The Versatility of the Helicobacter pylori Vacuolating Cytotoxin VacA in Signal Transduction and Molecular Crosstalk

Abstract
:1. Introduction
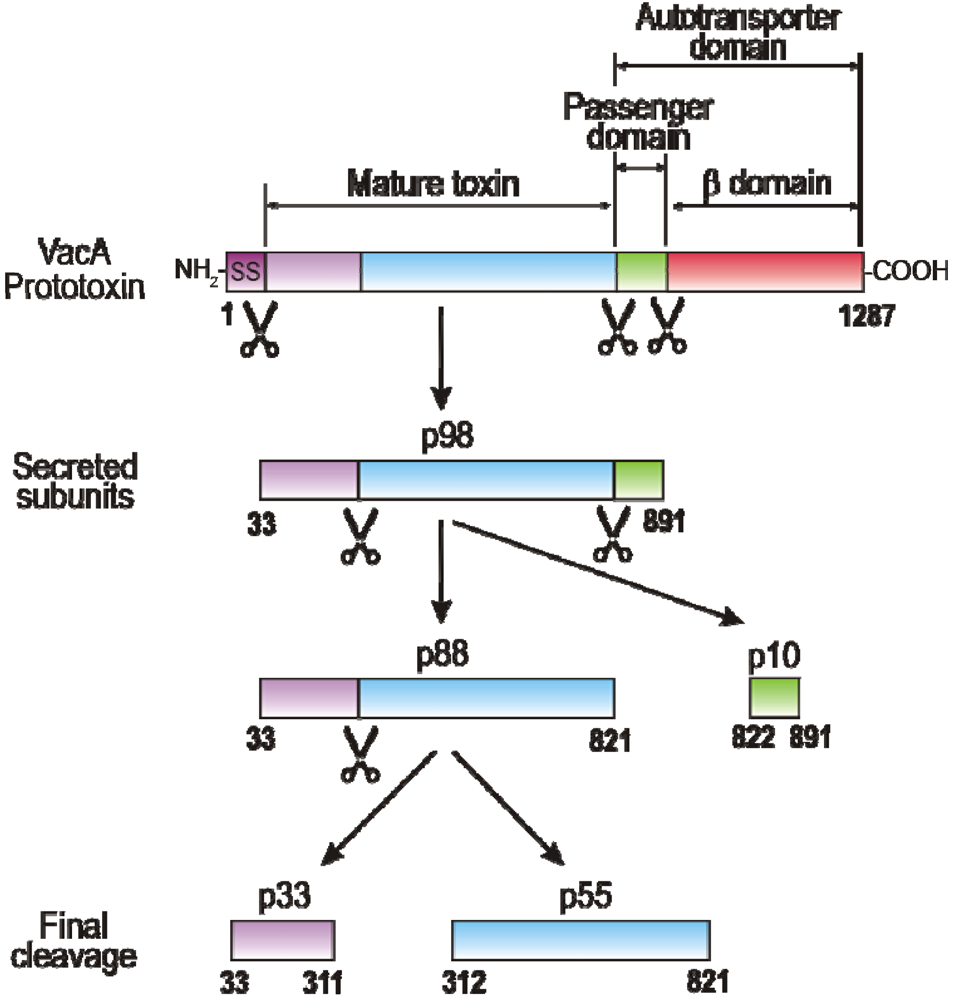
2. VacA Expression, Secretion, Maturation and Allelic Variation
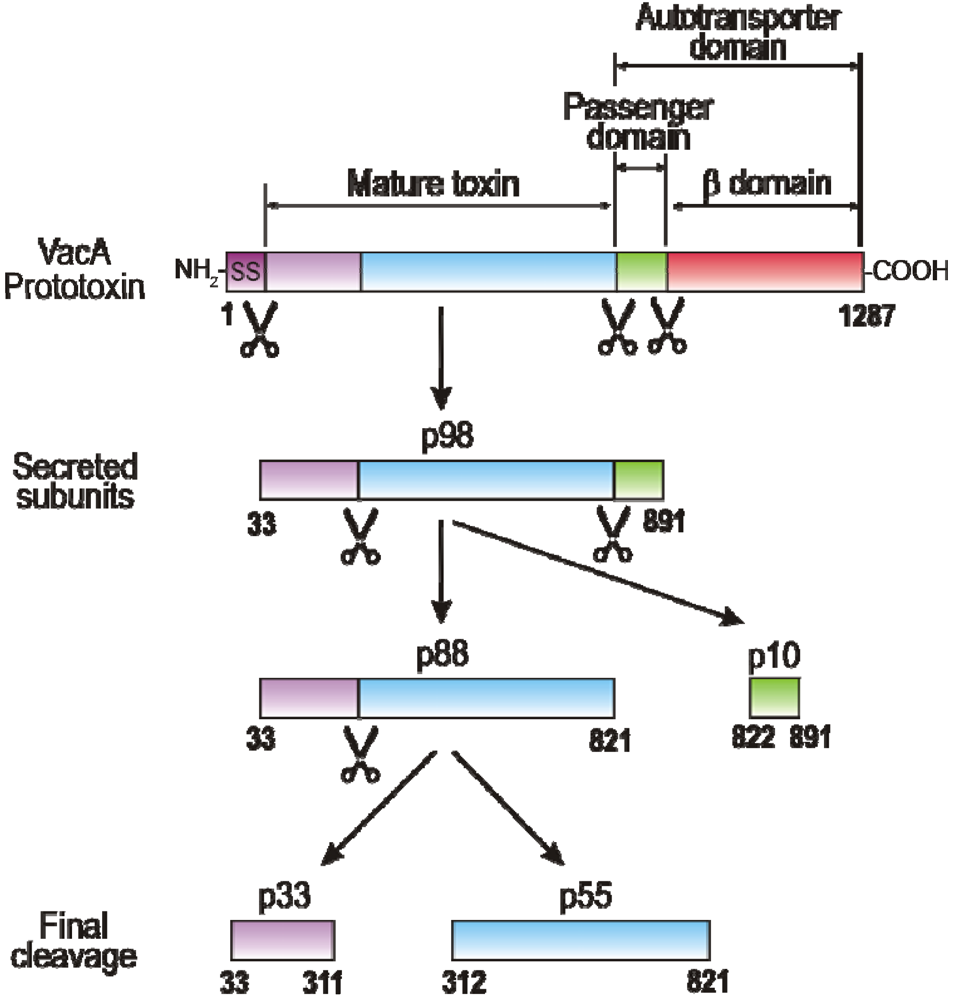
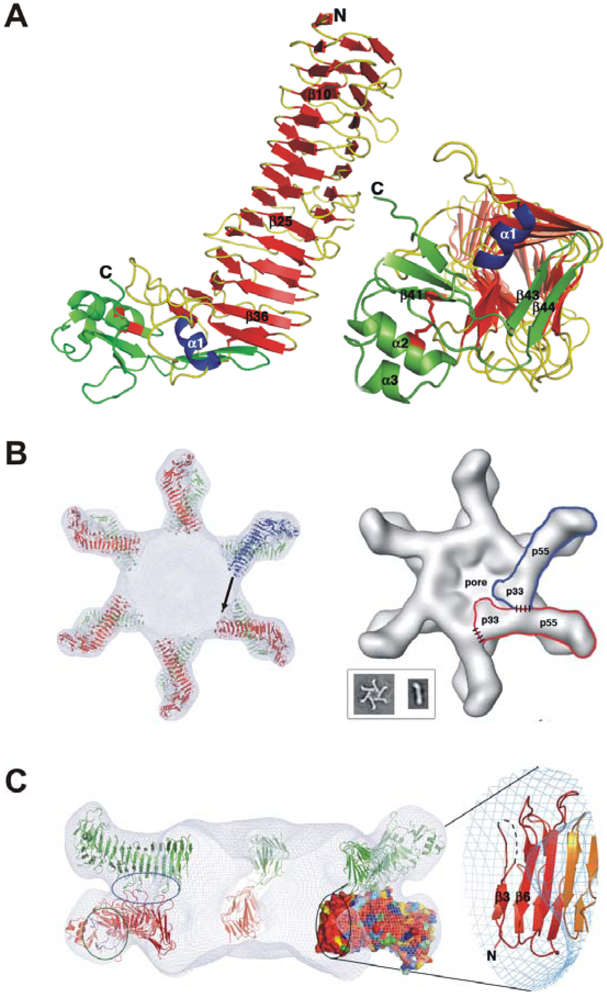
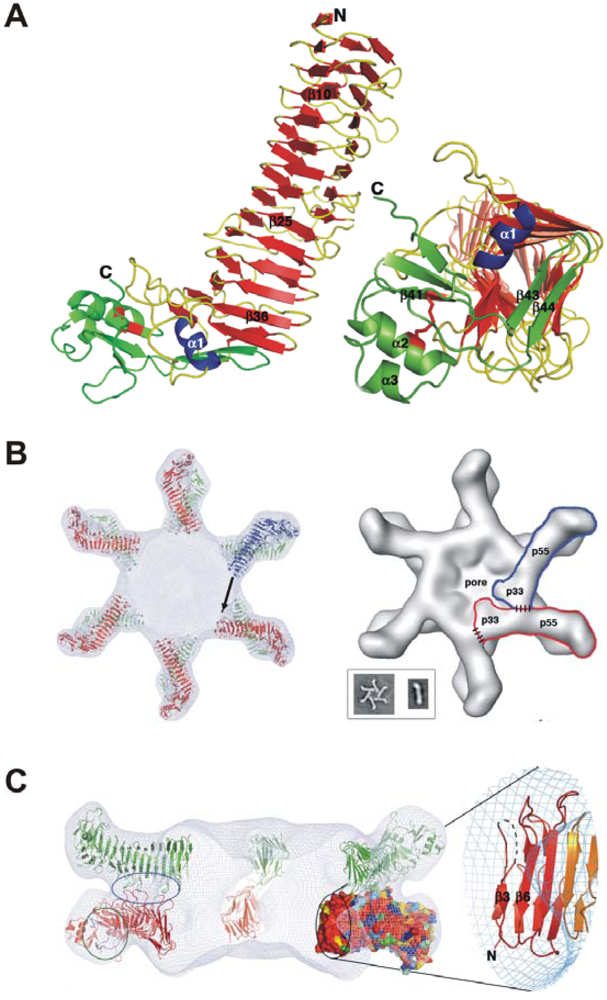
3. Domains of VacA and Crystal Structure of the p55 Subunit
4. VacA Targets Various Surface Factors Both on Epithelial and Immune Cells

{kind=link}
{kind=link}
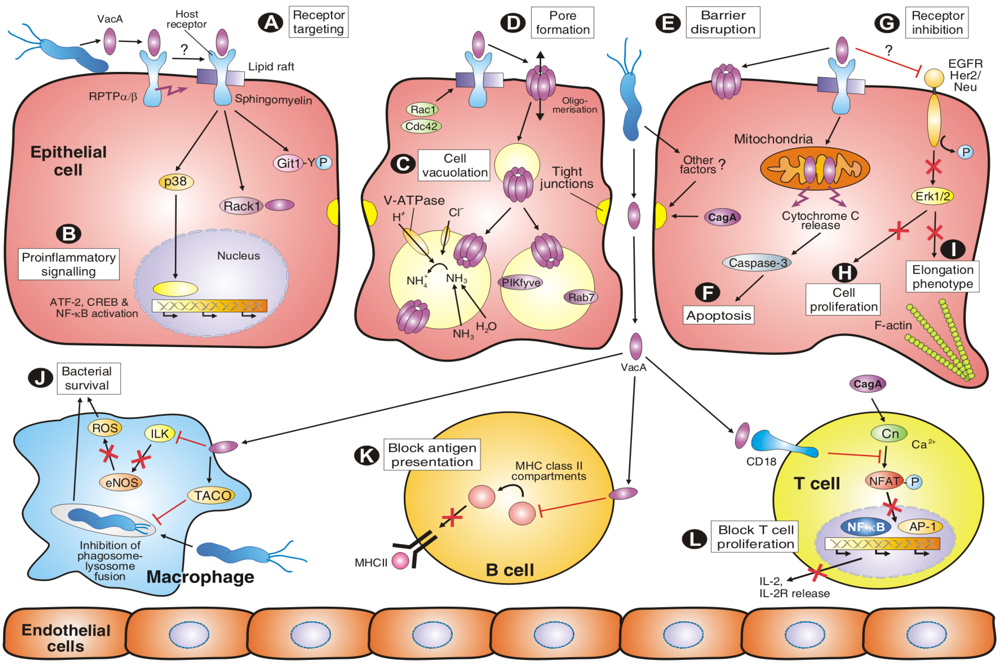{kind=link}
| VacA binding partners | Proposed role in VacA-host interactions | Methods used | Experimental evidenceb | Reference |
|---|---|---|---|---|
| EGF receptor | Receptor on epithelial cells | Antibody blocking, IP | Infection in vitro | [61] |
| Fibronectin | Binding of H. pylori to host cells | Binding and cell adhesion and assays | Binding in vitro | [60] |
| Glycosphingolipids | Binding partner for VacA internalization | Binding assays, Chromatography, MS | Binding in vitro | [65] |
| Heparin sulphate | Receptor/co-receptor on epithelial cells | SPR-based biosensor studies | Binding in vitro | [58] |
| Integrin β2 (CD18) | Receptor on T cells | Flow cytometry, IF, live cell imaging, use of knockout cells | Treatment of cells in vitro, Infection in vitro | [62] |
| Lipid bilayers | Low pH-triggered pore formation | AFM | Binding in vitro | [41] |
| Lipid rafts | Putative docking and entering site at cell surface | Cell fractionation, flow cytometry, IF, MCD inhibitor studies | Treatment of cells in vitro | [66,67,64] |
| Lipid vesicles | Binding and host cell entry of the toxin | Light scattering, energy transfer studies | Binding in vitro | [56] |
| Phospho-lipids | Binding and host cell entry of the toxin | ANS-binding studies, Photolabelling | Binding in vitro | [57] |
| RPTP-alpha | Receptor on epithelial cells | IP, MS, antisense silencing studies | Infection in vitro, Treatment of cells in vitro | [54,68] |
| RPTP-beta | Receptor on epithelial cells, receptor for Git1 phosphorylation | Flow cytometry, IF, IP, siRNA, use of knockout mice and cells | Binding in vitro, Infection of mice and cells | [39,63,55] |
| RACK-1 | Yet unknown | PD, Y2H | Binding in vitro | [69] |
| Sphingo-myelin | Receptor on epithelial cells | Binding studies, ELISA, flow cytometry, treatment with sphingomyelinase | Binding and treatment of cells in vitro | [59] |
5. VacA Activities on Endosomal Maturation and Cell Vacuolation
6. Amino-Terminal VacA Targets Mitochondria to Induce Apoptosis
7. VacA and Other Factors Affecting the Transepithelial Resistance of Polarized Cell Monolayers
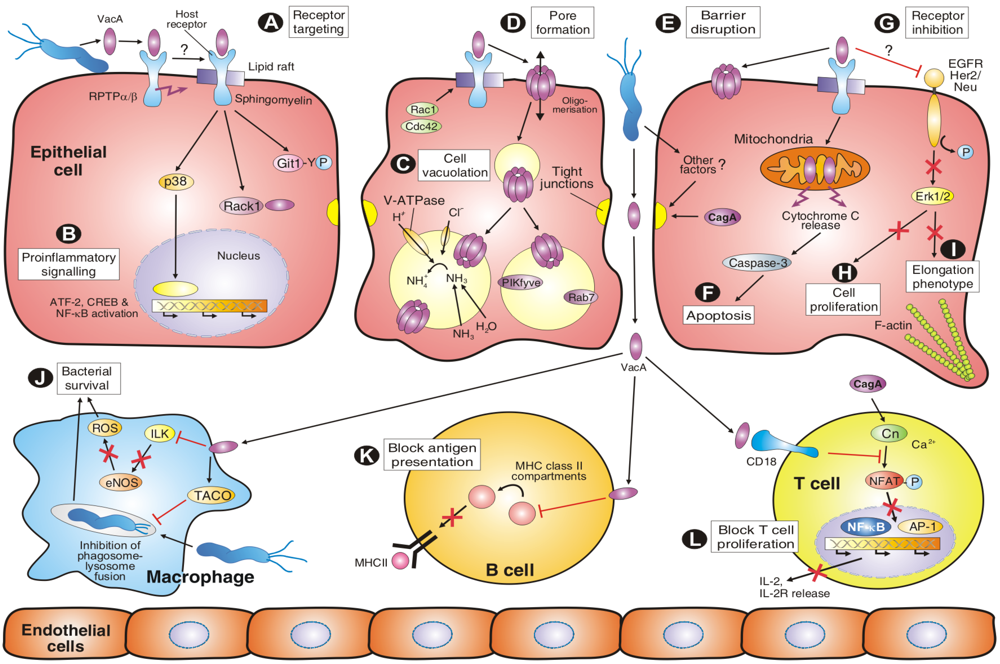
8. VacA Elicits Suppressive Activities on Immune Cell Proliferation
9. VacA-Dependent Pro-Inflammatory Signalling Activities
10. Molecular Crosstalk between VacA and CagA Signalling Pathways
10.1. Role of VacA and CagA in Apoptotic and Anti-Apoptotic Signalling
10.2. Role of VacA and CagA in NFAT Signalling
10.3. Role of VacA and CagA in the Vacuolation and Cell Elongation Phenotypes
10.4. Role of VacA and T4SS in Epidermal Growth Factor Receptor (EGFR) Signalling
11. Other VacA-Dependent Signalling Activities
12. Conclusions
Acknowledgements
References
- Montecucco, C.; Rappuoli, R. Living dangerously: How Helicobacter pylori survives in the human stomach. Nat. Rev. Mol. Cell Biol. 2001, 2, 457–466. [Google Scholar]
- Peek, R.M.; Blaser, M.J. Helicobacter pylori and gastrointestinal tract adenocarcinomas. Nat. Rev. Cancer 2002, 2, 28–37. [Google Scholar]
- Sepulveda, A.R.; Graham, D.Y. Role of Helicobacter pylori in gastric carcinogenesis. Gastroenterol. Clin. North Amer. 2002, 31, 517–535. [Google Scholar]
- Monack, D.M.; Mueller, A.; Falkow, S. Persistent bacterial infections: The interface of the pathogen and the host immune system. Nat. Rev. Microbiol. 2004, 2, 747–765. [Google Scholar]
- Kusters, J.G.; van Vliet, A.H.; Kuipers, E.J. Pathogenesis of Helicobacter pylori infection. Clin. Microbiol. Rev. 2006, 19, 449–490. [Google Scholar]
- Correa, P.; Houghton, J. Carcinogenesis of Helicobacter pylori. Gastroenterology 2007, 133, 659–672. [Google Scholar]
- Blaser, M.J.; Atherton, J.C. Helicobacter pylori persistence: Biology and disease. J. Clin. Invest. 2004, 113, 321–333. [Google Scholar]
- Cover, T.L.; Blanke, S.R. Helicobacter pylori VacA, a paradigm for toxin multifunctionality. Nat. Rev. Microbiol. 2005, 3, 320–332. [Google Scholar] [CrossRef] [PubMed]
- Rieder, G.; Fischer, W.; Haas, R. Interaction of Helicobacter pylori with host cells: Function of secreted and translocated molecules. Curr. Opin. Microbiol. 2005, 8, 67–73. [Google Scholar]
- Wilson, K.T.; Crabtree, J.E. Immunology of Helicobacter pylori: Insights into the failure of the immune response and perspectives on vaccine studies. Gastroenterology 2007, 133, 288–308. [Google Scholar]
- Backert, S.; Selbach, M. Role of type IV secretion in Helicobacter pylori pathogenesis. Cell Microbiol. 2008, 10, 1573–1581. [Google Scholar]
- Amieva, M.R.; El-Omar, E.M. Host-bacterial interactions in Helicobacter pylori infection. Gastroenterology 2008, 134, 306–323. [Google Scholar]
- Graham, D.Y.; Yamaoka, Y. Disease-specific Helicobacter pylori virulence factors: The unfulfilled promise. Helicobacter 2000, 5, 3–9. [Google Scholar]
- Viala, J.; Chaput, C.; Boneca, I.G.; Cardona, A.; Girardin, S.E.; Moran, A.P.; Athman, R.; Mémet, S.; Huerre, M.R.; Coyle, A.J.; DiStefano, P.S.; Sansonetti, P.J.; Labigne, A.; Bertin, J.; Philpott, D.J.; Ferrero, R.L. Nod1 responds to peptidoglycan delivered by the Helicobacter pylori cag pathogenicity island. Nat. Immunol. 2004, 5, 1166–1174. [Google Scholar]
- Dubois, A.; Borén, T. Helicobacter pylori is invasive and it may be a facultative intracellular organism. Cell. Microbiol. 2007, 9, 1108–1116. [Google Scholar]
- Suerbaum, S.; Josenhans, C. Helicobacter pylori evolution and phenotypic diversification in a changing host. Nat. Rev. Microbiol. 2007, 5, 441–452. [Google Scholar]
- Boquet, P.; Ricci, V.; Galmiche, A.; Gauthier, N.C. Gastric cell apoptosis and H. pylori: Has the main function of VacA finally been identified? Trends Microbiol. 2003, 11, 410–413. [Google Scholar] [CrossRef] [PubMed]
- Backert, S.; Meyer, T.F. Type IV secretion systems and their effectors in bacterial pathogenesis. Curr. Opin. Microbiol. 2006, 9, 207–217. [Google Scholar]
- Kwok, T.; Zabler, D.; Urman, S.; Rohde, M.; Hartig, R.; Wessler, S.; Misselwitz, R.; Berger, J.; Sewald, N.; König, W.; Backert, S. Helicobacter exploits integrin for type IV secretion and kinase activation. Nature 2007, 449, 862–866. [Google Scholar]
- Hatakeyama, M. SagA of CagA in Helicobacter pylori pathogenesis. Curr. Opin. Microbiol. 2008, 11, 30–37. [Google Scholar]
- Brandt, S.; Kwok, T.; Hartig, R.; König, W.; Backert, S. NF-κB activation and potentiation of proinflammatory responses by the Helicobacter pylori CagA protein. Proc. Natl. Acad. Sci. USA 2005, 102, 9300–9305. [Google Scholar]
- Mimuro, H.; Suzuki, T.; Nagai, S.; Rieder, G.; Suzuki, M.; Nagai, T.; Fujita, Y.; Nagamatsu, K.; Ishijima, N.; Koyasu, S.; Haas, R.; Sasakawa, C. Helicobacter pylori dampens gut epithelial self-renewal by inhibiting apoptosis, a bacterial strategy to enhance colonization of the stomach. Cell Host Microbe 2007, 2, 250–263. [Google Scholar] [CrossRef] [PubMed]
- Selbach, M.; Paul, F.E.; Brandt, S.; Guye, P.; Daumke, O.; Backert, S.; Dehio, C.; Mann, M. Host cell interactome of tyrosine-phosphorylated bacterial proteins. Cell Host Microbe 2009, 5, 397–403. [Google Scholar]
- Keates, S.; Sougioultzis, S.; Keates, A.C.; Zhao, D.; Peek, R.M.; Shaw, L.M.; Kelly, C.P. cag+ Helicobacterpylori induce transactivation of the epidermal growth factor receptor in AGS gastric epithelial cells. J. Biol. Chem. 2001, 276, 48127–48134. [Google Scholar] [PubMed]
- Wallasch, C.; Crabtree, J.E.; Bevec, D.; Robinson, P.A.; Wagner, H.; Ullrich, A. Helicobacter pylori-stimulated EGF receptor transactivation requires metalloprotease cleavage of HB-EGF. Biochem. Biophys. Res. Commun. 2002, 29, 695–701. [Google Scholar]
- Churin, Y.; Kardalinou, E.; Meyer, T.F.; Naumann, M. Pathogenicity island-dependent activation of Rho GTPases Rac1 and Cdc42 in Helicobacter pylori infection. Mol. Microbiol. 2001, 40, 815–823. [Google Scholar]
- Suzuki, M.; Mimuro, H.; Suzuki, T.; Park, M.; Yamamoto, T.; Sasakawa, C. Interaction of CagA-Crk plays an important role in Helicobacter pylori-induced loss of gastric epithelial cell adhesion. J. Exp. Med. 2005, 202, 1235–1247. [Google Scholar]
- Brandt, S.; Shafikhani, S.; Balachandran, P.; Jin, S.; Hartig, R.; König, W.; Engel, J.; Backert, S. Use of a novel coinfection system reveals a role for Rac1, H-Ras, and CrkII phosphorylation in Helicobacter pylori-induced host cell actin cytoskeletal rearrangements. FEMS Immunol. Med. Microbiol. 2007, 50, 190–205. [Google Scholar] [CrossRef] [PubMed]
- Leunk, R.D.; Johnson, P.T.; David, B.C.; Kraft, W.G.; Morgan, D.R. Cytotoxic activity in broth-culture filtrates of Campylobacter pylori. J. Med. Microbiol. 1988, 26, 93–99. [Google Scholar]
- Bumann, D.; Aksu, S.; Wendland, M.; Janek, K.; Zimny-Arndt, U.; Sabarth, N.; Meyer, T.F.; Jungblut, P.R. Proteome analysis of secreted proteins of the gastric pathogen Helicobacter pylori. Infect. Immun. 2002, 70, 3396–3403. [Google Scholar]
- Backert, S.; Kwok, T.; Schmid, M.; Selbach, M.; Moese, S.; Peek, R.M.; König, W.; Meyer, T.F.; Jungblut, P.R. Subproteomes of soluble and structure-bound Helicobacter pylori proteins analyzed by two-dimensional gel electrophoresis and mass spectrometry. Proteomics 2005, 5, 1331–1345. [Google Scholar]
- Telford, J.L.; Ghiara, P.; Dell'Orco, M.; Comanducci, M.; Burroni, D.; Bugnoli, M.; Tecce, M.F.; Censini, S.; Covacci, A.; Xiang, Z. Gene structure of the Helicobacter pylori cytotoxin and evidence of its key role in gastric disease. J. Exp. Med. 1994, 179, 1653–1658. [Google Scholar]
- Lupetti, P.; Heuser, J.E.; Manetti, R.; Massari, P.; Lanzavecchia, S.; Bellon, P.L.; Dallai, R.; Rappuoli, R.; Telford, J.L. Oligomeric and subunit structure of the Helicobacter pylori vacuolating cytotoxin. J. Cell Biol. 1996, 133, 801–807. [Google Scholar]
- Ricci, V.; Sommi, P.; Fiocca, R.; Romano, M.; Solcia, E.; Ventura, U. Helicobacter pylori vacuolating toxin accumulates within the endosomal-vacuolar compartment of cultured gastric cells and potentiates the vacuolating activity of ammonia. J. Pathol. 1997, 183, 453–459. [Google Scholar]
- Rhead, J.L.; Letley, D.P.; Mohammadi, M.; Hussein, N.; Mohagheghi, M.A.; Eshagh Hosseini, M.; Atherton, J.C. A new Helicobacter pylori vacuolating cytotoxin determinant, the intermediate region, is associated with gastric cancer. Gastroenterology 2007, 133, 926–936. [Google Scholar] [CrossRef] [PubMed]
- Pagliaccia, C.; de Bernard, M.; Lupetti, P.; Ji, X.; Burroni, D.; Cover, T.L.; Papini, E.; Rappuoli, R.; Telford, J.L.; Reyrat, J.M. The m2 form of the Helicobacter pylori cytotoxin has cell type-specific vacuolating activity. Proc. Natl. Acad. Sci. USA 1998, 95, 10212–10217. [Google Scholar]
- Reyrat, J.M.; Lanzavecchia, S.; Lupetti, P.; de Bernard, M.; Pagliaccia, C.; Pelicic, V.; Charrel, M.; Ulivieri, C.; Norais, N.; Ji, X.; Cabiaux, V.; Papini, E.; Rappuoli, R.; Telford, J.L. 3D imaging of the 58 kDa cell binding subunit of the Helicobacter pylori cytotoxin. J. Mol. Biol. 1999, 290, 459–470. [Google Scholar]
- de Bernard, M.; Papini, E.; de Filippis, V.; Gottardi, E.; Telford, J.; Manetti, R.; Fontana, A.; Rappuoli, R.; Montecucco, C. Low pH activates the vacuolating toxin of Helicobacter pylori, which becomes acid and pepsin resistant. J. Biol. Chem. 1995, 270, 23937–23940. [Google Scholar] [PubMed]
- Yahiro, K.; Niidome, T.; Kimura, M.; Hatakeyama, T.; Aoyagi, H.; Kurazono, H.; Imagawa, K.; Wada, A.; Moss, J.; Hirayama, T. Activation of Helicobacter pylori VacA toxin by alkaline or acid conditions increases its binding to a 250-kDa receptor protein-tyrosine phosphatase β. J. Biol. Chem. 1999, 274, 36693–36699. [Google Scholar]
- Cover, T.L.; Hanson, P.I.; Heuser, J.E. Acid-induced dissociation of VacA, the Helicobacter pylori vacuolating cytotoxin, reveals its pattern of assembly. J. Cell Biol. 1997, 138, 759–769. [Google Scholar] [CrossRef] [PubMed]
- Czajkowsky, D.M.; Iwamoto, H.; Cover, T.L.; Shao, Z. The vacuolating toxin from Helicobacter pylori forms hexameric pores in lipid bilayers at low pH. Proc. Natl. Acad. Sci. USA 1999, 96, 2001–2006. [Google Scholar]
- Tombola, F.; Carlesso, C.; Szabó, I.; de Bernard, M.; Reyrat, J.M.; Telford, J.L.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Helicobacter pylori vacuolating toxin forms anion-selective channels in planar lipid bilayers: Possible implications for the mechanism of cellular vacuolation. Biophys. J. 1999, 76, 1401–1409. [Google Scholar] [CrossRef] [PubMed]
- Iwamoto, H.; Czajkowsky, D.M.; Cover, T.L.; Szabo, G.; Shao, Z. VacA from Helicobacter pylori: A hexameric chloride channel. FEBS Lett. 1999, 450, 101–104. [Google Scholar]
- El-Bez, C.; Adrian, M.; Dubochet, J.; Cover, T.L. High resolution structural analysis of Helicobacter pylori VacA toxin oligomers by cryo-negative staining electron microscopy. J. Struct. Biol. 2005, 151, 215–228. [Google Scholar]
- Ye, D.; Willhite, D.C.; Blanke, S.R. Identification of the minimal intracellular vacuolating domain of the Helicobacter pylori vacuolating toxin. J. Biol. Chem. 1999, 274, 9277–9282. [Google Scholar]
- Torres, V.J.; McClain, M.S.; Cover, T.L. Interactions between p-33 and p-55 domains of the Helicobacter pylori vacuolating cytotoxin (VacA). J. Biol. Chem. 2004, 279, 2324–2331. [Google Scholar]
- Torres, V.J.; Ivie, S.E.; McClain, M.S.; Cover, T.L. Functional properties of the p33 and p55 domains of the Helicobacter pylori vacuolating cytotoxin. J. Biol. Chem. 2005, 280, 21107–21114. [Google Scholar]
- Vinion-Dubiel, A.D.; McClain, M.S.; Czajkowsky, D.M.; Iwamoto, H.; Ye, D.; Cao, P.; Schraw, W.; Szabo, G.; Blanke, S.R.; Shao, Z.; Cover, T.L. A dominant negative mutant of Helicobacter pylori vacuolating toxin (VacA) inhibits VacA-induced cell vacuolation. J. Biol. Chem. 1999, 274, 37736–37742. [Google Scholar]
- McClain, M.S.; Iwamoto, H.; Cao, P.; Vinion-Dubiel, A.D.; Li, Y.; Szabo, G.; Shao, Z.; Cover, T.L. Essential role of a GXXXG motif for membrane channel formation by Helicobacter pylori vacuolating toxin. J. Biol. Chem. 2003, 278, 12101–12108. [Google Scholar]
- Wang, H.J.; Wang, W.C. Expression and binding analysis of GST-VacA fusions reveals that the C-terminal approximately 100-residue segment of exotoxin is crucial for binding in HeLa cells. Biochem. Biophys. Res. Commun. 2000, 278, 449–454. [Google Scholar]
- Gangwer, K.A.; Mushrush, D.J.; Stauff, D.L.; Spiller, B.; McClain, M.S.; Cover, T.L.; Lacy, D.B. Crystal structure of the Helicobacter pylori vacuolating toxin p55 domain. Proc. Natl. Acad. Sci. USA 2007, 104, 16293–16298. [Google Scholar]
- Ricci, V.; Galmiche, A.; Doye, A.; Necchi, V.; Solcia, E.; Boquet, P. High cell sensitivity to Helicobacter pylori VacA toxin depends on a GPI-anchored protein and is not blocked by inhibition of the clathrin-mediated pathway of endocytosis. Mol. Biol. Cell. 2000, 11, 3897–3909. [Google Scholar]
- Gauthier, N.C.; Monzo, P.; Kaddai, V.; Doye, A.; Ricci, V.; Boquet, P. Helicobacter pylori VacA cytotoxin: A probe for a clathrin-independent and Cdc42-dependent pinocytic pathway routed to late endosomes. Mol. Biol. Cell. 2005, 16, 4852–4866. [Google Scholar]
- Yahiro, K.; Wada, A.; Nakayama, M.; Kimura, T.; Ogushi, K.; Niidome, T.; Aoyagi, H.; Yoshino, K.; Yonezawa, K.; Moss, J.; Hirayama, T. Protein-tyrosine phosphatase alpha, RPTPα, is a Helicobacter pylori VacA receptor. J. Biol. Chem. 2003, 278, 19183–19189. [Google Scholar] [PubMed]
- Fujikawa, A.; Shirasaka, D.; Yamamoto, S.; Ota, H.; Yahiro, K.; Fukada, M.; Shintani, T.; Wada, A.; Aoyama, N.; Hirayama, T.; Fukamachi, H.; Noda, M. Mice deficient in protein tyrosine phosphatase receptor type Z are resistant to gastric ulcer induction by VacA of Helicobacter pylori. Nat. Genet. 2003, 33, 375–381. [Google Scholar]
- Moll, G.; Papini, E.; Colonna, R.; Burroni, D.; Telford, J.; Rappuoli, R.; Montecucco, C. Lipid interaction of the 37-kDa and 58-kDa fragments of the Helicobacter pylori cytotoxin. Eur. J. Biochem. 1995, 234, 947–952. [Google Scholar]
- Molinari, M.; Galli, C.; de Bernard, M.; Norais, N.; Ruysschaert, J.M.; Rappuoli, R.; Montecucco, C. The acid activation of Helicobacter pylori toxin VacA: Structural and membrane binding studies. Biochem. Biophys. Res. Commun. 1998, 248, 334–340. [Google Scholar]
- Utt, M.; Danielsson, B.; Wadström, T. Helicobacter pylori vacuolating cytotoxin binding to a putative cell surface receptor, heparan sulfate, studied by surface plasmon resonance. FEMS Immunol. Med. Microbiol. 2001, 30, 109–113. [Google Scholar] [CrossRef] [PubMed]
- Gupta, V.R.; Patel, H.K.; Kostolansky, S.S.; Ballivian, R.A.; Eichberg, J.; Blanke, S.R. Sphingomyelin functions as a novel receptor for Helicobacter pylori VacA. PLoS Pathog. 2008, 4. [Google Scholar]
- Hennig, E.E.; Godlewski, M.M.; Butruk, E.; Ostrowski, J. Helicobacter pylori VacA cytotoxin interacts with fibronectin and alters HeLa cell adhesion and cytoskeletal organization in vitro. FEMS Immunol. Med. Microbiol. 2005, 44, 143–150. [Google Scholar]
- Seto, K.; Hayashi-Kuwabara, Y.; Yoneta, T.; Suda, H.; Tamaki, H. Vacuolation induced by cytotoxin from Helicobacter pylori is mediated by the EGF receptor in HeLa cells. FEBS Lett. 1998, 431, 347–350. [Google Scholar]
- Sewald, X.; Gebert-Vogl, B.; Prassl, S.; Barwig, I.; Weiss, E.; Fabbri, M.; Osicka, R.; Schiemann, M.; Busch, D.H.; Semmrich, M.; Holzmann, B.; Sebo, P.; Haas, R. Integrin subunit CD18 Is the T-lymphocyte receptor for the Helicobacter pylori vacuolating cytotoxin. Cell Host Microbe 2008, 3, 20–29. [Google Scholar]
- Padilla, P.I.; Wada, A.; Yahiro, K.; Kimura, M.; Niidome, T.; Aoyagi, H.; Kumatori, A.; Anami, M.; Hayashi, T.; Fujisawa, J.; Saito, H.; Moss, J.; Hirayama, T. Morphologic differentiation of HL-60 cells is associated with appearance of RPTPβ and induction of Helicobacter pylori VacA sensitivity. J. Biol. Chem. 2000, 275, 15200–15206. [Google Scholar] [PubMed]
- Nakayama, M.; Hisatsune, J.; Yamasaki, E.; Nishi, Y.; Wada, A.; Kurazono, H.; Sap, J.; Yahiro, K.; Moss, J.; Hirayama, T. Clustering of Helicobacter pylori VacA in lipid rafts, mediated by its receptor, receptor-like protein tyrosine phosphatase β, is required for intoxication in AZ-521 Cells. Infect. Immun. 2006, 74, 6571–6580. [Google Scholar] [CrossRef] [PubMed]
- Roche, N.; Ilver, D.; Angstrom, J.; Barone, S.; Telford, J.L.; Teneberg, S. Human gastric glycosphingolipids recognized by Helicobacter pylori vacuolating cytotoxin VacA. Microbes Infect. 2007, 9, 605–614. [Google Scholar]
- Schraw, W.; Li, Y.; McClain, M.S.; van der Goot, F.G.; Cover, T.L. Association of Helicobacter pylori vacuolating toxin (VacA) with lipid rafts. J. Biol. Chem. 2002, 277, 34642–34650. [Google Scholar]
- Kuo, C.H.; Wang, W.C. Binding and internalization of Helicobacter pylori VacA via cellular lipid rafts in epithelial cells. Biochem. Biophys. Res. Commun. 2003, 303, 640–644. [Google Scholar]
- De Guzman, B.B.; Hisatsune, J.; Nakayama, M.; Yahiro, K.; Wada, A.; Yamasaki, E.; Nishi, Y.; Yamazaki, S.; Azuma, T.; Ito, Y.; Ohtani, M.; van der Wijk, T.; den Hertog, J.; Moss, J.; Hirayama, T. Cytotoxicity and recognition of receptor-like protein tyrosine phosphatases, RPTPα and RPTPβ, by Helicobacter pylori m2Va. Cell. Microbiol. 2005, 7, 1285–1293. [Google Scholar]
- Hennig, E.E.; Butruk, E.; Ostrowski, J. RACK1 protein interacts with Helicobacter pylori VacA cytotoxin: The yeast two-hybrid approach. Biochem. Biophys. Res. Commun. 2001, 289, 103–110. [Google Scholar]
- Molinari, M.; Galli, C.; Norais, N.; Telford, J.L.; Rappuoli, R.; Luzio, J.P.; Montecucco, C. Vacuoles induced by Helicobacter pylori toxin contain both late endosomal and lysosomal markers. J. Biol. Chem. 1997, 272, 25339–25344. [Google Scholar]
- Gauthier, N.C.; Monzo, P.; Gonzalez, T.; Doye, A.; Oldani, A.; Gounon, P.; Ricci, V.; Cormont, M.; Boquet, P. Early endosomes associated with dynamic F-actin structures are required for late trafficking of H. pylori VacA toxin. J. Cell Biol. 2007, 177, 343–354. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; de Bernard, M.; Bugnoli, M.; Milia, E.; Rappuoli, R.; Montecucco, C. Cell vacuolization induced by Helicobacter pylori: Inhibition by bafilomycins A1, B1, C1 and D. FEMS Microbiol. Lett. 1993, 113, 155–159. [Google Scholar] [CrossRef] [PubMed]
- Papini, E.; Satin, B.; Bucci, C.; de Bernard, M.; Telford, J.L.; Manetti, R.; Rappuoli, R.; Zerial, M.; Montecucco, C. The small GTP binding protein rab7 is essential for cellular vacuolation induced by Helicobacter pylori cytotoxin. EMBO J. 1997, 16, 15–24. [Google Scholar]
- Hotchin, N.A.; Cover, T.L.; Akhtar, N. Cell vacuolation induced by the VacA cytotoxin of Helicobacter pylori is regulated by the Rac1 GTPase. J. Biol. Chem. 2000, 275, 14009–14012. [Google Scholar]
- Ikonomov, O.C.; Sbrissa, D.; Yoshimori, T.; Cover, T.L.; Shisheva, A. PIKfyve Kinase and SKD1 AAA ATPase define distinct endocytic compartments. Only PIKfyve expression inhibits the cell-vacuolating activity of Helicobacter pylori VacA toxin. J. Biol. Chem. 2002, 277, 46785–46790. [Google Scholar] [PubMed]
- de Bernard, M.; Moschioni, M.; Habermann, A.; Griffiths, G.; Montecucco, C. Cell vacuolization induced by Helicobacter pylori VacA cytotoxin does not depend on late endosomal SNAREs. Cell. Microbiol. 2002, 4, 11–18. [Google Scholar]
- Suzuki, J.; Ohnishi, H.; Wada, A.; Hirayama, T.; Ohno, H.; Ueda, N.; Yasuda, H.; Iiri, T.; Wada, Y.; Futai, M.; Mashima, H. Involvement of syntaxin 7 in human gastric epithelial cell vacuolation induced by the Helicobacter pylori-produced cytotoxin VacA. J. Biol. Chem. 2003, 278, 25585–25590. [Google Scholar]
- Suzuki, J.; Ohnsihi, H.; Shibata, H.; Wada, A.; Hirayama, T.; Iiri, T.; Ueda, N.; Kanamaru, C.; Tsuchida, T.; Mashima, H.; Yasuda, H.; Fujita, T. Dynamin is involved in human epithelial cell vacuolation caused by the Helicobacter pylori-produced cytotoxin VacA. J. Clin. Invest. 2001, 107, 363–370. [Google Scholar]
- de Bernard, M.; Burroni, D.; Papini, E.; Rappuoli, R.; Telford, J.; Montecucco, C. Identification of the Helicobacter pylori VacA toxin domain active in the cell cytosol. Infect. Immun. 1998, 66, 6014–6016. [Google Scholar]
- Ye, D.; Blanke, S.R. Functional complementation reveals the importance of intermolecular monomer interactions for Helicobacter pylori VacA vacuolating activity. Mol. Microbiol. 2002, 43, 1243–1253. [Google Scholar]
- Willhite, D.C.; Ye, D.; Blanke, S.R. Fluorescence resonance energy transfer microscopy of the Helicobacter pylori vacuolating cytotoxin within mammalian cells. Infect. Immun. 2002, 70, 3824–3832. [Google Scholar]
- Kim, S.; Chamberlain, A.K.; Bowie, J.U. Membrane channel structure of Helicobacter pylori vacuolating toxin: Role of multiple GXXXG motifs in cylindrical channels. Proc. Natl. Acad. Sci. USA 2004, 101, 5988–5991. [Google Scholar]
- Szabó, I.; Brutsche, S.; Tombola, F.; Moschioni, M.; Satin, B.; Telford, J.L.; Rappuoli, R.; Montecucco, C.; Papini, E.; Zoratti, M. Formation of anion-selective channels in the cell plasma membrane by the toxin VacA of Helicobacter pylori is required for its biological activity. EMBO J. 1999, 18, 5517–5527. [Google Scholar]
- Kimura, M.; Goto, S.; Wada, A.; Yahiro, K.; Niidome, T.; Hatakeyama, T.; Aoyagi, H.; Hirayama, T.; Kondo, T. Vacuolating cytotoxin purified from Helicobacter pylori causes mitochondrial damage in human gastric cells. Microb. Pathog. 1999, 26, 45–52. [Google Scholar]
- Galmiche, A.; Rassow, J.; Doye, A.; Cagnol, S.; Chambard, J.C.; Contamin, S.; de Thillot, V.; Just, I.; Ricci, V.; Solcia, E.; Van Obberghen, E.; Boquet, P. The N-terminal 34 kDa fragment of Helicobacter pylori vacuolating cytotoxin targets mitochondria and induces cytochrome c release. EMBO J. 2000, 19, 6361–6370. [Google Scholar]
- Kuck, D.; Kolmerer, B.; Iking-Konert, C.; Krammer, P.H.; Stremmel, W.; Rudi, J. Vacuolating cytotoxin of Helicobacter pylori induces apoptosis in the human gastric epithelial cell line AGS. Infect. Immun. 2001, 69, 5080–5087. [Google Scholar]
- Cover, T.L.; Krishna, U.S.; Israel, D.A.; Peek, R.M. Induction of gastric epithelial cell apoptosis by Helicobacter pylori vacuolating cytotoxin. Cancer Res. 2003, 63, 951–957. [Google Scholar]
- Chiozzi, V.; Mazzini, G.; Oldani, A.; Sciullo, A.; Ventura, U.; Romano, M.; Boquet, P.; Ricci, V. Relationship between Vac A toxin and ammonia in Helicobacter pylori-induced apoptosis in human gastric epithelial cells. J. Physiol. Pharmacol. 2009, 60, 23–30. [Google Scholar]
- Papini, E.; Satin, B.; Norais, N.; de Bernard, M.; Telford, J.L.; Rappuoli, R.; Montecucco, C. Selective increase of the permeability of polarized epithelial cell monolayers by Helicobacter pylori vacuolating toxin. J. Clin. Invest. 1998, 102, 813–820. [Google Scholar]
- Pelicic, V.; Reyrat, J.M.; Sartori, L.; Pagliaccia, C.; Rappuoli, R.; Telford, J.L.; Montecucco, C.; Papini, E. Helicobacter pylori VacA cytotoxin associated with the bacteria increases epithelial permeability independently of its vacuolating activity. Microbiology 1999, 145, 2043–2050. [Google Scholar]
- Tombola, F.; Morbiato, L.; Del Giudice, G.; Rappuoli, R.; Zoratti, M.; Papini, E. The Helicobacter pylori VacA toxin is a urea permease that promotes urea diffusion across epithelia. J. Clin. Invest. 2001, 108, 929–937. [Google Scholar]
- Amieva, M.R.; Vogelmann, R.; Covacci, A.; Tompkins, L.S.; Nelson, W.J.; Falkow, S. Disruption of the epithelial apical-junctional complex by Helicobacter pylori CagA. Science 2003, 300, 1430–1434. [Google Scholar]
- Hofman, V.; Ricci, V.; Galmiche, A.; Brest, P.; Auberger, P.; Rossi, B.; Boquet, P.; Hofman, P. Effect of Helicobacter pylori on polymorphonuclear leukocyte migration across polarized T84 epithelial cell monolayers: Role of vacuolating toxin VacA and cag pathogenicity island. Infect. Immun. 2000, 68, 5225–5233. [Google Scholar]
- Wroblewski, L.E.; Shen, L.; Ogden, S.; Romero-Gallo, J.; Lapierre, L.A.; Israel, D.A.; Turner, J.R.; Peek, R.M. Helicobacter pylori dysregulation of gastric epithelial tight junctions by urease-mediated myosin II activation. Gastroenterology 2009, 136, 236–246. [Google Scholar]
- Wessler, S.; Backert, S. Molecular mechanisms of epithelial-barrier disruption by Helicobacter pylori. Trends Microbiol. 2008, 16, 397–405. [Google Scholar]
- Molinari, M.; Salio, M.; Galli, C.; Norais, N.; Rappuoli, R.; Lanzavecchia, A.; Montecucco, C. Selective inhibition of Ii-dependent antigen presentation by Helicobacter pylori toxin VacA. J. Exp. Med. 1998, 187, 135–140. [Google Scholar]
- Zheng, P.Y.; Jones, N.L. Helicobacter pylori strains expressing the vacuolating cytotoxin interrupt phagosome maturation in macrophages by recruiting and retaining TACO (coronin 1) protein. Cell. Microbiol. 2003, 5, 25–40. [Google Scholar]
- Yuan, J.; Li, P.; Tao, J.; Shi, X.; Hu, B.; Chen, H.; Guo, X. H. pylori escape host immunoreaction through inhibiting ILK expression by VacA. Cell. Mol. Immunol. 2009, 6, 191–197. [Google Scholar] [CrossRef] [PubMed]
- Ramarao, N.; Gray-Owen, S.D.; Backert, S.; Meyer, T.F. Helicobacter pylori inhibits phagocytosis by professional phagocytes involving type IV secretion components. Mol. Microbiol. 2000, 37, 1389–1404. [Google Scholar]
- Rittig, M.G.; Shaw, B.; Letley, D.P.; Thomas, R.J.; Argent, R.H.; Atherton, J.C. Helicobacter pylori-induced homotypic phagosome fusion in human monocytes is independent of the bacterial vacA and cag status. Cell. Microbiol. 2003, 5, 887–899. [Google Scholar]
- Gebert, B.; Fischer, W.; Weiss, E.; Hoffmann, R.; Haas, R. Helicobacter pylori vacuolating cytotoxin inhibits T lymphocyte activation. Science 2003, 301, 1099–1102. [Google Scholar]
- Boncristiano, M.; Paccani, S.R.; Barone, S.; Ulivieri, C.; Patrussi, L.; Ilver, D.; Amedei, A.; D'Elios, M.M.; Telford, J.L.; Baldari, C.T. The Helicobacter pylori vacuolating toxin inhibits T cell activation by two independent mechanisms. J. Exp. Med. 2003, 198, 1887–1897. [Google Scholar]
- Sundrud, M.S.; Torres, V.J.; Unutmaz, D.; Cover, T.L. Inhibition of primary human T cell proliferation by Helicobacter pylori vacuolating toxin (VacA) is independent of VacA effects on IL-2 secretion. Proc. Natl. Acad. Sci. USA 2004, 101, 7727–7732. [Google Scholar]
- Nakayama, M.; Kimura, M.; Wada, A.; Yahiro, K.; Ogushi, K.; Niidome, T.; Fujikawa, A.; Shirasaka, D.; Aoyama, N.; Kurazono, H.; Noda, M.; Moss, J.; Hirayama, T. Helicobacter pylori VacA activates the p38/activating transcription factor 2-mediated signal pathway in AZ-521 cells. J. Biol. Chem. 2004, 279, 7024–7028. [Google Scholar]
- Hisatsune, J.; Nakayama, M.; Isomoto, H.; Kurazono, H.; Mukaida, N.; Mukhopadhyay, A.K.; Azuma, T.; Yamaoka, Y.; Sap, J.; Yamasaki, E.; Yahiro, K.; Moss, J.; Hirayama, T. Molecular characterization of Helicobacter pylori VacA induction of IL-8 in U937 cells reveals a prominent role for p38MAPK in activating transcription factor-2, cAMP response element binding protein, and NF-kappaB activation. J. Immunol. 2008, 180, 5017–5027. [Google Scholar] [PubMed]
- Takeshima, E.; Tomimori, K.; Takamatsu, R.; Ishikawa, C.; Kinjo, F.; Hirayama, T.; Fujita, J.; Mori, N. Helicobacter pylori VacA activates NF-kappaB in T cells via the classical but not alternative pathway. Helicobacter 2009, 14, 271–279. [Google Scholar]
- de Bernard, M.; Cappon, A.; Pancotto, L.; Ruggiero, P.; Rivera, J.; Del Giudice, G.; Montecucco, C. The Helicobacter pylori VacA cytotoxin activates RBL-2H3 cells by inducing cytosolic calcium oscillations. Cell. Microbiol. 2005, 7, 191–198. [Google Scholar]
- Supajatura, V.; Ushio, H.; Wada, A.; Yahiro, K.; Okumura, K.; Ogawa, H.; Hirayama, T.; Ra, C. Cutting edge: VacA, a vacuolating cytotoxin of Helicobacter pylori, directly activates mast cells for migration and production of proinflammatory cytokines. J. Immunol. 2002, 168, 2603–2607. [Google Scholar] [PubMed]
- Oldani, A.; Cormont, M.; Hofman, V.; Chiozzi, V.; Oregioni, O; Canonici, A; Sciullo, A; Sommi, P.; Fabbri, A.; Ricci, V.; Boquet, P. Helicobacter pylori counteracts the apoptotic action of its VacA toxin by injecting the CagA protein into gastric epithelial cells. PLoS Pathog. 2009, 5. [Google Scholar]
- Yokoyama, K.; Higashi, H.; Ishikawa, S.; Fujii, Y.; Kondo, S.; Kato, H.; Azuma, T.; Wada, A.; Hirayama, T.; Aburatani, H.; Hatakeyama, M. Functional antagonism between Helicobacter pylori CagA and vacuolating toxin VacA in control of the NFAT signalling pathway in gastric epithelial cells. Proc. Natl. Acad. Sci. USA 2005, 102, 9661–9666. [Google Scholar]
- Argent, R.H.; Thomas, R.J.; Letley, D.P.; Rittig, M.G.; Hardie, K.R.; Atherton, J.C. Functional association between the Helicobacter pylori virulence factors VacA and CagA. J Med. Microbiol. 2008, 57, 145–150. [Google Scholar]
- Tegtmeyer, N.; Zabler, D.; Schmidt, D.; Hartig, R.; Brandt, S.; Backert, S. Importance of EGF receptor, HER2/Neu and Erk1/2 kinase signalling for host cell elongation and scattering induced by the Helicobacter pylori CagA protein: Antagonistic effects of the vacuolating cytotoxin VacA. Cell. Microbiol. 2009, 11, 488–505. [Google Scholar] [CrossRef] [PubMed]
- Ricci, V.; Ciacci, C.; Zarrilli, R.; Sommi, P.; Tummuru, M.K.; Del Vecchio Blanco, C.; Bruni, C.B.; Cover, T.L.; Blaser, M.J.; Romano, M. Effect of Helicobacter pylori on gastric epithelial cell migration and proliferation in vitro: Role of VacA and CagA. Infect. Immun. 1996, 64, 2829–2833. [Google Scholar]
- Pai, R.; Wyle, F.A.; Cover, T.L.; Itani, R.M.; Domek, M.J.; Tarnawski, A.S. Helicobacter pylori culture supernatant interferes with epidermal growth factor-activated signal transduction in human gastric KATO III cells. Am. J. Pathol. 1998, 152, 1617–1624. [Google Scholar]
- Pai, R.; Sasaki, E.; Tarnawski, A.S. Helicobacter pylori vacuolating cytotoxin (VacA) alters cytoskeleton-associated proteins and interferes with re-epithelialization of wounded gastric epithelial monolayers. Cell Biol. Int. 2000, 24, 291–301. [Google Scholar]
- Tabel, G.; Hoa, N.T.; Tarnawski, A.; Chen, J.; Domek, M.; Ma, T.Y. Helicobacter pylori infection inhibits healing of the wounded duodenal epithelium in vitro. J. Lab. Clin. Med. 2003, 142, 421–430. [Google Scholar] [CrossRef] [PubMed]
- Satin, B.; Norais, N.; Telford, J.; Rappuoli, R.; Murgia, M.; Montecucco, C.; Papini, E. Effect of Helicobacter pylori vacuolating toxin on maturation and extracellular release of procathepsin D and on epidermal growth factor degradation. J. Biol. Chem. 1997, 272, 25022–25028. [Google Scholar]
- Nakayama, M.; Hisatsune, J.; Yamasaki, E.; Isomoto, H.; Kurazono, H.; Hatakeyama, M.; Azuma, T.; Yamaoka, Y.; Yahiro, K.; Moss, J.; Hirayama, T. Helicobacter pylori VacA-induced inhibition of GSK3 through the PI3K/Akt signalling pathway. J. Biol. Chem. 2009, 284, 1612–1619. [Google Scholar]
© 2010 by the authors; licensee Molecular Diversity Preservation International, Basel, Switzerland This article is an open-access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Backert, S.; Tegtmeyer, N. The Versatility of the Helicobacter pylori Vacuolating Cytotoxin VacA in Signal Transduction and Molecular Crosstalk. Toxins 2010, 2, 69-92. https://doi.org/10.3390/toxins2010069
Backert S, Tegtmeyer N. The Versatility of the Helicobacter pylori Vacuolating Cytotoxin VacA in Signal Transduction and Molecular Crosstalk. Toxins. 2010; 2(1):69-92. https://doi.org/10.3390/toxins2010069
Chicago/Turabian StyleBackert, Steffen, and Nicole Tegtmeyer. 2010. "The Versatility of the Helicobacter pylori Vacuolating Cytotoxin VacA in Signal Transduction and Molecular Crosstalk" Toxins 2, no. 1: 69-92. https://doi.org/10.3390/toxins2010069