Stereoselective Luche Reduction of Deoxynivalenol and Three of Its Acetylated Derivatives at C8

 , , and
, , and
Abstract
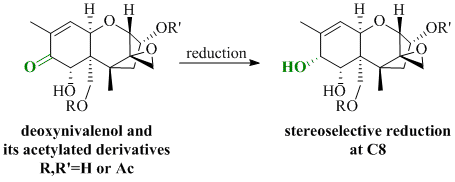
:
1. Introduction
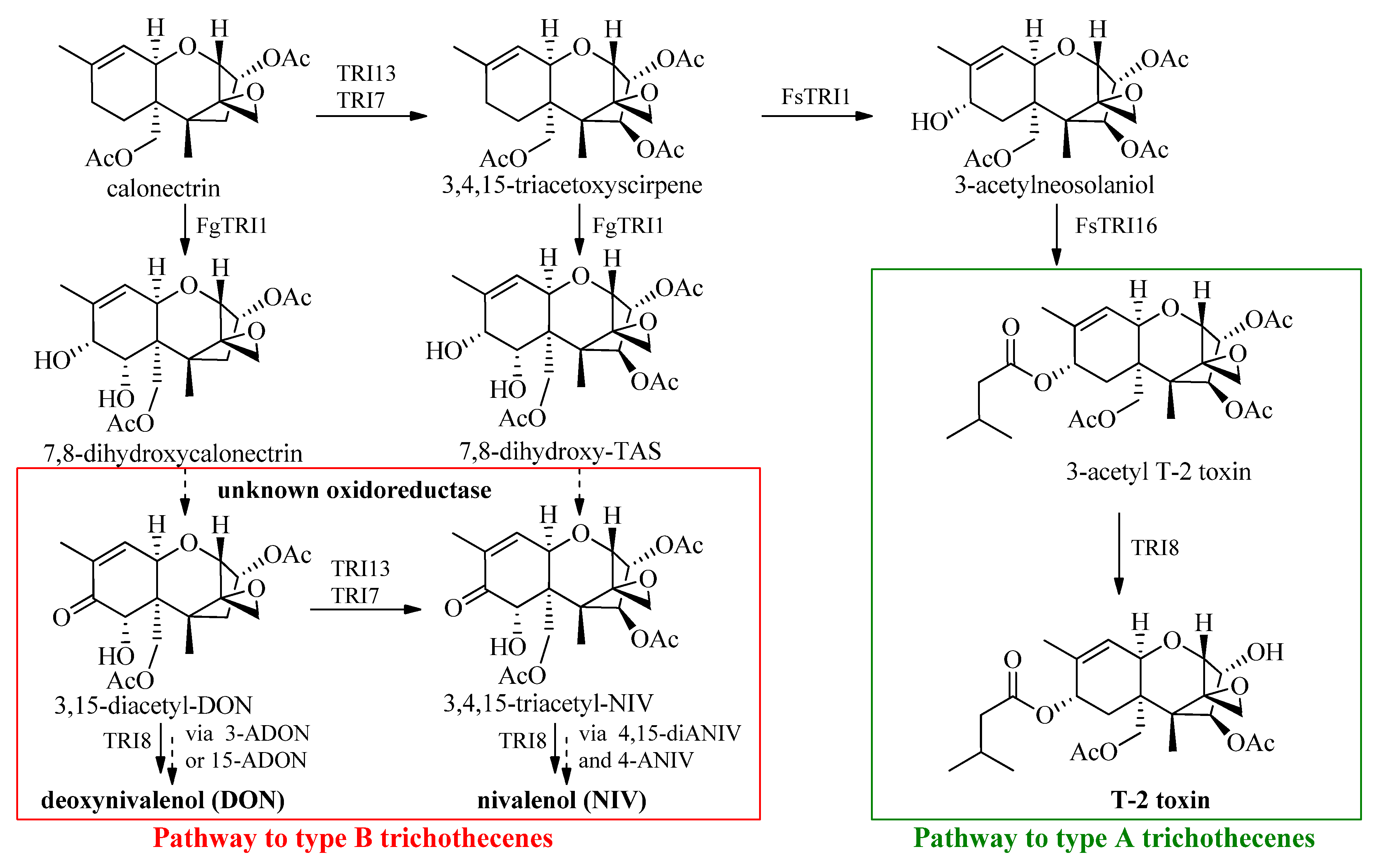
2. Synthetic Approach
2.1. General Aspects
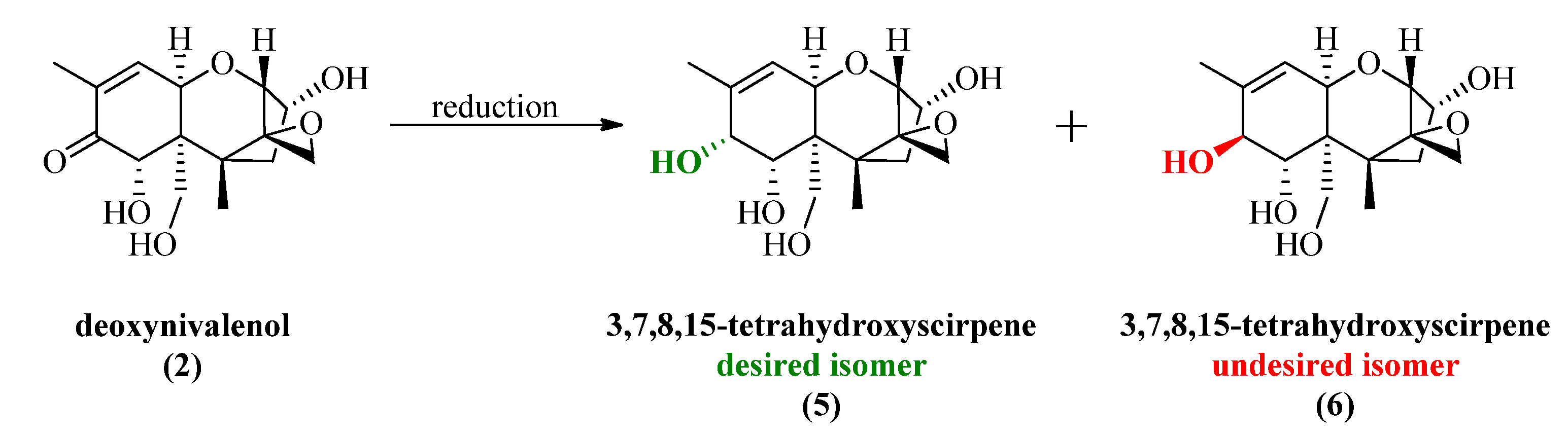
2.2. Luche Reduction
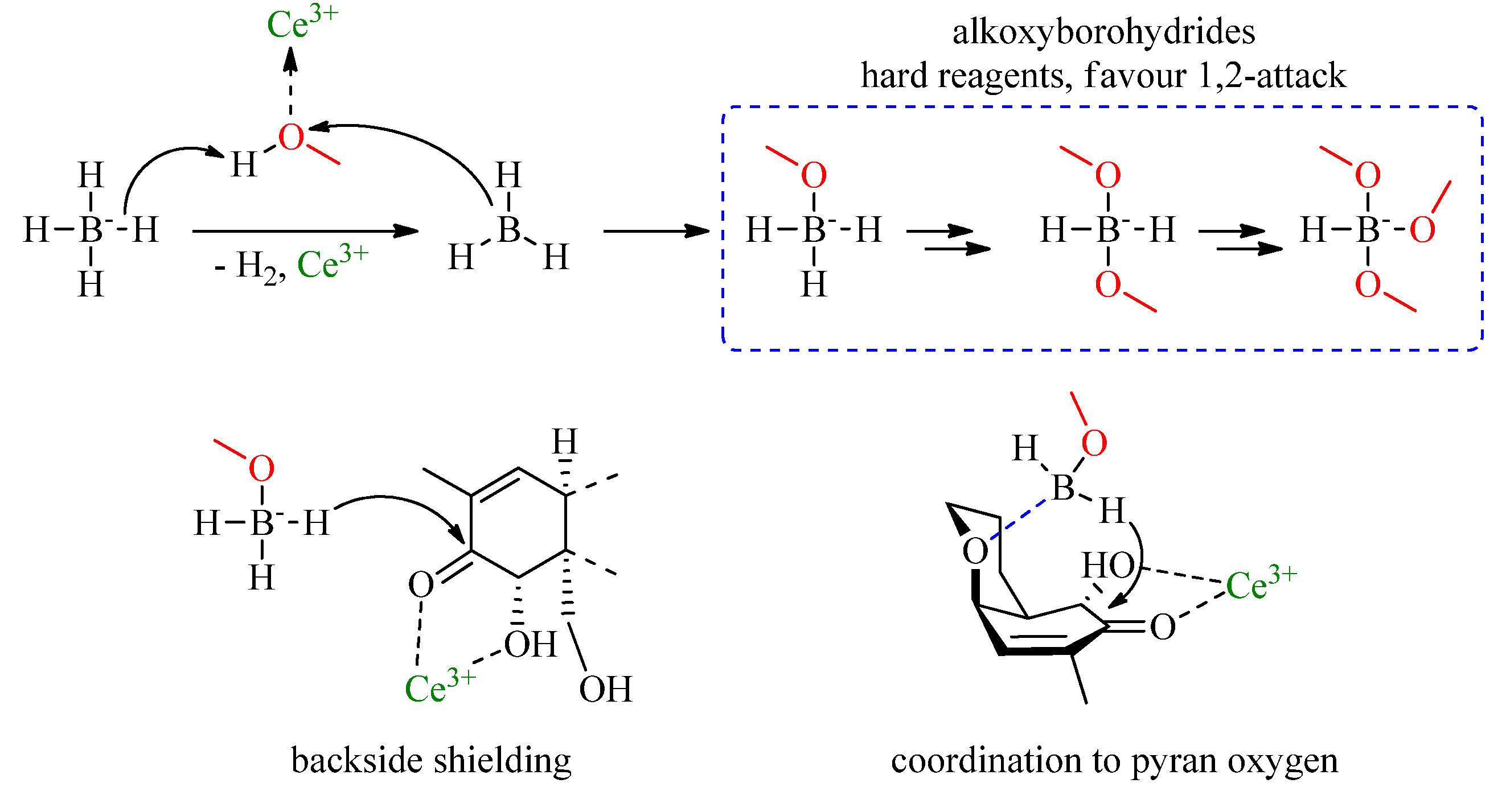
3. Results and Discussion
3.1. Method Development

3.2. Synthesis of 3-ADON, 15-ADON, 3,15-diADON and Their Reduction
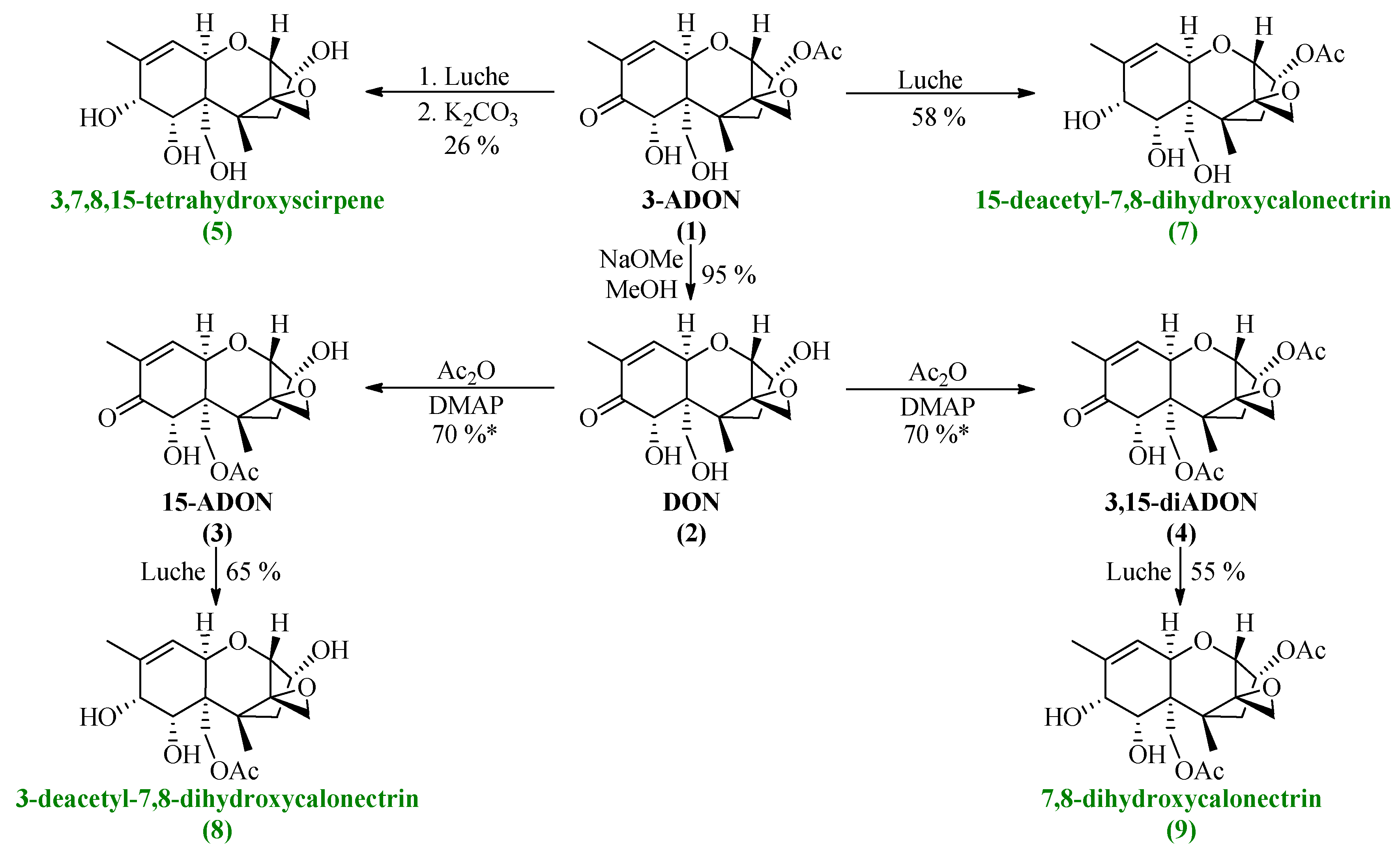
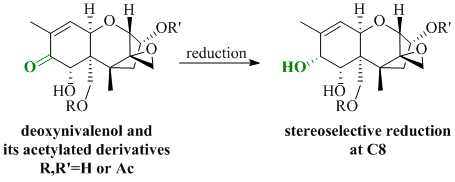
All synthesized DON derivatives were reduced applying an optimized Luche protocol (See 3.3. Reduction Protocol) leading to the desired 7,8-dihydroxycalonectrin derivatives in moderate to good yields.
3.3. Reduction Protocol
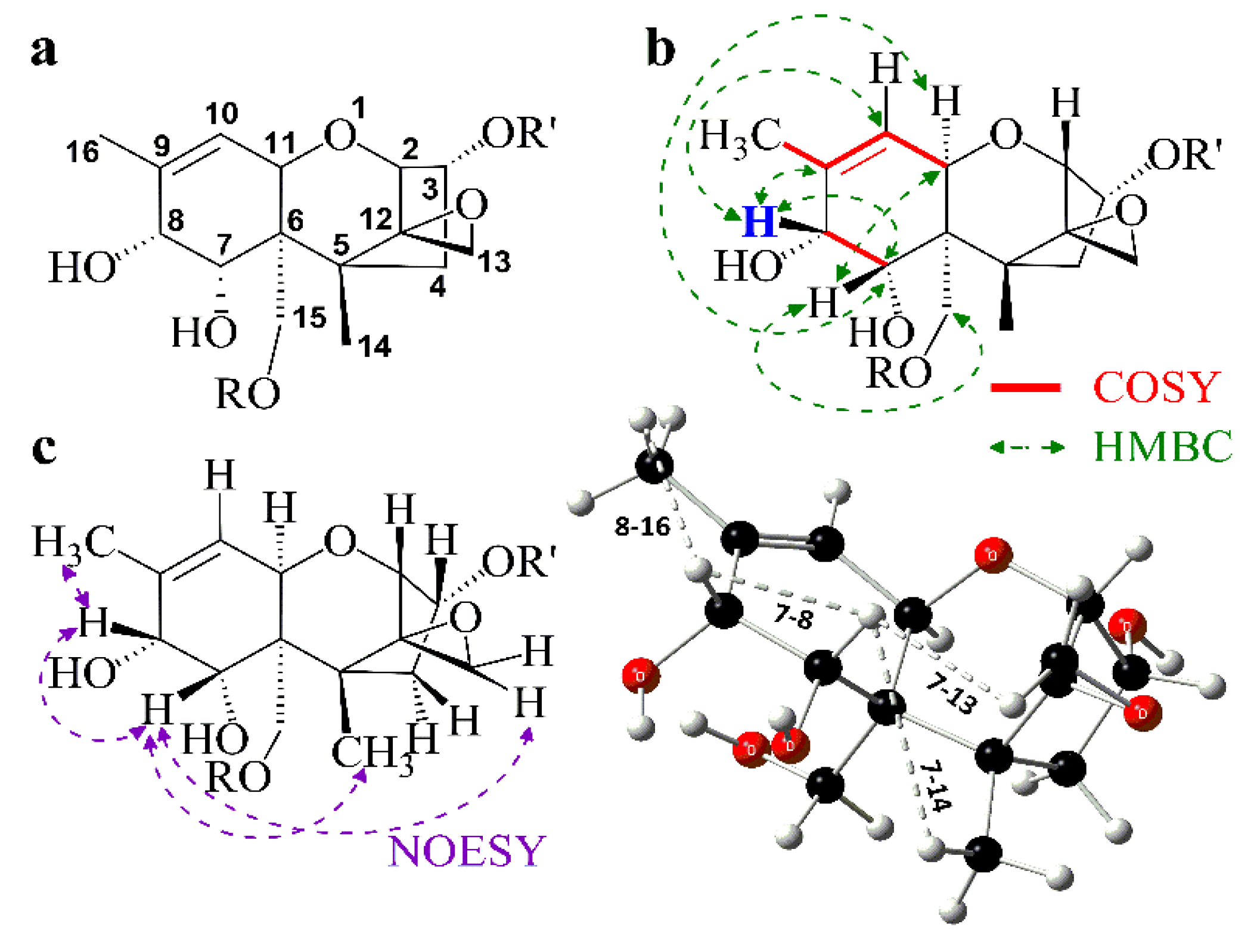
3.4. Spectroscopic Investigation

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Product | 2 (d) | 3 (dt) | 4α (dd) | 4β (dd) | 7β (d) | 8β (d) | 10 (dq) | 11 (d) |
|---|---|---|---|---|---|---|---|---|
| (5) | 3.38 [4.5] | 4.31 [10.7, 4.5] | 2.21 [14.6, 4.5] | 1.97 [14.6, 10.7] | 4.43 [4.7] | 3.91 [4.7] | 5.57 [5.6] (b) | 4.40 [5.6] |
| (7) | 3.68 [4.4] | 5.05 [11.1, 4.4] | 2.48 [15.0, 4.4] | 2.04 [15.0, 11.1] | 4.40 [5.0] | 3.91 [5.0] | 5.54 [5.5, 1.4] | 4.33 [5.5] |
| (8) | 3.41 [4.4] | 4.30 [11.1, 4.4] | 2.49 [14.6, 4.4] | 1.97 [14.6, 11.1] | 4.45 [5.6] | 3.92 [5.6] | 5.57 [5.8, 1.4] | 4.64 [5.8] |
| (9) | 3.72 [4.4] | 5.05 [11.2, 4.4] | 2.68 [15.1, 4,4] | 1.95–2.15(m) | 4.42 [5.2] | 3.91 [5.2] | 5.53 [5.9, 1.5] | 4.57 [5.9] |
| Product | 13a (d) | 13b (d) | 14 (s) | 15a (d) | 15b (d) | 16 (s) | 3 R' (s) | 15 R (s) |
| (5) | 3.02 [4.4] | 3.16 [4.4] | 1.14 | 3.64 [12.6] | 3.88 [12.6] | 1.84 | --- | --- |
| (7) | 3.08 [4.1] | 3.20 [4.1] | 1.17 | 3.67 [12.6] | 3.90 [12.6] | 1.84 | --- | 2.10 |
| (8) | 3.04 [4.4] | 3.18 [4.4] | 1.15 | 4.34 [12.6] | 4.38 [12.6] | 1.85 | 2.04 | --- |
| (9) | 3.09 [4.3] | 3.22 [4.3] | 1.18 | 4.37(s), 2H | 1.85 | 2.04 | 2.10 | |
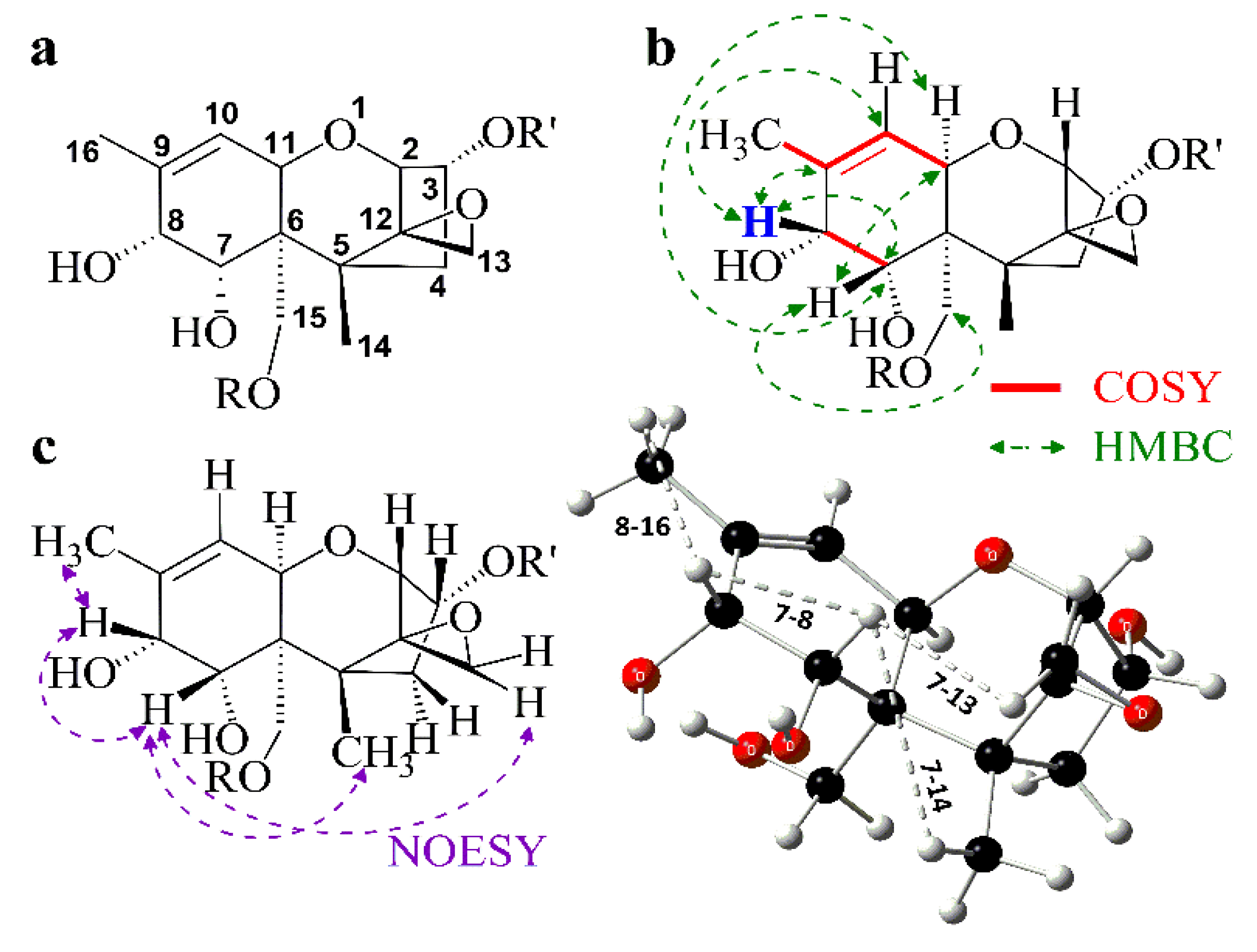
4. Experimental Section
4.1. General
4.2. Deoxynivalenol (2)
4.3. 15-ADON (3) and 3,15-diADON (4)
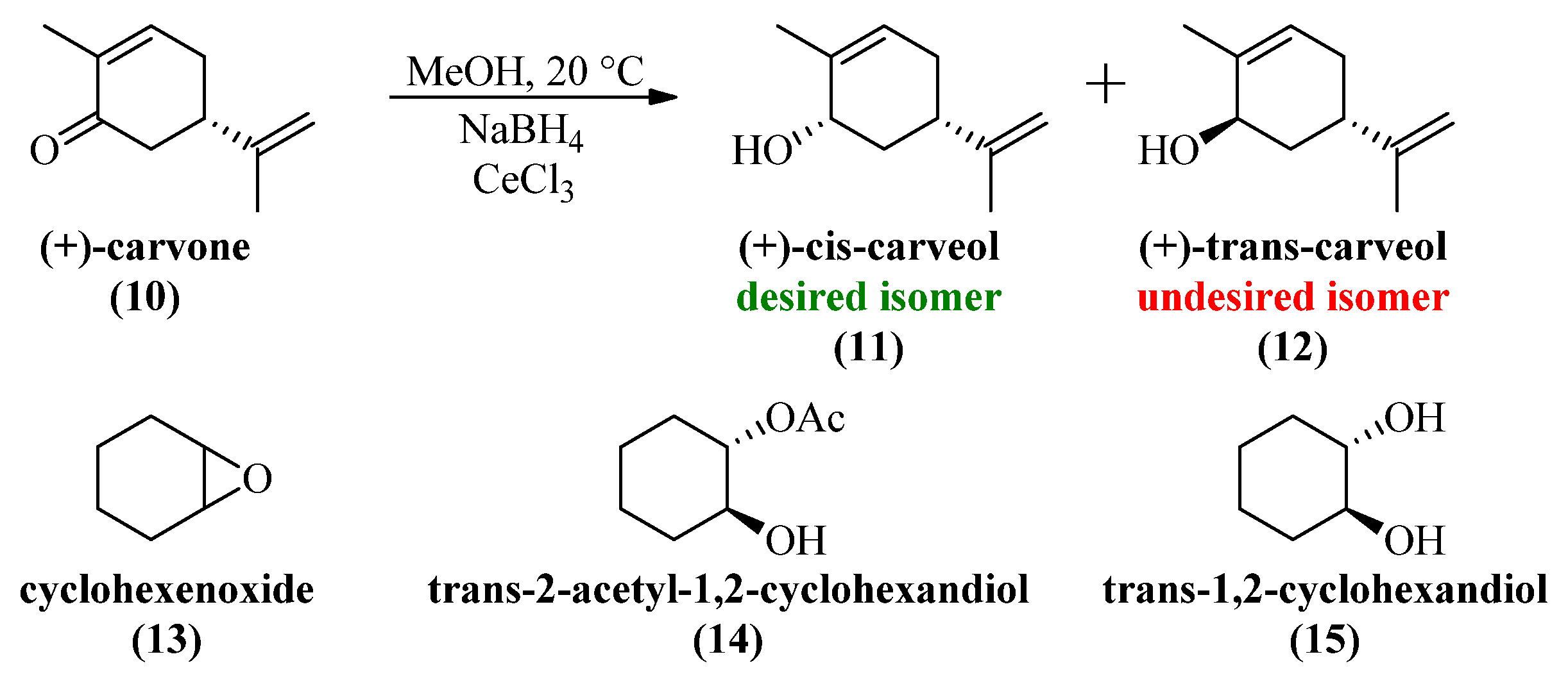
4.4. (+)-cis-Carveol (11) (Large Scale Luche Reduction)
4.5. Conversion of 3-ADON (1) to 15-Deacetyl-7,8-dihydroxycalonectrin (7) and 3,7,8,15-Tetrahydroxyscirpene (5)
4.6. Reduction of 15-ADON (3) and 3,15-diADON (4) to 3-Deacetyl-7,8-dihydroxycalonectrin (8) and 7,8-Dihydroxycalonectrin (9)
5. Conclusions
Acknowledgments
Conflicts of Interest
References
- D’Mello, J.P.F.; Macdonald, A.M.C. Mycotoxins. Anim. Feed Sci. Technol. 1997, 69, 155–166. [Google Scholar] [CrossRef]
- Cundliffe, E.; Cannon, M.; Davies, J. Mechanism of inhibition of eukaryotic protein synthesis by trichothecene fungal toxins. Proc. Natl. Acad. Sci. USA 1974, 71, 30–34. [Google Scholar] [CrossRef]
- Pestka, J. Deoxynivalenol: Mechanisms of action, human exposure, and toxicological relevance. Arch. Toxicol. 2010, 84, 663–679. [Google Scholar]
- Arunachalam, C.; Doohan, F.M. Trichothecene toxicity in eukaryotes: Cellular and molecular mechanisms in plants and animals. Toxicol. Lett. 2013, 217, 149–158. [Google Scholar] [CrossRef]
- Rotter, B.A. Invited review: Toxicology of deoxynivalenol (vomitoxin). J. Toxicol. Environ. Health 1996, 48, 1–34. [Google Scholar] [CrossRef]
- Pestka, J.J. Deoxynivalenol-induced proinflammatory gene expression: Mechanisms and pathological sequelae. Toxins 2010, 2, 1300–1317. [Google Scholar] [CrossRef]
- Maresca, M. From the gut to the brain: Journey and pathophysiological effects of the food-associated trichothecene mycotoxin deoxynivalenol. Toxins 2013, 5, 784–820. [Google Scholar] [CrossRef]
- Ueno, Y. The toxicology of mycotoxins. Crit. Rev. Toxicol. 1985, 14, 99–132. [Google Scholar] [CrossRef]
- Grovey, J.F. The trichothecenes and their biosynthesis. In Progress in the Chemistry of Organic Natural Products; Herz, W., Falk, H., Kirby, G.W., Eds.; Springer Vienna: Vienna, Austria, 2007; Volume 88, pp. 63–130. [Google Scholar]
- McCormick, S.P.; Stanley, A.M.; Stover, N.A.; Alexander, N.J. Trichothecenes: From simple to complex mycotoxins. Toxins 2011, 3, 802–814. [Google Scholar] [CrossRef]
- Fruhmann, P.; Mikula, H.; Wiesenberger, G.; Varga, E.; Lumpi, D.; Stöger, B.; Häubl, G.; Lemmens, M.; Berthiller, F.; Krska, R.; et al. Isolation and structure elucidation of pentahydroxyscirpene, a trichothecene fusarium mycotoxin. J. Nat. Prod. 2014. [Google Scholar] [CrossRef]
- Greenhalgh, R.; Levandier, D.; Adams, W.; Miller, J.D.; Blackwell, B.A.; McAlees, A.J.; Taylor, A. Production and characterization of deoxynivalenol and other secondary metabolites of fusarium culmorum (cmi 14764, hlx 1503). J. Agric. Food Chem. 1986, 34, 98–102. [Google Scholar] [CrossRef]
- Greenhalgh, R.; Meier, R.M.; Blackwell, B.A.; Miller, J.D.; Taylor, A.; ApSimon, J.W. Minor metabolites of fusarium roseum (atcc 28114). J. Agric. Food Chem. 1984, 32, 1261–1264. [Google Scholar] [CrossRef]
- Hanson, A. The structure of a trichothecene from fusarium roseum. Acta Crystallogr. Sect. C 1986, 42, 503–505. [Google Scholar] [CrossRef]
- Hesketh, A.R.; Gledhill, L.; Marsh, D.C.; Bycroft, B.W.; Dewick, P.M.; Gilbert, J. Biosynthesis of trichothecene mycotoxins: Identification of isotrichodiol as a post-trichodiene intermediate. Phytochemistry 1991, 30, 2237–2243. [Google Scholar] [CrossRef]
- Hesketh, A.R.; gledhill, L.; Bycroft, B.W.; Dewick, P.M.; Gilbert, J. Potential inhibitors of trichothecene biosynthesis in fusarium culmorum: Epoxidation of a trichodiene derivative. Phytochemistry 1992, 32, 93–104. [Google Scholar] [CrossRef]
- Kononenko, G.P.; Soboleva, N.A.; Leonov, A.N. 3,7,8,15-tetrahydroxy-12,13-epoxytrichothec-9-en in a culture of fusarium graminearum. Chem. Nat. Compd. 1990, 26, 219–220. [Google Scholar] [CrossRef]
- Gemal, A.L.; Luche, J.L. Lanthanoids in organic synthesis. 6. Reduction of Alpha.-enones by sodium borohydride in the presence of lanthanoid chlorides: Synthetic and mechanistic aspects. J. Am. Chem. Soc. 1981, 103, 5454–5459. [Google Scholar] [CrossRef]
- Luche, J.L. Lanthanides in organic chemistry. 1. Selective 1,2 reductions of conjugated ketones. J. Am. Chem. Soc. 1978, 100, 2226–2227. [Google Scholar]
- Šťastná, E.; Černý, I.; Pouzar, V.; Chodounská, H. Stereoselectivity of sodium borohydride reduction of saturated steroidal ketones utilizing conditions of luche reduction. Steroids 2010, 75, 721–725. [Google Scholar] [CrossRef]
- Cram, D.J.; Elhafez, F.A.A. Studies in stereochemistry. X. The rule of “steric control of asymmetric induction” in the syntheses of acyclic systems. J. Am. Chem. Soc. 1952, 74, 5828–5835. [Google Scholar] [CrossRef]
- Savard, M.E.; Blackwell, B.A.; Greenhalgh, R. An 1 H nuclear magnetic resonance study of derivatives of 3-hydroxy-12,13-epoxytrichothec-9-enes. Can. J. Chem. 1987, 65, 2254–2262. [Google Scholar] [CrossRef]
- Grove, J.F.; McAlees, A.J.; Taylor, A. Preparation of 10-g quantities of 15-O-acetyl-4-deoxynivalenol. J. Org. Chem. 1988, 53, 3860–3862. [Google Scholar] [CrossRef]
- Pirrung, M.C. Appendix 3: Recipes for TLC stains. In The Synthetic Organic Chemist’s Companion; John Wiley & Sons, Inc.: Hoboken, NJ, USA, 2006; pp. 171–172. [Google Scholar]
- Fulmer, G.R.; Miller, A.J.M.; Sherden, N.H.; Gottlieb, H.E.; Nudelman, A.; Stoltz, B.M.; Bercaw, J.E.; Goldberg, K.I. NMR chemical shifts of trace impurities: Common laboratory solvents, organics, and gases in deuterated solvents relevant to the organometallic chemist. Organometallics 2010, 29, 2176–2179. [Google Scholar] [CrossRef]
- Blackwell, B.A.; Greenhalgh, R.; Bain, A.D. Carbon-13 and proton nuclear magnetic resonance spectral assignments of deoxynivalenol and other mycotoxins from fusarium graminearum. J. Agric. Food Chem. 1984, 32, 1078–1083. [Google Scholar] [CrossRef]
- Muñoz, L.; Castro, J.L.; Cardelle, M.; Castedo, L.; Riguera, R. Acetylated mycotoxins from fusarium graminearum. Phytochemistry 1989, 28, 83–85. [Google Scholar]
- Cravero, R.M.; González-Sierra, M.; Labadie, G.R. Convergent approaches to saudin intermediates. Helv. Chim. Acta 2003, 86, 2741–2753. [Google Scholar] [CrossRef]
- Rafiński, Z.; Ścianowski, J. Synthesis and reactions of enantiomerically pure dialkyl diselenides from the p-menthane group. Tetrahedron 2008, 19, 1237–1244. [Google Scholar]
Supplementary Files
© 2014 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Fruhmann, P.; Hametner, C.; Mikula, H.; Adam, G.; Krska, R.; Fröhlich, J. Stereoselective Luche Reduction of Deoxynivalenol and Three of Its Acetylated Derivatives at C8. Toxins 2014, 6, 325-336. https://doi.org/10.3390/toxins6010325
Fruhmann P, Hametner C, Mikula H, Adam G, Krska R, Fröhlich J. Stereoselective Luche Reduction of Deoxynivalenol and Three of Its Acetylated Derivatives at C8. Toxins. 2014; 6(1):325-336. https://doi.org/10.3390/toxins6010325
Chicago/Turabian StyleFruhmann, Philipp, Christian Hametner, Hannes Mikula, Gerhard Adam, Rudolf Krska, and Johannes Fröhlich. 2014. "Stereoselective Luche Reduction of Deoxynivalenol and Three of Its Acetylated Derivatives at C8" Toxins 6, no. 1: 325-336. https://doi.org/10.3390/toxins6010325