, M.D.; Wallace, J.L. Proteinase-activated receptors 1 and 4 counter-regulate endostatin and VEGF release from human platelets. Proc. Natl. Acad. Sci. USA 2005, 102, 216–220. [Google Scholar] [CrossRef]
- Ma, L.; Elliott, S.N.; Cirino, G.; Buret, A.; Ignarro, L.J.; Wallace, J.L. Platelets modulate gastric ulcer healing: role of endostatin and vascular endothelial growth factor release. Proc. Natl. Acad. Sci. USA 2001, 98, 6470–6475. [Google Scholar]
- Ashford, T.P.; Freiman, D.G. Platelet aggregation at sites of minimal endothelial injury. An electron microscopic study. Am. J. Pathol. 1968, 53, 599–607. [Google Scholar]
- Cleator, J.H.; Zhu, W.Q.; Vaughan, D.E.; Hamm, H.E. Differential regulation of endothelial exocytosis of P-selectin and von Willebrand factor by protease-activated receptors and cAMP. Blood 2006, 107, 2736–2744. [Google Scholar] [CrossRef]
- Heijnen, H.F.; Debili, N.; Vainchencker, W.; Breton-Gorius, J.; Geuze, H.J.; Sixma, J.J. Multivesicular bodies are an intermediate stage in the formation of platelet alpha-granules. Blood 1998, 91, 2313–2325. [Google Scholar]
- Ma, L.; Hollenberg, M.D.; Wallace, J.L. Thrombin-induced platelet endostatin release is blocked by a proteinase activated receptor-4 (PAR4) antagonist. Br. J. Pharmacol. 2001, 134, 701–704. [Google Scholar] [CrossRef]
- Perini, R.; Wallace, J.L.; Ma, L. Roles of platelets and proteinase-activated receptors in gastric ulcer healing. Dig. Dis. Sci. 2005, 50 Suppl. 1, S12–S15. [Google Scholar] [CrossRef]
- Mohle, R.; Green, D.; Moore, M.A.; Nachman, R.L.; Rafii, S. Constitutive production and thrombin-induced release of vascular endothelial growth factor by human megakaryocytes and platelets. Proc. Natl. Acad. Sci. USA 1997, 94, 663–668. [Google Scholar]
- Brill, A.; Dashevsky, O.; Rivo, J.; Gozal, Y.; Varon, D. Platelet-derived microparticles induce angiogenesis and stimulate post-ischemic revascularization. Cardiovasc Res. 2005, 67, 30–38. [Google Scholar] [CrossRef]
- Brill, A.; Elinav, H.; Varon, D. Differential role of platelet granular mediators in angiogenesis. Cardiovasc Res. 2004, 63, 226–235. [Google Scholar] [CrossRef]
- Pintucci, G.; Froum, S.; Pinnell, J.; Mignatti, P.; Rafii, S.; Green, D. Trophic effects of platelets on cultured endothelial cells are mediated by platelet-associated fibroblast growth factor-2 (FGF-2) and vascular endothelial growth factor (VEGF). Thromb. Haemost. 2002, 88, 834–842. [Google Scholar]
- Adams, J.; Carder, P.J.; Downey, S.; Forbes, M.A.; MacLennan, K.; Allgar, V.; Kaufman, S.; Hallam, S.; Bicknell, R.; Walker, J.J.; Cairnduff, F.; Selby, P.J.; Perren, T.J.; Lansdown, M.; Banks, R.E. Vascular endothelial growth factor (VEGF) in breast cancer: comparison of plasma, serum, and tissue VEGF and microvessel density and effects of tamoxifen. Cancer Res. 2000, 60, 2898–2905. [Google Scholar]
- Fuhrmann-Benzakein, E.; Ma, M.N.; Rubbia-Brandt, L.; Mentha, G.; Ruefenacht, D.; Sappino, A.P.; Pepper, M.S. Elevated levels of angiogenic cytokines in the plasma of cancer patients. Int. J. Cancer 2000, 85, 40–45. [Google Scholar] [CrossRef]
- George, M.L.; Eccles, S.A.; Tutton, M.G.; Abulafi, A.M.; Swift, R.I. Correlation of plasma and serum vascular endothelial growth factor levels with platelet count in colorectal cancer: clinical evidence of platelet scavenging? Clin. Cancer Res. 2000, 6, 3147–3152. [Google Scholar]
- Lee, J.K.; Hong, Y.J.; Han, C.J.; Hwang, D.Y.; Hong, S.I. Clinical usefulness of serum and plasma vascular endothelial growth factor in cancer patients: which is the optimal specimen? Int. J. Oncol. 2000, 17, 149–152. [Google Scholar]
- Nguyen, M. Angiogenic factors as tumor markers. Invest. New Drugs 1997, 15, 29–37. [Google Scholar] [CrossRef]
- Wynendaele, W.; Derua, R.; Hoylaerts, M.F.; Pawinski, A.; Waelkens, E.; de Bruijn, E.A.; Paridaens, R.; Merlevede, W.; van Oosterom, A.T. Vascular endothelial growth factor measured in platelet poor plasma allows optimal separation between cancer patients and volunteers: a key to study an angiogenic marker in vivo? Ann. Oncol. 1999, 10, 965–971. [Google Scholar] [CrossRef]
- Dosquet, C.; Coudert, M.C.; Lepage, E.; Cabane, J.; Richard, F. Are angiogenic factors, cytokines, and soluble adhesion molecules prognostic factors in patients with renal cell carcinoma? Cli. Cancer Res. 1997, 3, 2451–2458. [Google Scholar]
- Abendstein, B.; Daxenbichler, G.; Windbichler, G.; Zeimet, A.G.; Geurts, A.; Sweep, F.; Marth, C. Predictive value of uPA, PAI-1, HER-2 and VEGF in the serum of ovarian cancer patients. Anticancer Res. 2000, 20, 569–572. [Google Scholar]
- Wong, A.K.; Alfert, M.; Castrillon, D.H.; Shen, Q.; Holash, J.; Yancopoulos, G.D.; Chin, L. Excessive tumor-elaborated VEGF and its neutralization define a lethal paraneoplastic syndrome. Proc. Natl. Acad. Sci. USA 2001, 98, 7481–7486. [Google Scholar]
- Gonzalez, F.J.; Rueda, A.; Sevilla, I.; Alonso, L.; Villarreal, V.; Torres, E.; Alba, E. Shift in the balance between circulating thrombospondin-1 and vascular endothelial growth factor in cancer patients: relationship to platelet alpha-granule content and primary activation. Int. J. Biol. Markers 2004, 19, 221–228. [Google Scholar]
- Spence, G.M.; Graham, A.N.; Mulholland, K.; McAllister, I.; Sloan, J.M.; Armstrong, M.A.; Campbell, F.C.; McGuigan, J.A. Vascular endothelial growth factor levels in serum and plasma following esophageal cancer resection--relationship to platelet count. Int. J. Biol. Markers 2002, 17, 119–124. [Google Scholar]
- Verheul, H.M.; Hoekman, K.; Luykx-de Bakker, S.; Eekman, C.A.; Folman, C.C.; Broxterman, H.J.; Pinedo, H.M. Platelet: transporter of vascular endothelial growth factor. Clin. Cancer Res. 1997, 3, 2187–2190. [Google Scholar]
- Akerblom, B.; Lindahl, T.L.; Larsson, A. ADP activation induces bFGF binding to platelets in vitro. Ups. J. Med. Sci. 2002, 107, 165–171. [Google Scholar] [CrossRef]
- Eddahibi, S.; Humbert, M.; Sediame, S.; Chouaid, C.; Partovian, C.; Maitre, B.; Teiger, E.; Rideau, D.; Simonneau, G.; Sitbon, O.; Adnot, S. Imbalance between platelet vascular endothelial growth factor and platelet-derived growth factor in pulmonary hypertension. Effect of prostacyclin therapy. Am. J. Respir. Crit. Care Med. 2000, 162, 1493–1499. [Google Scholar] [CrossRef]
- Solanilla, A.; Villeneuve, J.; Auguste, P.; Hugues, M.; Alioum, A.; Lepreux, S.; Ducroix, J.P.; Duhaut, P.; Conri, C.; Viallard, J.F.; Nurden, A.T.; Constans, J.; Ripoche, J. The transport of high amounts of vascular endothelial growth factor by blood platelets underlines their potential contribution in systemic sclerosis angiogenesis. Rheumatology (Oxford) 2009, 48, 1036–1044. [Google Scholar] [CrossRef]
- Peterson, J.; Zurakowski, D.; Italiano, J.; Michel, L.; Fox, L.; Klement, G.; Folkman, J. Normal ranges of angiogenesis regulatory proteins in human platelets. Am. J. Hematol. 2010, in press. [Google Scholar]
© 2010 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Almog, N.; Klement, G.L. Platelet Proteome and Tumor Dormancy: Can Platelets Content Serve as Predictive Biomarkers for Exit of Tumors from Dormancy? Cancers 2010, 2, 842-858. https://doi.org/10.3390/cancers2020842
Almog N, Klement GL. Platelet Proteome and Tumor Dormancy: Can Platelets Content Serve as Predictive Biomarkers for Exit of Tumors from Dormancy? Cancers. 2010; 2(2):842-858. https://doi.org/10.3390/cancers2020842
Chicago/Turabian StyleAlmog, Nava, and Giannoula Lakka Klement. 2010. "Platelet Proteome and Tumor Dormancy: Can Platelets Content Serve as Predictive Biomarkers for Exit of Tumors from Dormancy?" Cancers 2, no. 2: 842-858. https://doi.org/10.3390/cancers2020842