Management of Melanoma Families

Abstract
:1. Introduction to the Clinical Phenotype and Diagnostic Criteria
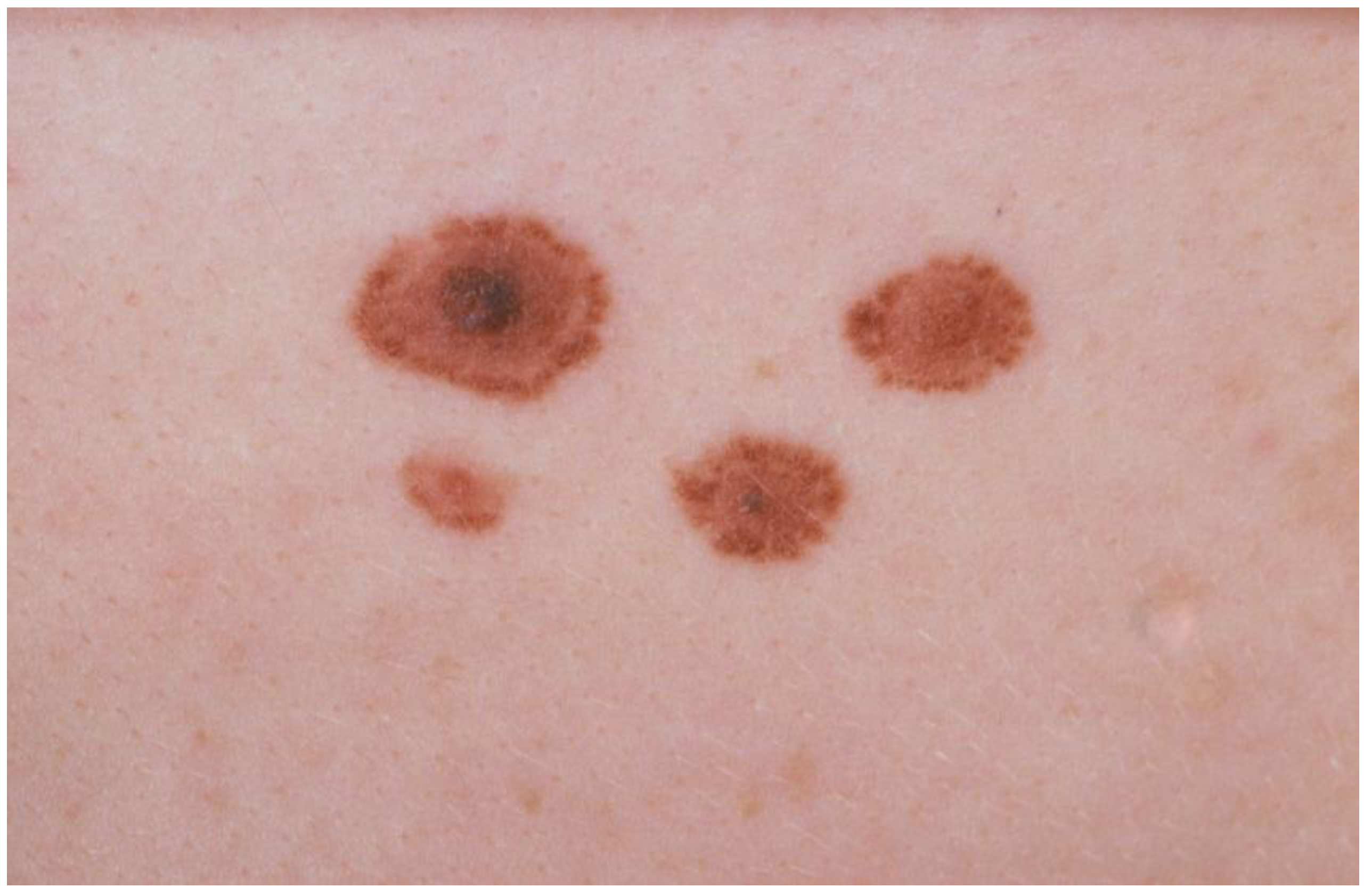
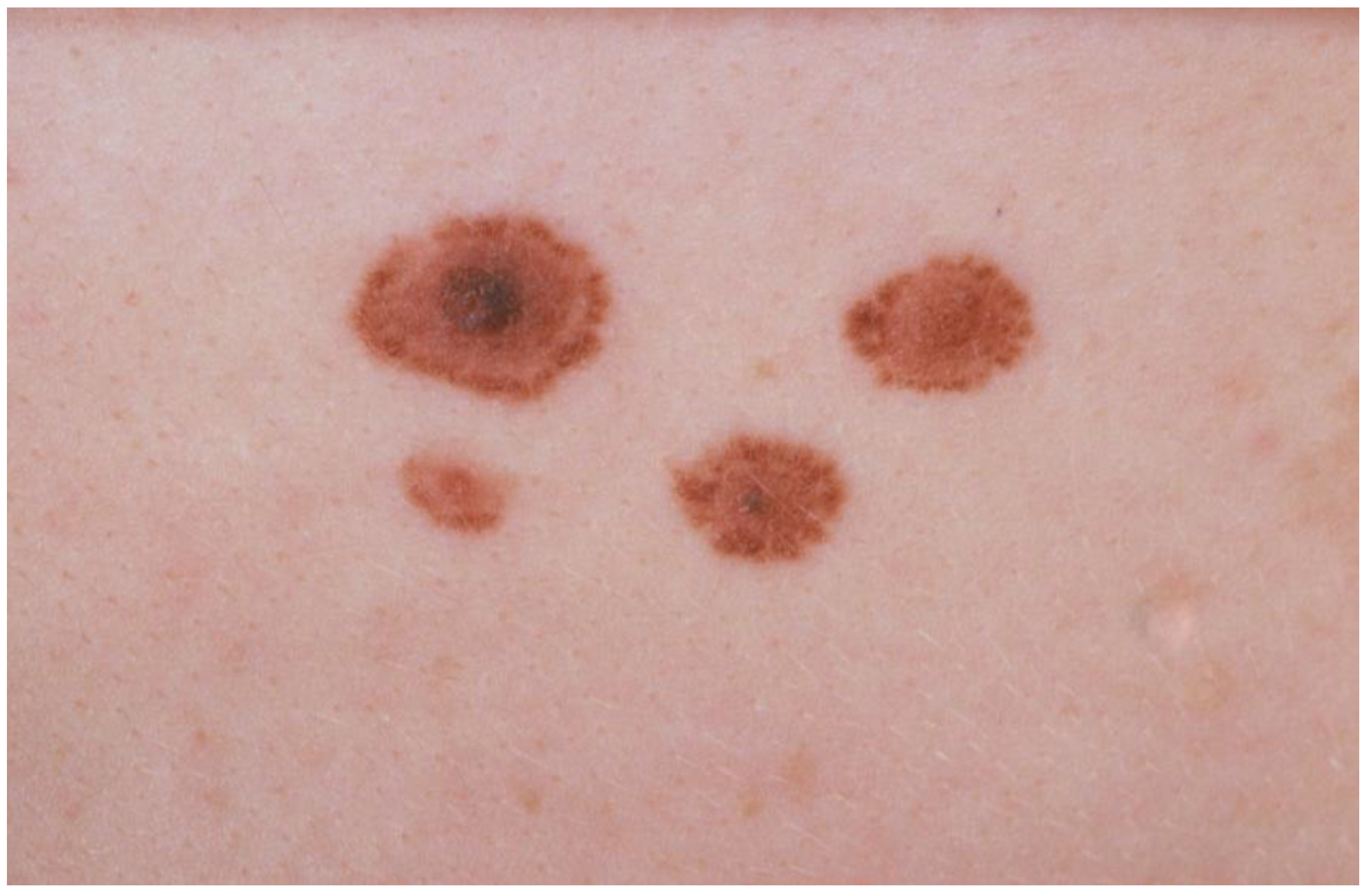
2. Management of FAMMM Families: Practical Guidelines and Review of the Evidence Thereof

{kind=link}
| Other family members with melanoma? |
|
| Youngest age at diagnosis of melanoma in the family | ……….. years |
| Multiple primary melanomas in the family | Yes / No |
| Other cancer types in the family? | Pancreatic carcinoma Yes / No Other: ………………….. |
| Atypical nevi in the patient? | Yes / No |
| Atypical nevi in first-degree relatives? | Yes / No |
| Skin type I and red hair in the family? | Yes / No |
| Reported cancer cases verified by medical documentation? | Yes / No |
| Referral to Clinical Geneticist | Yes / No |
| Likelihood of finding mutations in this family | Low / High |
3. Efficacy of Screening/Surveillance of FAMMM Families
| # hits | Relevant after screening title | Minus duplicate titles | Relevant after screening abstract | |
|---|---|---|---|---|
| Search 1 | 153 | 54 | 37 | 25 |
| Search 2 | 46 | 32 | 17 | 14 |
| Search 3 | 146 | 62 | 30 | 17 |
| Total | 345 | 148 | 84 | 56 |
| Titles on management of familial melanoma | N = 56 | References: |
| Reviews/guidelines | N = 25 | [12,16,17,18,19,20,21,22,23,24,25,26,27,28,29,30,31,323334,35,36,37,38,39] |
| National reports/founder populations | N = 9 | [40,41,42,43,44,45,46,47,48] |
| Regarding (preventive) DNA testing | N = 8 | [49,50,51,52,53,54,55,56] |
| Regarding pancreatic carcinoma (risk/screening) | N = 5 | [57,58,59,60,61] |
| Clinical features: sun sensitivity, early onset, age, metastasis, body site, survival, MC1R variants | N = 5 | [62,63,64,65,66] |
| Management/screening | N = 4 | [4,67,68,69] |
4. FAMMM: Phenotype and Genetic Variability
5. High Risk Melanoma Gene Search Reflects Complexity
6. Conclusions
References
- Ford, D.; Bliss, J.M.; Swerdlow, A.J.; Armstrong, B.K.; Franceschi, S.; Green, A.; Holly, E.A.; Mack, T.; MacKie, R.M.; Osterlind, A. Risk of cutaneous melanoma associated with a family history of the disease. The International Melanoma Analysis Group (IMAGE). Int. J. Cancer 1995, 62, 377–381. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Tucker, M.A. Genetic epidemiology of familial melanoma. Dermatol. Clin. 1995, 13, 605–6123. [Google Scholar]
- Lynch, H.T.; Frichot, B.C.; Lynch, J.F. Familial atypical multiple mole-melanoma syndrome. J. Med. Genet. 1978, 15, 352–356. [Google Scholar] [CrossRef]
- Tucker, M.A.; Fraser, M.C.; Goldstein, A.M.; Struewing, J.P.; King, M.A.; Crawford, J.T.; Chiazze, E.A.; Zametkin, D.P.; Fontaine, L.S.; Clark, W.H., Jr. A natural history of melanomas and dysplastic nevi: an atlas of lesions in melanoma-prone families. Cancer 2002, 94, 3192–3209. [Google Scholar] [CrossRef]
- Bishop, J.A.; Wachsmuth, R.C.; Harland, M.; Bataille, V.; Pinney, E.; Mack, P.; Baglietto, L.; Cuzick, J.; Bishop, D.T. Genotype/phenotype and penetrance studies in melanoma families with germline CDKN2A mutations. J. Invest. Dermatol. 2000, 114, 28–33. [Google Scholar] [CrossRef]
- Clark, W.H., Jr.; Tucker, M.A. Problems with lesions related to the development of malignant melanoma: common nevi, dysplastic nevi, malignant melanoma in situ, and radial growth phase malignant melanoma. Hum. Pathol. 1998, 29, 8–14. [Google Scholar] [CrossRef]
- Rabkin, M.S. The limited specificity of histological examination in the diagnosis of dysplastic nevi. J. Cutan. Pathol. 2008, 35, 20–23. [Google Scholar] [CrossRef]
- Hussein, M.R. Melanocytic dysplastic naevi occupy the middle ground between benign melanocytic naevi and cutaneous malignant melanomas: emerging clues. J. Clin. Pathol. 2005, 58, 453–456. [Google Scholar] [CrossRef]
- Miller, A.J.; Mihm, M.C., Jr. Melanoma. N. Engl. J. Med. 2006, 355, 51–65. [Google Scholar] [CrossRef]
- Vasen, H.F.; Bergman, W.; van Haeringen, A.; Scheffer, E.; van Slooten, E.A. The familial dysplastic nevus syndrome. Natural history and the impact of screening on prognosis. A study of nine families in the Netherlands. Eur. J. Cancer Clin. Oncol. 1989, 25, 337–341. [Google Scholar] [CrossRef]
- Masri, G.D.; Clark, W.H., Jr.; Guerry, D.; Halpern, A.; Thompson, C.J.; Elder, D.E. Screening and surveillance of patients at high risk for malignant melanoma result in detection of earlier disease. J. Am. Acad. Dermatol. 1990, 22, 1042–1048. [Google Scholar] [CrossRef]
- de Snoo, F.A.; Bergman, W.; Gruis, N.A. Familial melanoma: a complex disorder leading to controversy on DNA testing. Fam. Cancer 2003, 2, 109–116. [Google Scholar] [CrossRef]
- Halpern, A.C.; Marghoob, A.A.; Bialoglow, T.W.; Witmer, W.; Slue, W. Standardized positioning of patients (poses) for whole body cutaneous photography. J. Am. Acad. Dermatol. 2003, 49, 593–598. [Google Scholar] [CrossRef]
- Bafounta, M.L.; Beauchet, A.; Aegerter, P.; Saiag, P. Is dermoscopy (epiluminescence microscopy) useful for the diagnosis of melanoma? Results of a meta-analysis using techniques adapted to the evaluation of diagnostic tests. Arch. Dermatol. 2001, 137, 1343–1350. [Google Scholar] [CrossRef]
- Whiteman, D.C.; Whiteman, C.A.; Green, A.C. Childhood sun exposure as a risk factor for elanoma: a systematic review of epidemiologic studies. Cancer Causes Contr. 2001, 12, 69–82. [Google Scholar] [CrossRef]
- Hershock, D. Genetics, prevention and screening for melanoma. Cancer Chemother. Biol. Response Modif. 2005, 22, 707–728. [Google Scholar] [CrossRef]
- Hayward, N. New developments in melanoma genetics. Curr. Oncol. Rep. 2000, 2, 300–306. [Google Scholar] [CrossRef]
- Kefford, R.F.; Mann, G.J. Is there a role for genetic testing in patients with melanoma? Curr. Opin. Oncol. 2003, 15, 157–161. [Google Scholar] [CrossRef]
- Rivers, J.K. Is there more than one road to melanoma? Lancet 2004, 363, 728–730. [Google Scholar] [CrossRef]
- Lynch, H.T.; Fusaro, R.M.; Lynch, J.F. Hereditary cancer syndrome diagnosis: molecular genetic clues and cancer control. Future Oncol. 2007, 3, 169–181. [Google Scholar] [CrossRef]
- Bishop, J.N.; Harland, M.; Randerson-Moor, J.; Bishop, D.T. Management of familial melanoma. Lancet Oncol. 2007, 8, 46–54. [Google Scholar] [CrossRef]
- Dalle, S.; Martin-Denavit, T.; Thomas, L. Genotypic hypervariability of melanoma: a therapeutic challenge. Med. Sci. 2006, 22, 178–182. [Google Scholar]
- Lange, J.R.; Balch, C.M. Screening for cutaneous melanoma. Surg. Oncol. Clin. N. Am. 2005, 14, 799–811. [Google Scholar] [CrossRef]
- Tsao, H.; Niendorf, K. Genetic testing in hereditary melanoma. J. Am. Acad. Dermatol. 2004, 51, 803–808. [Google Scholar] [CrossRef]
- Hansen, C.B.; Wadge, L.M.; Lowstuter, K.; Boucher, K.; Leachman, S.A. Clinical germline genetic testing for melanoma. Lancet Oncol. 2004, 5, 314–319. [Google Scholar] [CrossRef]
- Czajkowski, R.; Placek, W.; Drewa, G.; Czajkowska, A.; Uchanska, G. FAMMM syndrome: pathogenesis and management. Dermatol. Surg. 2004, 30, 291–296. [Google Scholar] [CrossRef]
- Gibbs, P.; Brady, B.M.; Robinson, W.A. The genes and genetics of malignant melanoma. J. Cutan. Med. Surg. 2002, 6, 229–235. [Google Scholar] [CrossRef]
- Gruis, N.A.; Bergman, W. From gene to disease; from p16 to melanoma. Ned. Tijdschr. Geneeskd. 2000, 144, 2100–2102. [Google Scholar]
- Fusaro, R.M.; Lynch, H.T. The FAMMM syndrome: epidemiology and surveillance strategies. Cancer Invest. 2000, 18, 670–680. [Google Scholar] [CrossRef]
- Itin, P.H. Skin check-up—who and when? Ther. Umsch. 2000, 57, 22–25. [Google Scholar] [CrossRef]
- Platz, A.; Ringborg, U.; Hansson, J. Hereditary cutaneous melanoma. Semin. Cancer Biol. 2000, 10, 319–326. [Google Scholar] [CrossRef]
- Oliveria, S.; Dusza, S.; Berwick, M. Issues in the epidemiology of melanoma. Expert. Rev. Anticancer Ther. 2001, 1, 453–459. [Google Scholar] [CrossRef]
- Tucker, M.A.; Goldstein, A.M. Melanoma etiology: where are we? Oncogene 2003, 22, 3042–3052. [Google Scholar] [CrossRef]
- Stahl, J.M.; Sharma, A.; Cheung, M.; Zimmerman, M.; Cheng, J.Q.; Bosenberg, M.W.; Kester, M.; Sandirasegarane, L.; Robertson, G.P. Deregulated Akt3 activity promotes development of malignant melanoma. Cancer Res. 2004, 64, 7002–7010. [Google Scholar] [CrossRef]
- Pho, L.; Grossman, D.; Leachman, S.A. Melanoma genetics: a review of genetic factors and clinical phenotypes in familial melanoma. Curr. Opin. Oncol. 2006, 18, 173–179. [Google Scholar] [CrossRef]
- Kefford, R.; Bishop, J.N.; Tucker, M.; Bressac-de, P.B.; Bianchi-Scarra, G.; Bergman, W.; Goldstein, A.; Puig, S.; Mackie, R.; Elder, D.; Hansson, J.; Hayward, N.; Hogg, D.; Olsson, H. Genetic testing for melanoma. Lancet Oncol. 2002, 3, 653–654. [Google Scholar] [CrossRef]
- Fraser, M.C.; Goldstein, A.M.; Tucker, M.A. Genetic testing for inherited predisposition to melanoma: has the time come? J. Drugs Dermatol. 2004, 3, 93–95. [Google Scholar]
- Hansson, J. Familial melanoma. Surg. Clin. North Am. 2008, 88, 897–916. [Google Scholar] [CrossRef]
- Santillan, A.A.; Cherpelis, B.S.; Glass, L.F.; Sondak, V.K. Management of familial melanoma and nonmelanoma skin cancer syndromes. Surg. Oncol. Clin. North. Am. 2009, 18, 73–98. [Google Scholar] [CrossRef]
- Mantelli, M.; Pastorino, L.; Ghiorzo, P.; Barile, M.; Bruno, W.; Gargiulo, S.; Sormani, M.P.; Gliori, S.; Vecchio, S.; Ciotti, P.; Sertoli, M.R.; Queirolo, P.; Goldstein, A.M.; Bianchi-Scarra, G. Early onset may predict G101W CDKN2A founder mutation carrier status in Ligurian melanoma patients. Melanoma Res. 2004, 14, 443–448. [Google Scholar] [CrossRef]
- Eldon, B.J.; Thorlacius, S.; Jonsson, T.; Jonasson, J.G.; Kjartansson, J.; Bodvarsson, S.; Steingrimsson, E.; Rafnar, T. A population-based study on the familial aggregation of cutaneous malignant melanoma in Iceland. Eur. J. Cancer 2006, 42, 922–926. [Google Scholar] [CrossRef]
- Lamperska, K.; Karezewska, A.; Kwiatkowska, E.; Mackiewicz, A. Analysis of mutations in the p16/CDKN2A gene in sporadic and familial melanoma in the Polish population. Acta Biochim. Pol. 2002, 49, 369–376. [Google Scholar]
- Pjanova, D.; Engele, L.; Randerson-Moor, J.A.; Harland, M.; Bishop, D.T.; Newton Bishop, J.A.; Taylor, C.; Debniak, T.; Lubinski, J.; Kleina, R.; Heisele, O. CDKN2A and CDK4 variants in Latvian melanoma patients: analysis of a clinic-based population. Melanoma Res. 2007, 17, 185–191. [Google Scholar] [CrossRef]
- Ashton-Prolla, P.; Bakos, L.; Junqueira, G., Jr.; Giugliani, R.; Azevedo, S.J.; Hogg, D. Clinical and molecular characterization of patients at risk for hereditary melanoma in southern Brazil. J. Invest. Dermatol. 2008, 128, 421–425. [Google Scholar]
- Gensini, F.; Sestini, R.; Piazzini, M.; Vignoli, M.; Chiarugi, A.; Brandani, P.; Ghiorzo, P.; Salvini, C.; Borgognoni, L.; Palli, D.; Bianchi-Scarra, G.; Carli, P.; Genuardi, M. The p.G23S CDKN2A founder mutation in high-risk melanoma families from Central Italy. Melanoma Res. 2007, 17, 387–392. [Google Scholar] [CrossRef]
- Borges, A.L.; Cuellar, F.; Puig-Butille, J.A.; Scarone, M.; Delgado, L.; Badenas, C.; Mila, M.; Malvehy, J.; Barquet, V.; Nunez, J.; Laporte, M.; Fernandez, G.; Levrero, P.; Martinez-Asuaga, M.; Puig, S. CDKN2A mutations in melanoma families from Uruguay. Br. J. Dermatol. 2009, 161, 536–541. [Google Scholar] [CrossRef]
- Peric, B.; Cerkovnik, P.; Novakovic, S.; Zgajnar, J.; Besic, N.; Hocevar, M. Prevalence of variations in melanoma susceptibility genes among Slovenian melanoma families. Med. Genet. 2008, 9, 86–89. [Google Scholar]
- Nagore, E.; Botella-Estrada, R.; Garcia-Casado, Z.; Requena, C.; Serra-Guillen, C.; Llombart, B.; Sanmartin, O.; Guillen, C. Comparison between familial and sporadic cutaneous melanoma in Valencia, Spain. J. Eur. Acad. Dermatol. Venereol. 2008, 22, 931–936. [Google Scholar] [CrossRef]
- Leachman, S.A.; Carucci, J.; Kohlmann, W.; Banks, K.C.; Asgari, M.M.; Bergman, W.; Bianchi-Scarra, G.; Brentnall, T.; Bressac-de, P.B.; Bruno, W.; Curiel-Lewandrowski, C.; de Snoo, F.A.; Debniak, T.; Demierre, M.F.; Elder, D.; Goldstein, A.M.; Grant-Kels, J.; Halpern, A.C.; Ingvar, C.; Kefford, R.F.; Lang, J.; MacKie, R.M.; Mann, G.J.; Mueller, K.; Newton-Bishop, J.; Olsson, H.; Petersen, G.M.; Puig, S.; Rigel, D.; Swetter, S.M.; Tucker, M.A.; Yakobson, E.; Zitelli, J.A.; Tsao, H. Selection criteria for genetic assessment of patients with familial melanoma. J. Am. Acad. Dermatol. 2009, 61, 677–684. [Google Scholar]
- Kasparian, N.A.; Meiser, B.; Butow, P.N.; Soames Job, R.F.; Mann, G.J. Anticipated uptake of genetic testing for familial melanoma in an Australian sample: An exploratory study. Psychooncology. 2007, 16, 69–78. [Google Scholar] [CrossRef]
- Kasparian, N.A.; Butow, P.N.; Meiser, B.; Mann, G.J. High- and average-risk individuals' beliefs about, and perceptions of, malignant melanoma: an Australian perspective. Psychooncology. 2008, 17, 270–279. [Google Scholar] [CrossRef]
- Riedijk, S.R.; de Snoo, F.A.; van Dijk, S.; Bergman, W.; van Haeringen, A.; Silberg, S.; van Elderen, T.M.; Tibben, A. Hereditary melanoma and predictive genetic testing: why not? Psychooncology. 2005, 14, 738–745. [Google Scholar]
- de Snoo, F.A.; Riedijk, S.R.; van Mil, A.M.; Bergman, W.; ter Huurne, J.A.; Timman, R.; Bertina, W.; Gruis, N.A.; Vasen, H.F.; van Haeringen, A.; Breuning, M.H.; Tibben, A. Genetic testing in familial melanoma: uptake and implications. Psychooncology 2008, 17, 790–796. [Google Scholar] [CrossRef]
- Kasparian, N.A.; Meiser, B.; Butow, P.N.; Simpson, J.M.; Mann, G.J. Predictors of psychological distress among individuals with a strong family history of malignant melanoma. Clin. Genet. 2008, 73, 121–131. [Google Scholar]
- Bergenmar, M.; Hansson, J.; Brandberg, Y. Family members' perceptions of genetic testing for malignant melanoma—a prospective interview study. Eur. J. Oncol. Nurs. 2009, 13, 74–80. [Google Scholar] [CrossRef]
- Aspinwall, L.G.; Leaf, S.L.; Dola, E.R.; Kohlmann, W.; Leachman, S.A. CDKN2A/p16 genetic test reporting improves early detection intentions and practices in high-risk melanoma families. Cancer Epidemiol. Biomarkers Prev. 2008, 17, 1510–1519. [Google Scholar] [CrossRef]
- Parker, J.F.; Florell, S.R.; Alexander, A.; DiSario, J.A.; Shami, P.J.; Leachman, S.A. Pancreatic carcinoma surveillance in patients with familial melanoma. Arch. Dermatol. 2003, 139, 1019–1025. [Google Scholar] [CrossRef]
- Bishop, J.A.; Wachsmuth, R.C.; Harland, M.; Bataille, V.; Pinney, E.; Mac, K.P.; Baglietto, L.; Cuzick, J.; Bishop, D.T. Genotype/Phenotype and penetrance studies in melanoma families with germline CDKN2A mutations. J. Invest. Dermatol 2000, 114, 28–33. [Google Scholar] [CrossRef]
- Lynch, H.T.; Brand, R.E.; Hogg, D.; Deters, C.A.; Fusaro, R.M.; Lynch, J.F.; Liu, L.; Knezetic, J.; Lassam, N.J.; Goggins, M.; Kern, S. Phenotypic variation in eight extended CDKN2A germline mutation familial atypical multiple mole melanoma-pancreatic carcinoma-prone families: the familial atypical mole melanoma-pancreatic carcinoma syndrome. Cancer 2002, 94, 84–96. [Google Scholar] [CrossRef]
- Kluijt, I.; Cats, A.; Fockens, P.; Nio, Y.; Gouma, D.J.; Bruno, M.J. Atypical Familial Presentation of FAMMM Syndrome With a High Incidence of Pancreatic Cancer: Case Finding of Asymptomatic Individuals by EUS Surveillance. J. Clin. Gastroenterol. 2009, 43, 853–857. [Google Scholar] [CrossRef]
- Poley, J.W.; Kluijt, I.; Gouma, D.J.; Harinck, F.; Wagner, A.; Aalfs, C.; van Eijck, C.H.; Cats, A.; Kuipers, E.J.; Nio, Y.; Fockens, P.; Bruno, M.J. The yield of first-time endoscopic ultrasonography in screening individuals at a high risk of developing pancreatic cancer. Am. J. Gastroenterol. 2009, 104, 2175–2181. [Google Scholar] [CrossRef]
- Briollais, L.; Chompret, A.; Guilloud-Bataille, M.; Bressac-de, P.B.; Avril, M.F.; Demenais, F. Patterns of familial aggregation of three melanoma risk factors: great number of naevi, light phototype and high degree of sun exposure. Int. J. Epidemiol. 2000, 29, 408–415. [Google Scholar] [CrossRef] [Green Version]
- Hoiom, V.; Tuominen, R.; Kaller, M.; Linden, D.; Ahmadian, A.; Mansson-Brahme, E.; Egyhazi, S.; Sjoberg, K.; Lundeberg, J.; Hansson, J. MC1R variation and melanoma risk in the Swedish population in relation to clinical and pathological parameters. Pigment Cell Melanoma Res. 2009, 22, 196–204. [Google Scholar] [CrossRef]
- Hornbuckle, J.; Culjak, G.; Jarvis, E.; Gebski, V.; Coates, A.; Mann, G.; Kefford, R. Patterns of metastases in familial and non-familial melanoma. Melanoma Res. 2003, 13, 105–109. [Google Scholar] [CrossRef]
- Gillgren, P.; Brattstrom, G.; Frisell, J.; Palmgren, J.; Ringborg, U.; Hansson, J. Body site of cutaneous malignant melanoma--a study on patients with hereditary and multiple sporadic tumours. Melanoma Res. 2003, 13, 279–286. [Google Scholar] [CrossRef]
- Florell, S.R.; Boucher, K.M.; Garibotti, G.; Astle, J.; Kerber, R.; Mineau, G.; Wiggins, C.; Noyes, R.D.; Tsodikov, A.; Cannon-Albright, L.A.; Zone, J.J.; Samlowski, W.E.; Leachman, S.A. Population-based analysis of prognostic factors and survival in familial melanoma. J. Clin. Oncol. 2005, 23, 7168–7177. [Google Scholar] [CrossRef]
- Hansson, J.; Bergenmar, M.; Hofer, P.A.; Lundell, G.; Mansson-Brahme, E.; Ringborg, U.; Synnerstad, I.; Bratel, A.T.; Wennberg, A.M.; Rosdahl, I. Monitoring of kindreds with hereditary predisposition for cutaneous melanoma and dysplastic nevus syndrome: results of a Swedish preventive program. J. Clin. Oncol. 2007, 25, 2819–2824. [Google Scholar] [CrossRef]
- Mesters, I.; Jonkman, L.; Vasen, H., de Vries. Skin self-examination of persons from families with familial atypical multiple mole melanoma (FAMMM). Patient Educ. Couns. 2009, 75, 251–255. [Google Scholar] [CrossRef]
- Loescher, L.J.; Crist, J.D.; Siaki, L.A. Perceived intrafamily melanoma risk communication. Cancer Nurs. 2009, 32, 203–210. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Chan, M.; Harland, M.; Hayward, N.K.; Demenais, F.; Bishop, D.T.; Azizi, E.; Bergman, W.; Bianchi-Scarra, G.; Bruno, W.; Calista, D.; Albright, L.A.; Chaudru, V.; Chompret, A.; Cuellar, F.; Elder, D.E.; Ghiorzo, P.; Gillanders, E.M.; Gruis, N.A.; Hansson, J.; Hogg, D.; Holland, E.A.; Kanetsky, P.A.; Kefford, R.F.; Landi, M.T.; Lang, J.; Leachman, S.A.; MacKie, R.M.; Magnusson, V.; Mann, G.J.; Bishop, J.N.; Palmer, J.M.; Puig, S.; Puig-Butille, J.A.; Stark, M.; Tsao, H.; Tucker, M.A.; Whitaker, L.; Yakobson, E. Features associated with germline CDKN2A mutations: a GenoMEL study of melanoma-prone families from three continents. J. Med. Genet. 2007, 44, 99–106. [Google Scholar]
- Bruno, W.; Ghiorzo, P.; Battistuzzi, L.; Ascierto, P.A.; Barile, M.; Gargiulo, S.; Gensini, F.; Gliori, S.; Guida, M.; Lombardo, M.; Manoukian, S.; Menin, C.; Nasti, S.; Origone, P.; Pasini, B.; Pastorino, L.; Peissel, B.; Pizzichetta, M.A.; Queirolo, P.; Rodolfo, M.; Romanini, A.; Scaini, M. C.; Testori, A.; Tibiletti, M.G.; Turchetti, D.; Leachman, S.A.; Bianchi-Scarra, G. Clinical genetic testing for familial melanoma in Italy: a cooperative study. J. Am. Acad. Dermatol. 2009, 61, 775–782. [Google Scholar] [CrossRef]
- Bishop, D.T.; Demenais, F.; Goldstein, A.M.; Bergman, W.; Bishop, J.N.; Bressac-de, P.B.; Chompret, A.; Ghiorzo, P.; Gruis, N.; Hansson, J.; Harland, M.; Hayward, N.; Holland, E.A.; Mann, G.J.; Mantelli, M.; Nancarrow, D.; Platz, A.; Tucker, M.A. Geographical variation in the penetrance of CDKN2A mutations for melanoma. J. Natl. Cancer Inst. 2002, 94, 894–903. [Google Scholar] [CrossRef]
- Begg, C.B.; Orlow, I.; Hummer, A.J.; Armstrong, B.K.; Kricker, A.; Marrett, L.D.; Millikan, R.C.; Gruber, S.B.; Anton-Culver, H.; Zanetti, R.; Gallagher, R.P.; Dwyer, T.; Rebbeck, T.R.; Mitra, N.; Busam, K.; From, L.; Berwick, M. Lifetime risk of melanoma in CDKN2A mutation carriers in a population-based sample. J. Natl. Cancer Inst. 2005, 97, 1507–1515. [Google Scholar]
- Vasen, H.F.; Gruis, N.A.; Frants, R.R.; van der Velden, P.A.; Hille, E.T.; Bergman, W. Risk of developing pancreatic cancer in families with familial atypical multiple mole melanoma associated with a specific 19 deletion of p16 (p16-Leiden). Int. J. Cancer 2000, 8, 809–811. [Google Scholar]
- de Vos tot Nederveen Cappel, W.H.; Offerhaus, G.J.; van, P.M.; Caspers, E.; Gruis, N.A.; de Snoo, F.A.; Lamers, C.B.; Griffioen, G.; Bergman, W.; Vasen, H.F.; Morreau, H. Pancreatic carcinoma in carriers of a specific 19 base pair deletion of CDKN2A/p16 (p16-leiden). Clin. Cancer Res. 2003, 9, 3598–3605. [Google Scholar]
- Hille, E.; van Duijn, E.; Gruis, N.A.; Rosendaal, F.R.; Bergman, W.; Vandenbroucke, J.P. Excess cancer mortality in six Dutch pedigrees with the familial atypical multiple mole-melanoma syndrome from 1830 to 1994. J. Invest. Dermatol. 1998, 110, 788–92. [Google Scholar] [CrossRef]
- Lynch, H.T.; Fusaro, R.M.; Albano, W.A.; Pester, J.; Kimberling, W.J.; Lynch, J.F. Phenotypic variation in the familial atypical multiple mole-melanoma syndrome (FAMMM). J. Med. Genet. 1983, 20, 25–29. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Chan, M.; Harland, M.; Gillanders, E.M.; Hayward, N.K.; Avril, M.F.; Azizi, E.; Bianchi-Scarra, G.; Bishop, D.T.; Bressac-de, P.B.; Bruno, W.; Calista, D.; Cannon Albright, L.A.; Demenais, F.; Elder, D.E.; Ghiorzo, P.; Gruis, N.A.; Hansson, J.; Hogg, D.; Holland, E.A.; Kanetsky, P.A.; Kefford, R.F.; Landi, M.T.; Lang, J.; Leachman, S.A.; Mackie, R.M.; Magnusson, V.; Mann, G.J.; Niendorf, K.; Newton, B.J.; Palmer, J.M.; Puig, S.; Puig-Butille, J.A.; de Snoo, F.A.; Stark, M.; Tsao, H.; Tucker, M.A.; Whitaker, L.; Yakobson, E. High-risk melanoma susceptibility genes and pancreatic cancer, neural system tumors, and uveal melanoma across GenoMEL. Cancer Res. 2006, 66, 9818–9828. [Google Scholar] [CrossRef]
- de Snoo, F.A.; Bishop, D.T.; Bergman, W.; van Leeuwen-Cornelissen, I; van der Drift, C.; van Nieuwpoort, F.A.; Out-Luiting, C.J.; Vasen, H.F.; ter Huurne, J.A.; Frants, R.R.; Willemze, R.; Breuning, M.H.; Gruis, N.A. Increased risk of cancer other than melanoma in CDKN2A founder mutation (p16-Leiden)-positive melanoma families. Clin. Cancer Res. 2008, 14, 7151–7157. [Google Scholar] [CrossRef]
- Cannon-Albright, L.A.; Goldgar, D.E.; Neuhausen, S.; Gruis, N.A.; Anderson, D.E.; Lewis, C. M.; Jost, M.; Tran, T.D.; Nyguen, K.; Kamb, A.; et al. Localization of the 9p melanoma susceptibility locus (MLM) to a 2-cM region between D9S736 and D9S171. Genomics 1994, 23, 265–268. [Google Scholar] [CrossRef]
- Kamb, A.; Gruis, N.A.; Weaver-Feldhaus, J.; Liu, Q.; Harshman, K.; Tavtigian, S.V.; Stockert, E.; Day, R.S.; Johnson, B.E.; Skolnick, M.H. A cell cycle regulator potentially involved in genesis of many tumor types. Science 1994, 264, 436–440. [Google Scholar]
- Chin, L.; Pomerantz, J.; DePinho, R.A. The INK4a/ARF tumor suppressor: one gene--two products--two pathways. Trends Biochem. Sci. 1998, 23, 291–296. [Google Scholar] [CrossRef]
- Bennett, D.C. Human melanocyte senescence and melanoma susceptibility genes. Oncogene 2003, 22, 3063–3069. [Google Scholar] [CrossRef]
- Gray-Schopfer, V.C.; Cheong, S.C.; Chong, H.; Chow, J.; Moss, T.; Abdel-Malek, Z.A.; Marais, R.; Wynford-Thomas, D.; Bennett, D.C. Cellular senescence in naevi and immortalisation in melanoma: a role for p16? Br. J. Cancer 2006, 95, 496–505. [Google Scholar] [CrossRef]
- Rizos, H.; Woodruff, S.; Kefford, R.F. p14ARF interacts with the SUMO-conjugating enzyme Ubc9 and promotes the sumoylation of its binding partners. Cell Cycle 2005, 4, 597–603. [Google Scholar] [CrossRef]
- Rizos, H.; McKenzie, H.A.; Ayub, A.L.; Woodruff, S.; Becker, T.M.; Scurr, L.L.; Stahl, J.; Kefford, R.F. Physical and functional interaction of the p14ARF tumor suppressor with ribosomes. J. Biol. Chem. 2006, 281, 380–388. [Google Scholar]
- Harland, M.; Taylor, C.F.; Chambers, P.A.; Kukalizch, K.; Randerson-Moor, J.A.; Gruis, N.A.; de Snoo, F.A.; ter Huurne, J.A.C.; Goldstein, A.M.; Tucker, M.A.; Bishop, D.T.; Bishop, J.A. Mutation hotspot at the p14ARF splice site. Oncogene 2005, 24, 4604–4608. [Google Scholar] [CrossRef]
- Randerson-Moor, J.A.; Harland, M.; Williams, S.; Cuthbert-Heavens, D.; Sheridan, E.; Aveyard, J.; Sibley, K.; Whitaker, L.; Knowles, M.; Bishop, J.N.; Bishop, D.T. A germline deletion of p14(ARF) but not CDKN2A in a melanoma-neural system tumour syndrome family. Hum. Mol. Genet. 2001, 10, 55–62. [Google Scholar] [CrossRef]
- Zuo, L.; Weger, J.; Yang, Q.; Goldstein, A.M.; Tucker, M.A.; Walker, G.J.; Hayward, N.; Dracopoli, N.C. Germline mutations in the p16INK4a binding domain of CDK4 in familial melanoma. Nat. Genet. 1996, 12, 97–99. [Google Scholar] [CrossRef]
- Goldstein, A.M.; Chidambaram, A.; Halpern, A.; Holly, E.A.; Guerry, I.D.; Sagebiel, R.; Elder, D.E.; Tucker, M.A. Rarity of CDK4 germline mutations in familial melanoma. Melanoma Res. 2002, 12, 51–55. [Google Scholar] [CrossRef]
- Mistry, S.H.; Taylor, C.; Randerson-Moor, J.A.; Harland, M.; Turner, F.; Barrett, J.H.; Whitaker, L.; Jenkins, R.B.; Knowles, M.A.; Bishop, J.A.; Bishop, D.T. Prevalence of 9p21 deletions in UK melanoma families. Genes Chromosomes Cancer 2005, 44, 292–300. [Google Scholar] [CrossRef]
- Van Doorn, R.; Zoutman, W.H.; Gruis, N.A. Absence of germline epimutation of the CDKN2A gene in familial melanoma. J. Invest. Dermatol. 2009, 129, 781–784. [Google Scholar] [CrossRef]
- Gillanders, E.; Juo, S.H.; Holland, E.A.; Jones, M.; Nancarrow, D.; Freas-Lutz, D.; Sood, R.; Park, N.; Faruque, M.; Markey, C.; Kefford, R.F.; Palmer, J.; Bergman, W.; Bishop, D.T.; Tucker, M.A.; Bressac-de, P.B.; Hansson, J.; Stark, M.; Gruis, N.A.; Bishop, J.N.; Goldstein, A.M.; Bailey-Wilson, J.E.; Mann, G.J.; Hayward, N.; Trent, J. Localization of a novel melanoma susceptibility locus to 1p22. Am. J. Hum. Genet. 2003, 73, 301–313. [Google Scholar] [CrossRef]
- Kennedy, C.; ter, Huurne. J.A.; Berkhout, M.; Gruis, N.A.; Bastiaens, M.; Bergman, W.; Willemze, R.; Bouwes Bavinck, J.N. Melanocortin 1 receptor (MC1R) gene variants are associated with an increased risk for cutaneous melanoma which is largely independent of skin type and hair color. J. Invest. Dermatol. 2001, 117, 294–300. [Google Scholar] [CrossRef]
- Valverde, P.; Healy, E.; Sikkink, S.; Haldane, F.; Thody, A.J.; Carothers, A.; Jackson, I.J.; Rees, J.L. The Asp84Glu variant of the melanocortin 1 receptor (MC1R) is associated with melanoma. Hum. Mol. Genet. 1996, 5, 1663–1666. [Google Scholar] [CrossRef]
- Abdel-Malek, Z.A.; Scott, M.C.; Suzuki, I.; Tada, A.; Im, S.; Lamoreux, L.; Ito, S.; Barsh, G.; Hearing, V.J. The melanocortin-1 receptor is a key regulator of human cutaneous pigmentation. Pigment Cell Res. 2000, 13, 156–162. [Google Scholar]
- Suzuki, I.; Tada, A.; Ollmann, M.M.; Barsh, G.S.; Im, S.; Lamoreux, M.L.; Hearing, V.J.; Nordlund, J.J.; Abdel-Malek, Z.A. Agouti signaling protein inhibits melanogenesis and the response of human melanocytes to alpha-melanotropin. J. Invest. Dermatol. 1997, 108, 838–842. [Google Scholar] [CrossRef]
- Orlow, S.J.; Brilliant, M.H. The pink-eyed dilution locus controls the biogenesis of melanosomes and levels of melanosomal proteins in the eye. Exp. Eye Res. 1999, 68, 147–154. [Google Scholar] [CrossRef]
- Chen, K.; Manga, P.; Orlow, S.J. Pink-eyed dilution protein controls the processing of tyrosinase. Mol. Biol. Cell 2002, 13, 1953–1964. [Google Scholar] [CrossRef]
- Box, N.F.; Wyeth, J.R.; O'Gorman, L.E.; Martin, N.G.; Sturm, R.A. Characterization of melanocyte stimulating hormone receptor variant alleles in twins with red hair. Hum. Mol. Genet. 1997, 6, 1891–1897. [Google Scholar] [CrossRef]
- Rees, J.L. Genetics of hair and skin color. Annu. Rev. Genet. 2003, 37, 67–90. [Google Scholar] [CrossRef]
- Bastiaens, M.; ter Huurne, J.; Gruis, N.A.; Bergman, W.; Westendorp, R.; Vermeer, B.J.; Bouwes Bavinck, J.N. The melanocortin-1-receptor gene is the major freckle gene. Hum. Mol. Genet. 2001, 10, 1701–1708. [Google Scholar] [CrossRef]
- Bishop, D.T.; Demenais, F.; Iles, M.M.; Harland, M.; Taylor, J.C.; Corda, E.; Randerson-Moor, J.; Aitken, J.F.; Avril, M.F.; Azizi, E.; Bakker, B.; Bianchi-Scarra, G.; Bressac-de, P.B.; Calista, D.; Cannon-Albright, L.A.; Chin, A.W.; Debniak, T.; Galore-Haskel, G.; Ghiorzo, P.; Gut, I.; Hansson, J.; Hocevar, M.; Hoiom, V.; Hopper, J.L.; Ingvar, C.; Kanetsky, P.A.; Kefford, R.F.; Landi, M.T.; Lang, J.; Lubinski, J.; Mackie, R.M.; Malvehy, J.; Mann, G.J.; Martin, N.G.; Montgomery, G.W.; van Nieuwpoort, F.A.; Novakovic, S.; Olsson, H.; Puig, S.; Weiss, M.; van, W.W.; Zelenika, D.; Brown, K.M.; Goldstein, A.M.; Gillanders, E.M.; Boland, A.; Galan, P.; Elder, D.E.; Gruis, N.A.; Hayward, N.K.; Lathrop, G.M.; Barrett, J.H.; Bishop, J.A. Genome-wide association study identifies three loci associated with melanoma risk. Nat. Genet. 2009, 41, 920–925. [Google Scholar] [CrossRef]
- Brown, K.M.; Macgregor, S.; Montgomery, G.W.; Craig, D.W.; Zhao, Z.Z.; Iyadurai, K.; Henders, A.K.; Homer, N.; Campbell, M.J.; Stark, M.; Thomas, S.; Schmid, H.; Holland, E.A.; Gillanders, E.M.; Duffy, D.L.; Maskiell, J.A.; Jetann, J.; Ferguson, M.; Stephan, D.A.; Cust, A.E.; Whiteman, D.; Green, A.; Olsson, H.; Puig, S.; Ghiorzo, P.; Hansson, J.; Demenais, F.; Goldstein, A.M.; Gruis, N.A.; Elder, D.E.; Bishop, J.N.; Kefford, R.F.; Giles, G.G.; Armstrong, B.K.; Aitken, J.F.; Hopper, J. L.; Martin, N.G.; Trent, J.M.; Mann, G.J.; Hayward, N.K. Common sequence variants on 20q11.22 confer melanoma susceptibility. Nat. Genet. 2008, 40, 838–840. [Google Scholar] [CrossRef]
- Gudbjartsson, D.F.; Sulem, P.; Stacey, S.N.; Goldstein, A.M.; Rafnar, T.; Sigurgeirsson, B.; Benediktsdottir, K.R.; Thorisdottir, K.; Ragnarsson, R.; Sveinsdottir, S.G.; Magnusson, V.; Lindblom, A.; Kostulas, K.; Botella-Estrada, R.; Soriano, V.; Juberias, P.; Grasa, M.; Saez, B.; Andres, R.; Scherer, D.; Rudnai, P.; Gurzau, E.; Koppova, K.; Kiemeney, L.A.; Jakobsdottir, M.; Steinberg, S.; Helgason, A.; Gretarsdottir, S.; Tucker, M.A.; Mayordomo, J.I.; Nagore, E.; Kumar, R.; Hansson, J.; Olafsson, J.H.; Gulcher, J.; Kong, A.; Thorsteinsdottir, U.; Stefansson, K. ASIP and TYR pigmentation variants associate with cutaneous melanoma and basal cell carcinoma. Nat. Genet. 2008, 40, 886–891. [Google Scholar] [CrossRef]
- Falchi, M.; Bataille, V.; Hayward, N.K.; Duffy, D.L.; Bishop, J.A.; Pastinen, T.; Cervino, A.; Zhao, Z.Z.; Deloukas, P.; Soranzo, N.; Elder, D.E.; Barrett, J.H.; Martin, N.G.; Bishop, D.T.; Montgomery, G.W.; Spector, T.D. Genome-wide association study identifies variants at 9p21 and 22q13 associated with development of cutaneous nevi. Nat. Genet. 2009, 41, 915–919. [Google Scholar] [CrossRef]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Bergman, W.; Gruis, N.A. Management of Melanoma Families. Cancers 2010, 2, 549-566. https://doi.org/10.3390/cancers2020549
Bergman W, Gruis NA. Management of Melanoma Families. Cancers. 2010; 2(2):549-566. https://doi.org/10.3390/cancers2020549
Chicago/Turabian StyleBergman, Wilma, and Nelleke A. Gruis. 2010. "Management of Melanoma Families" Cancers 2, no. 2: 549-566. https://doi.org/10.3390/cancers2020549
APA StyleBergman, W., & Gruis, N. A. (2010). Management of Melanoma Families. Cancers, 2(2), 549-566. https://doi.org/10.3390/cancers2020549