The Relationship between Dietary Fatty Acids and Inflammatory Genes on the Obese Phenotype and Serum Lipids

Abstract
:1. Introduction
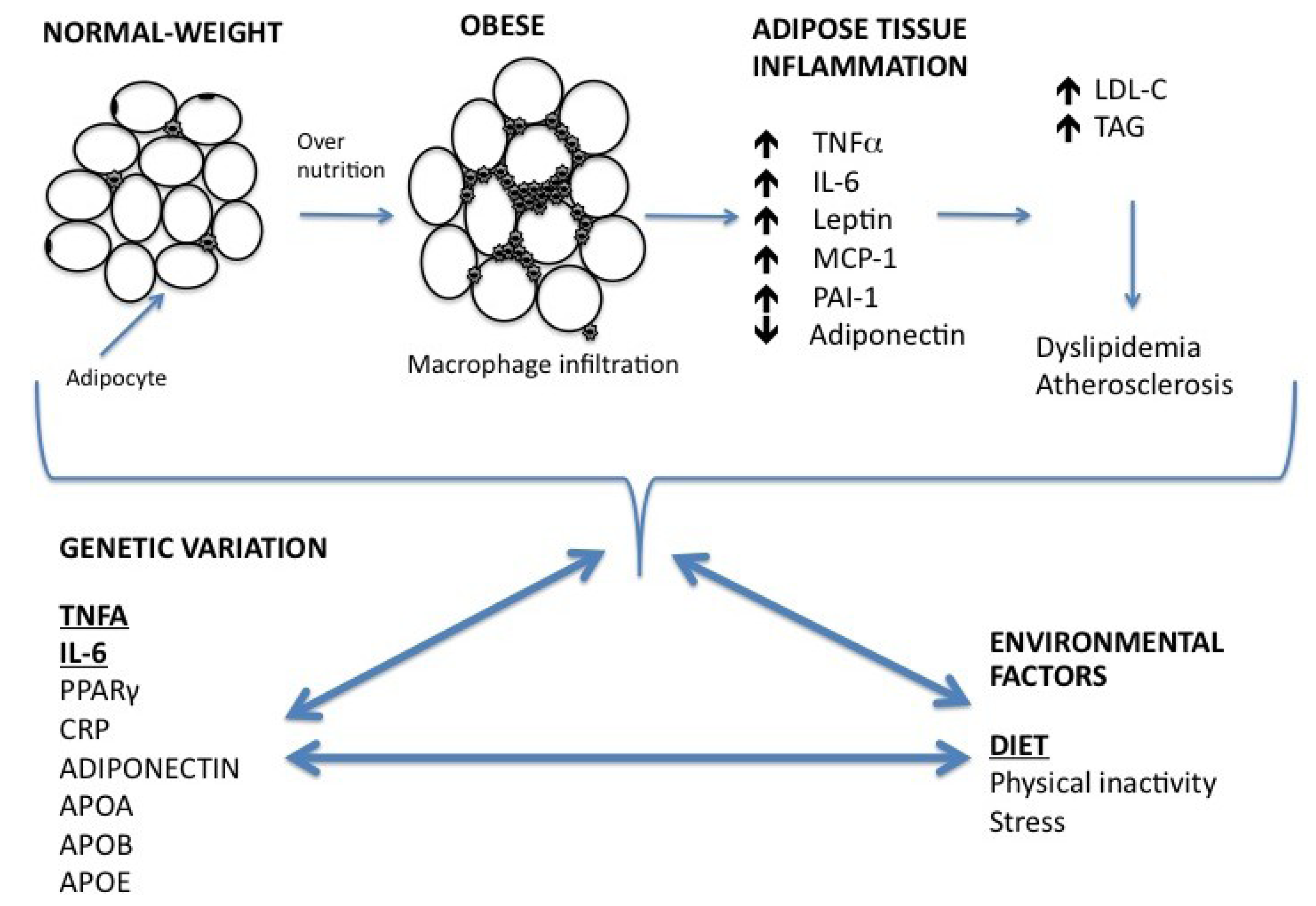
2. Relationship between Inflammation, Obesity and Lipid Metabolism
3. Dietary Fatty Acids and Inflammation
4. Tumor Necrosis Factor-α
4.1. TNFα and Dietary Fatty Acids
4.2. TNFA Gene Variants, Obesity and Serum Lipids

{kind=link}
{kind=link}
{kind=link}
| Study type | Dietary fatty acid | Effect on gene expression | Effect on plasma levels | Reference | |
|---|---|---|---|---|---|
| TNFα | |||||
| 3T3-L1 adipocytes. Incubation at 24 and 48 h with 50 or 500 μM fatty acid | Cell culture | SFA (PA) | Increase | Increase | [62] |
| MUFA (OA) | No effect | No effect | |||
| Human macrophages treated with n-3 PUFA | Cell culture | EPA & DHA | Decrease | Decrease | [77] |
| Male Wistar rats, high fat diet, 1 g/kg per day EPA, 5 weeks | Rodent | n-3 PUFA (EPA) | Prevent over expression | Not examined | [64] |
| NZB/NZW F1 Lupus-prone female mice, 10% fat, fed ad lib for lifespan | Rodent | n-3 PUFA | Decrease | Not examined | [63] |
| Caucasians. Supplemented normal diet with 18 g fish oil daily for 6 weeks | Human intervention (9) | n-3 PUFA | Decrease | [66] | |
| Caucasians. Supplemented normal diet with 6 g fish oil daily for 12 weeks | Human intervention (111) | n-3 PUFA | Decrease in subjects with lower levels of TNFα before supplementation | [67] | |
| Caucasians. Supplemented normal diet with flaxseed oil, and flaxseed oil and butter spread for 8 weeks. At week 4, diets were supplemented with fish oil, (1.62 g EPA, 1.08 g DHA)/day | Human intervention (28) | n-3 PUFA (EPA & DHA) | Decrease | [65] | |
| IL-6 | |||||
| 3T3-L1 adipocytes. Incubation at 24 h with 250 μM fatty acid | Cell culture | SFA (PA) | Increase | Increase | [31] |
| SFA (DA) | No effect | No effect | |||
| n-3 PUFA (DHA) | No effect | No effect | |||
| Human macrophages treated with n-3 PUFA | Cell culture | EPA & DHA | Decrease | Decrease | [77] |
| Male c57bl/10sCn mice fed ad lib either high fat control diet (soybean oil) or a high PA diet for 16 weeks | Rodent | SFA (PA) | Increase | Not examined | [78] |
| Male Sprague-Dawley rats fed ad lib one of 3 diets: SFA, MUFA, or PUFA for 4 weeks | Rodent | SFA (coconut oil) | Not examined | Increase IL-6 release from adipocytes | [79] |
| MUFA (olive oil) | Not examined | Decrease IL-6 release from adipocytes | |||
| PUFA (sunflower oil) | Not examined | Moderate IL-6 release from adipocytes | |||
| Abdominally overweight Caucasians. Fed either SFA-rich diet (19% SFA and 11% MUFA) or MUFA-rich diet (20% MUFA and 11% SFA) for 8 weeks | Human intervention (20) | SFA | Increase IL-6 gene expression | [80] | |
| MUFA | Decrease IL-6 gene expression | ||||
| African Americans, Caucasians, Chinese and Hispanics men and women. Relationship between dietary intake (food frequency questionnaire) and biomarkers of inflammation and endothelial activation | Human (5677) | n-3 PUFA | Decrease in IL-6 levels | [81] | |
| Relationship between plasma fatty acids and inflammatory marker levels | Human (1123) | n-3 PUFA (DHA) | Low plasma levels of DHA associated with increased IL-6 levels | [82] | |
| n-6 PUFA (AA) | Low plasma levels of AA associated with increased IL-6 levels | ||||
| SNP | Study cohort | Genotype frequency | Result | Reference |
|---|---|---|---|---|
| TNFA | ||||
| Obesity | ||||
| TNFA –308 G > A | Caucasian N-W (154) and obese (154) | N-W, GG: 75.8%; GA + AA: 24.2% Obese, GG: 70.8%; GA + AA: 29.2% |
| [71] |
| Caucasian women (378) | GG: 72.2%; GA + AA: 27.7% |
| [70] | |
| Caucasian BMI < 27.3 (44), BMI 27.3–31.9 (44), BMI 31.9–36.5 (44) and BMI > 36.5 (44) | BMI < 27.3, GG: 75%; GA + AA: 25% BMI 27.3–31.9, GG: 68.2%; GA + AA: 31.8% BMI 31.9–36.5, GG: 77%; GA + AA: 33% BMI > 36.5, GG: 47.7%; GA + AA: 52.3% |
| [68] | |
| Caucasian (1392) | GG: 67.6%; GA + AA: 32.3% |
| [69] | |
| Caucasian normal wt. (79) and obese (115) | N-W, GG: 73.4%; GA + AA: 24.2% Obese, GG: 75.6%; GA + AA: 26% |
| [83] | |
| Korean normal wt. (82) and obese (153) | N-W, GG: 82.9%; GA + AA: 17% Obese, GG: 84.3%; GA + AA: 15.7% |
| [84] | |
| Caucasian normotensive (113) and hypertensive (62) | Normotensive, GG: 84.8%; GA + AA: 15% Hypertensive, GG: 67.7%; GA + AA: 32.3% |
| [74] | |
| Caucasian normal weight (64) and overweight (65) | Not shown |
| [72] | |
| Caucasian men (262) | GG: 56.4%; GA + AA: 43.5% |
| [73] | |
| Meta-analysis (48 eligible studies) |
| [21] | ||
| TNFA –238 G > A | N-W (107) and obese (120) black, and N-W (89) and obese (62) white SA women | Black, GG: 60%; GA: 40%; AA: 0%; A allele: 20% White, GG: 78.5%; GA: 21%; AA: 0.5%; A allele: 11% |
| [5] |
| TNFA –308 G > A & –238 G > A | Iranian men and women (BMI < 25, 25 ≤ BMI < 30, BMI ≥ 30) (239) | Under 18 years, BMI < 85%, GG: 80.8%; GA: 19.2% BMI > 85%, GG: 81.2%; GA: 12.5%; AA: 6.2%. Above 18 years, BMI < 25, GG: 89.3%; GA: 10.7% 25 ≤ BMI < 30, GG: 87.7%; GA: 12.3% BMI ≥ 30, GG: 80.4%; 15.2%; 4.3% |
| [85] |
| Caucasian and African-American non-diabetics (424) | –308 G > A BMI < 25, GG: 73.2%; GA: 22.5%, AA: 4.3% BMI 25–29.9, GG: 66.7%; GA: 27%, AA: 6.3% BMI 30–40, GG: 67.2%; GA: 30.2%; AA: 2.6% BMI > 40, GG: 73.4%; GA: 24.3%; AA: 2.4% –238 G > A BMI < 25, GG: 90.4%; GA: 9.1%, AA: 0.5% BMI 25–29.9, GG: 90.5%; GA: 7.9%, AA: 1.6% BMI 30–40, GG: 85.9%; GA: 14.1% BMI > 40, GG: 91.1%; GA: 8.9% |
| [86] | |
| Serum lipids | ||||
| TNFA –308 G > A | Caucasian obese women (136) and obese men (34) | Obese women, GG: 57.3%; GA + AA: 42.6% Obese men, GG: 64.7%; GA + AA: 35.3% |
| [75] |
| Caucasian obese men (38) and obese women (83) | Obese men, GG: 50%; GA + AA: 50% Obese women, GG: 54.2%; GA + AA: 45.7% |
| [76] | |
| IL-6 | ||||
| Obesity | ||||
| IL-6 –174 G > C | Finnish men and women (1334) | GG: 19.3%; GC: 51.3%; CC: 29.3% | In men BMI was higher in the –174 CC genotype compared to GC and GG | [87] |
| Meta-analysis (48 eligible studies) |
| [21] | ||
| Meta-analysis Caucasians, diabetic and non-diabetic (25635) |
| [88] | ||
| IL-6 –174 G > C & IVS3 +281 G > T and IVS4 +869 A > G | Health men (980) and women (2255) and Meta-analysis (26944) |
| [89] | |
| IL-6 –174 G > C IVS3 +281 G > T & IVS4 +869 A > G | N-W (108) and obese (124) black, and N-W (89) and obese (63) white SA women | -174 G > C N-W black: GG: 97%; GC: 3% Obese black: GG: 95%; GC: 3%; CC: 2% N-W white: GG: 30%; GC: 58%; CC: 11% Obese white: GG: 32%; GC: 46%; CC: 22% IVS3 +281 G > T N-W black: GG: 54%; GT: 38%; TT: 8% Obese black: GG: 55%; GT: 37%; TT:9% N-W white: GG: 35%; GT: 48%; TT: 17% Obese white: GG: 30%; GT: 51%; TT: 19% IVS4 +869 A > G N-W black: AA: 51%; AG: 39%; GG: 10% Obese black: AA: 54%; AG: 40%; GG: 6% N-W white: AA: 42%; AG: 57%; GG: 1% Obese white: AA: 37%; AG: 63% |
| [90] |
| Serum lipids | ||||
| IL-6 –174 G > C | Caucasian men (245) and women (252) | Women, GG: 28%; GC: 47%; CC: 24% Men, GG: 30%; GC: 46%; CC: 24% |
| [91] |
| Finnish men and women (1334) | GG: 19.3%; GC: 51.3%; CC: 29.3% |
| [87] | |
| Spanish Caucasian men (15) and women (17) | GG: 25%; GC: 40.6%; CC: 34.4% |
| [92] | |
| Finnish men and women (2228) | GG: 20.8%; GC: 50.4%; CC: 28.8% |
| [93] | |
| IL-6 -174 G > C, IVS3 +281 G > T & IVS4 +869 A > G | N-W (108) and obese (124) black, and N-W (89) and obese (63) white SA women | –174 G > C N-W black: GG: 97%; GC: 3% Obese black: GG: 95%; GC: 3%; CC: 2% N-W white: GG: 30%; GC: 58%; CC: 11% Obese white: GG: 32%; GC: 46%; CC: 22% IVS3 +281 G > T N-W black: GG: 54%; GT: 38%; TT: 8% Obese black: GG: 55%; GT: 37%; TT:9% N-W white: GG: 35%; GT: 48%; TT: 17% Obese white: GG: 30%; GT: 51%; TT: 19% IVS4 +869 A > G N-W black: AA: 51%; AG: 39%; GG: 10% Obese black: AA: 54%; AG: 40%; GG: 6% N-W white: AA: 42%; AG: 57%; GG: 1% Obese white: AA: 37%; AG: 63% |
| [90] |
4.3. TNFA Gene and Diet Interactions on Obesity and Serum Lipids

| SNP | Study cohort | Genotype frequency | Diet assessment and fats | Diet-gene association | Reference |
|---|---|---|---|---|---|
| TNFA | |||||
| Obesity | |||||
| TNFA –308 G > A | Caucasian N-W (154) and obese (154) | Normal weight, GG: 75.8%; GA + AA: 24.2% Obese, GG: 70.8%; GA + AA: 29.2% | Food frequency questionnaire measured energy and dietary fatty acid intake. |
| [71] |
| Black South African N-W (105) and obese (118) | Black, GG: 69%; GA: 28%; AA: 3%; A allele: 17% | Food frequency questionnaire measured dietary fatty acid intake. |
| [3] | |
| TNFA –238 G > A | N-W (107) and obese (120) black, and N-W (89) and obese (62) white SA women | Black, GG: 60%; GA: 40%. A allele: 20% White, GG: 78.5%; GA: 21%; AA: 0.5%; A allele: 11% | Food frequency questionnaire measured dietary fatty acid intake. | In black women:
| [5] |
| Serum lipids | |||||
| TNFA –308 G > A | Black South African N-W (105) and obese (118) | Black, GG: 69%; GA: 28%; AA: 3%; A allele: 17% | Food frequency questionnaire measured dietary fatty acid intake. |
| [3] |
| Caucasian South African N-W (88) and obese (60) white SA women | White, GG: 56%; GA: 42%; AA: 2%; A allele: 23% | Food frequency questionnaire measured dietary fatty acid intake. |
| [4] | |
| TNFA –238 G > A | N-W (107) and obese (120) black, and N-W (89) and obese (62) white SA women | Black, GG: 60%; GA: 40%. A allele: 20% White, GG: 78.5%; GA: 21%; AA: 0.5%; A allele: 11% | Food frequency questionnaire measured dietary fatty acid intake. |
| [5] |
| TNFA –308 G > A & –238 G > A | Ethnoracially diverse Canadian diabetic men (53) and women (56) | –308 G > A GG: 63.3%; GA: 32%; AA: 0.05% –238 G > A GG: 75.2%; GA: 21.1%; AA: 0.04% | Three-day food record measured dietary fat intake. |
| [94] |
| TNFA –308 G > A & –238 G > A | Ethnoracially diverse Canadian healthy men (202) and women (393) | –308 G > A, 11% for A allele –238 G > A, 5% for A allele | Food frequency questionnaire measured dietary intake. |
| [95] |
| IL-6 | |||||
| Obesity | |||||
| IL-6 –174 G > C | Obese Caucasians men (181) and women (541) | GG: 28.8%; GC: 50.2%, CC: 18%. | Test meal consisted of 95%E from fat, of which 60% SFA. |
| [96] |
| 737 Spanish men and women | GG: 37.6%; GC: 46.8%; CC: 15.6% | Three years diet intervention assigned to low-fat diet; Mediterranean diet supplemented with virgin olive oil or with nuts. |
| [97] | |
| IL-6 –174 G > C, IVS3 +281 G > T & IVS4 +869 G > A | N-W (107) and obese (120) black, and N-W (89) and obese (62) white SA women | –174 G > C N-W black: GG: 97%; GC: 3% Obese black: GG: 95%; GC: 3%; CC: 2% N-W white: GG: 30%; GC: 58%; CC: 11% Obese white: GG: 32%; GC: 46%; CC: 22% IVS3 +281 G > T N-W black: GG: 54%; GT: 38%; TT: 8% Obese black: GG: 55%; GT: 37%; TT:9% N-W white: GG: 35%; GT: 48%; TT: 17% Obese white: GG: 30%; GT: 51%; TT: 19% IVS4 +869 A > G N-W black: AA: 51%; AG: 39%; GG: 10% Obese black: AA: 54%; AG: 40%; GG: 6% N-W white: AA: 42%; AG: 57%; GG: 1% Obese white: AA: 37%; AG: 63% | Food frequency questionnaire measured dietary fatty acid intake. |
| [98] |
| Serum lipids | |||||
| IL-6 –174 G > C | Spanish Caucasian men and women (32) | GG: 25%; GC: 40.6%; CC: 34.4% | Measured fasting and post-glucose load plasma lipids. |
| [92] |
| IL-6 –174 G > C, IVS3 +281 G > T & IVS4 +869 G > A | N-W (107) and obese (120) black, and N-W (89) and obese (62) white SA women | –174 G > C N-W black: GG: 97%; GC: 3% Obese black: GG: 95%; GC: 3%; CC: 2% N-W white: GG: 30%; GC: 58%; CC: 11% Obese white: GG: 32%; GC: 46%; CC: 22% IVS3 +281 G > T N-W black: GG: 54%; GT: 38%; TT: 8% Obese black: GG: 55%; GT: 37%; TT:9% N-W white: GG: 35%; GT: 48%; TT: 17% Obese white: GG: 30%; GT: 51%; TT: 19% IVS4 +869 A > G N-W black: AA: 51%; AG: 39%; GG: 10% Obese black: AA: 54%; AG: 40%; GG: 6% N-W white: AA: 42%; AG: 57%; GG: 1% Obese white: AA: 37%; AG: 63% | Food frequency questionnaire measured dietary fatty acid intake. |
| [98] |
5. Interleukin-6
5.1. IL-6 and Dietary Fatty Acids
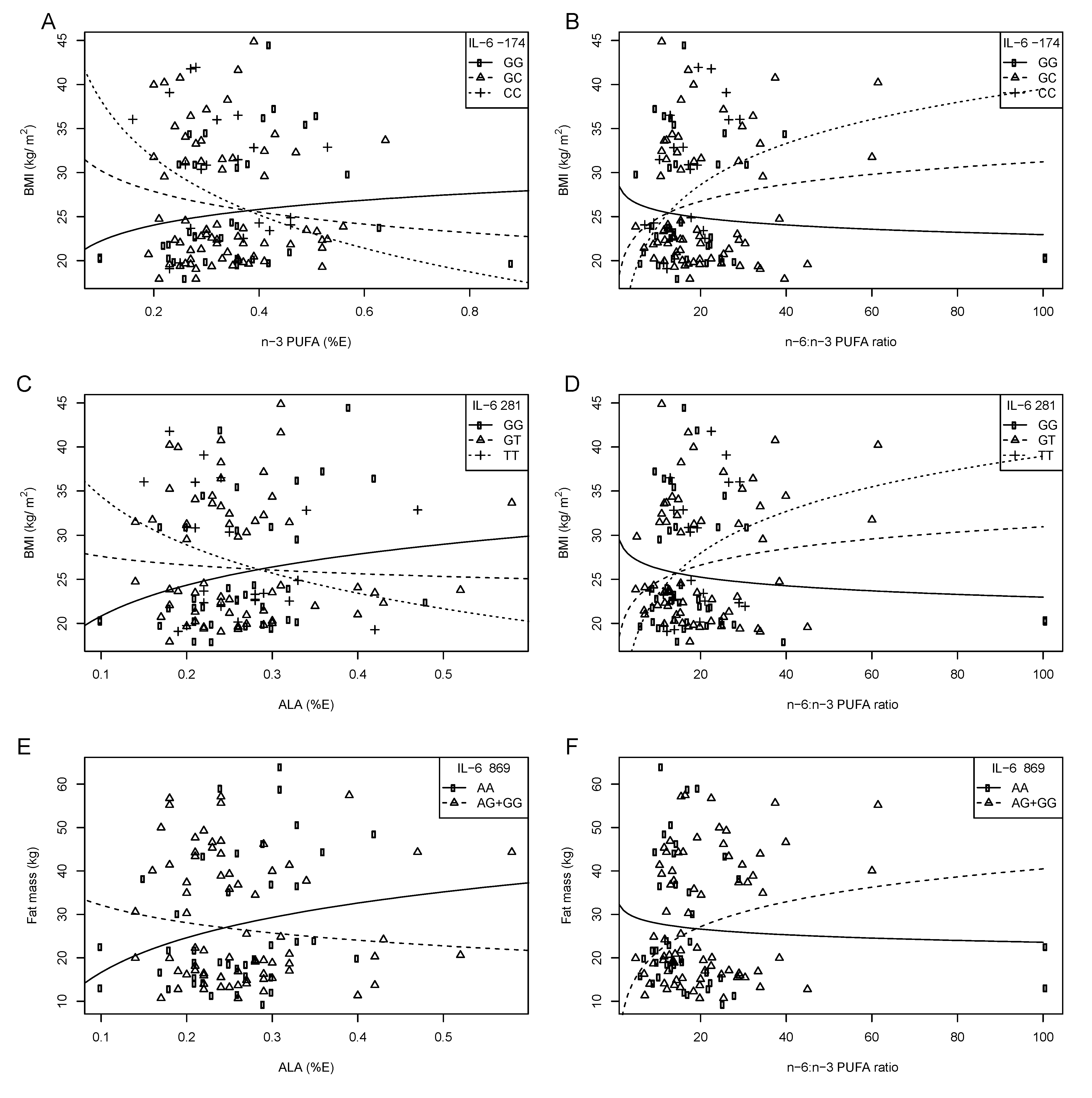
5.2. IL-6 Gene Variants, Obesity and Serum Lipids
5.3. IL-6 Gene and Diet Interactions on Obesity and Serum Lipids
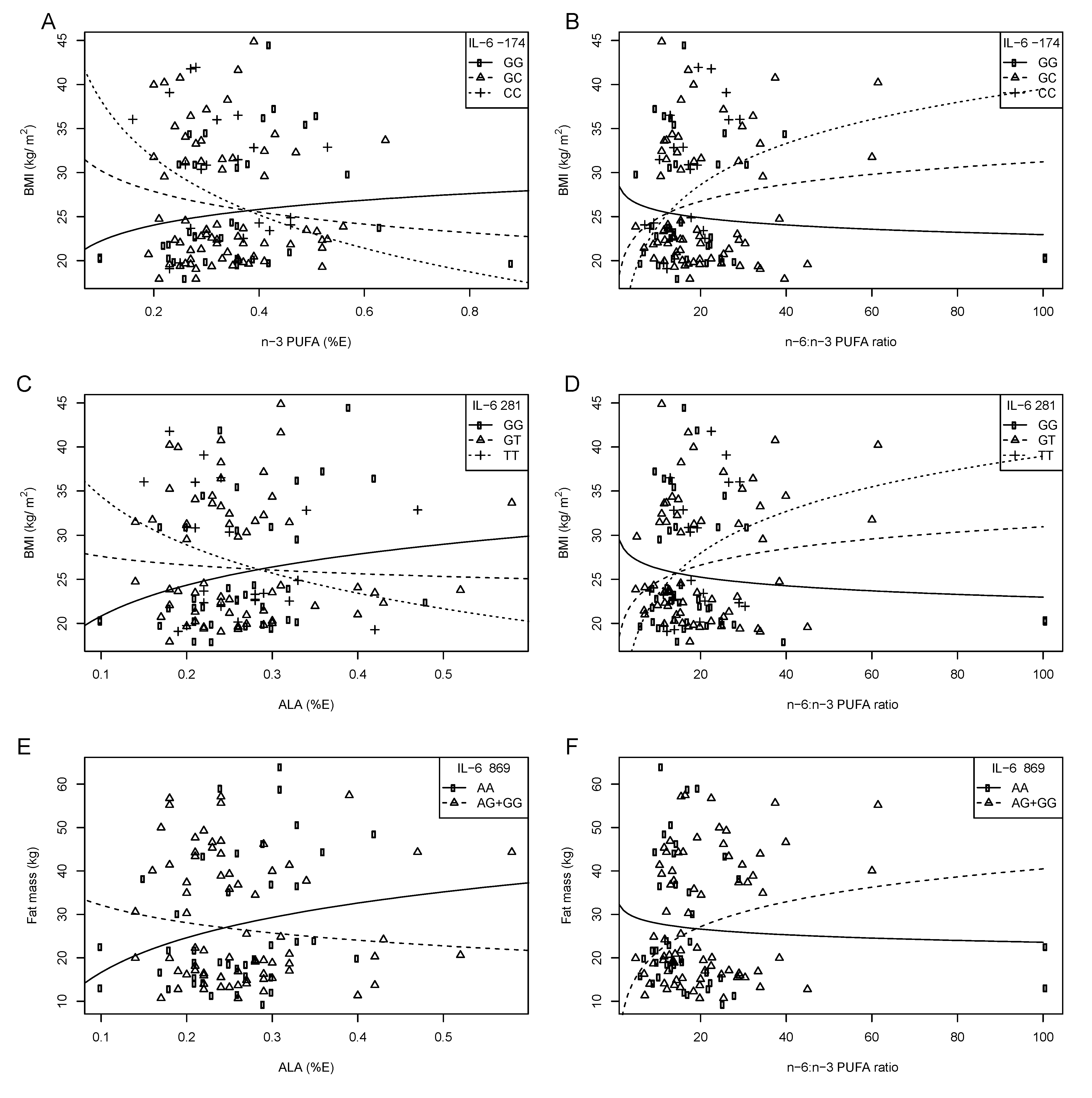
6. The Role of Ethnicity and Gender as Confounders
| Ensemble 1000 Genomes: phase 1 | ||||
|---|---|---|---|---|
| EUR | GBR | AFR | ASW | |
| TNFA –308 G > A rs1800629 | ||||
| GG | 0.75 | 0.80 | 0.81 | 0.87 |
| GA | 0.24 | 0.17 | 0.18 | 0.13 |
| AA | 0.02 | 0.03 | 0.00 | 0.00 |
| A allele | 0.14 | 0.12 | 0.10 | 0.10 |
| TNFA –238 G > A rs361525 | ||||
| GG | 0.87 | 0.81 | 0.93 | 0.90 |
| GA | 0.13 | 0.19 | 0.07 | 0.10 |
| AA | 0.00 | 0.00 | 0.00 | 0.00 |
| A allele | 0.07 | 0.10 | 0.03 | 0.05 |
| IL-6 –174 G > C rs1800795 | ||||
| GG | 0.36 | 0.39 | 0.95 | 0.78 |
| GC | 0.44 | 0.42 | 0.05 | 0.21 |
| CC | 0.20 | 0.19 | 0.0 | 0.0 |
| C allele | 0.41 | 0.40 | 0.02 | 0.11 |
| IL-6 IVS3 +281 G > T rs1554606 | ||||
| GG | 0.34 | 0.36 | 0.48 | 0.39 |
| GT | 0.45 | 0.44 | 0.46 | 0.57 |
| TT | 0.20 | 0.19 | 0.05 | 0.03 |
| T allele | 0.43 | 0.41 | 0.28 | 0.32 |
| IL-6 IVS4 +869 A > G rs2069845 | ||||
| AA | 0.34 | 0.36 | 0.46 | 0.37 |
| GA | 0.45 | 0.44 | 0.47 | 0.57 |
| GG | 0.20 | 0.19 | 0.05 | 0.04 |
| G allele | 0.43 | 0.41 | 0.29 | 0.33 |
7. Conclusions
Conflict of Interest
References
- Maury, E.; Brichard, S.M. Adipokine dysregulation, adipose tissue inflammation and metabolic syndrome. Mol. Cell. Endocrinol. 2010, 314, 1–16. [Google Scholar]
- Bray, M.S. Implications of gene-behavior interactions: Prevention and intervention for obesity. Obesity 2008, 16 (Suppl. 3), S72–S78. [Google Scholar] [CrossRef]
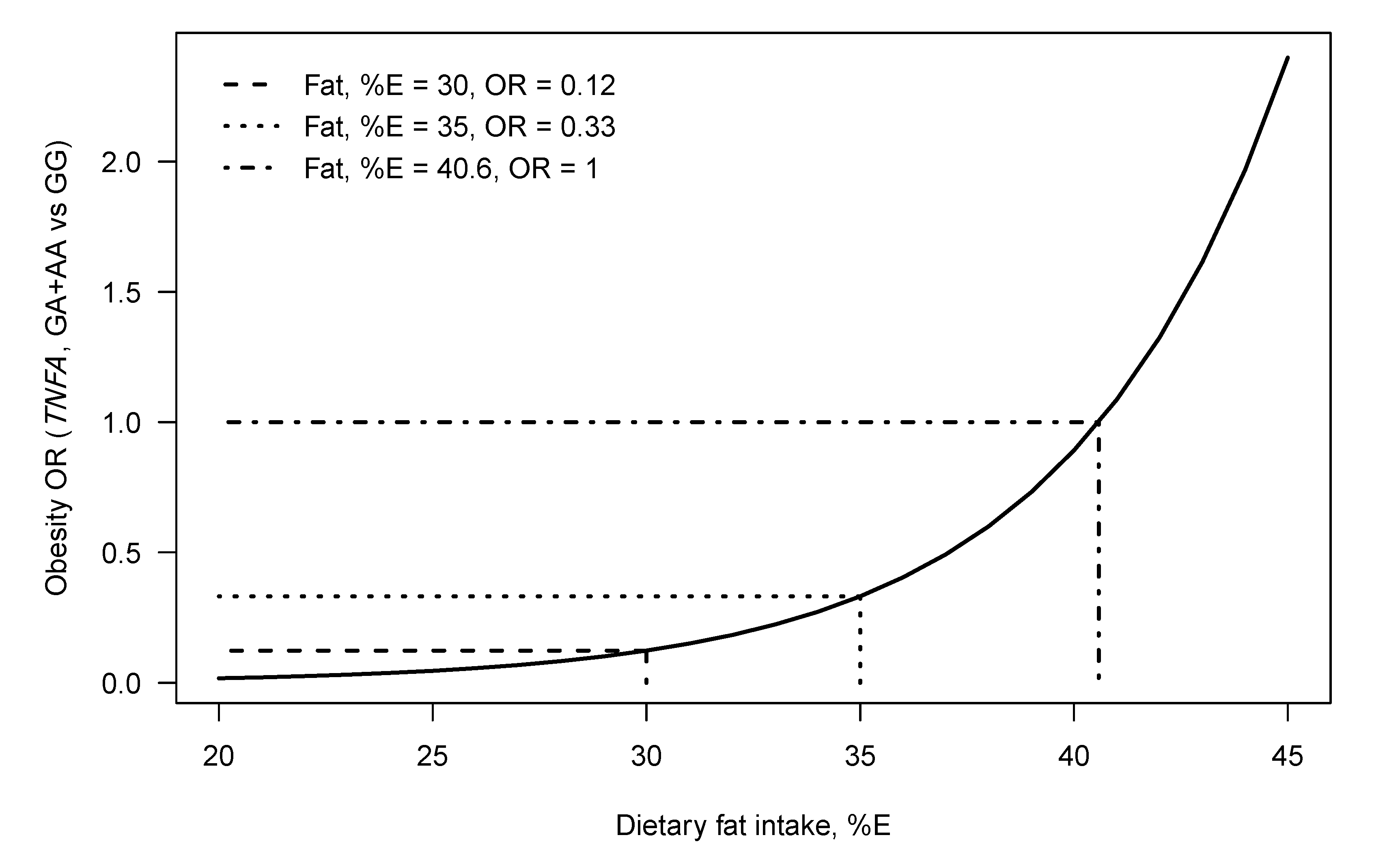
- Joffe, Y.T.; van der Merwe, L.; Carstens, M.; Collins, M.; Jennings, C.; Levitt, N.S.; Lambert, E.V.; Goedecke, J.H. Tumor necrosis factor-α gene –308 G/A polymorphism modulates the relationship between dietary fat intake, serum lipids, and obesity risk in black South African women. J. Nutr. 2010, 140, 901–907. [Google Scholar] [CrossRef]
- Joffe, Y.T.; van der Merwe, L.; Collins, M.; Carstens, M.; Evans, J.; Lambert, E.V.; Goedecke, J.H. The –308 G/A polymorphism of the tumour necrosis factor-α gene modifies the association between saturated fat intake and serum total cholesterol levels in white South African women. Genes Nutr. 2011, 6, 353–359. [Google Scholar]
- Joffe, Y.T.; van der Merwe, L.; Evans, J.; Collins, M.; Lambert, E.V.; September, A.; Goedecke, J.H. The tumor necrosis factor-α gene –238 G > A polymorphism, dietary fat intake, obesity risk and serum lipid concentrations in black and white South African women. Eur. J. Clin. Nutr. 2012, 66, 1295–1302. [Google Scholar] [CrossRef]
- Stryjecki, C.; Mutch, D.M. Fatty acid-gene interactions, adipokines and obesity. Eur. J. Clin. Nutr. 2011, 65, 285–297. [Google Scholar] [CrossRef]
- Fantuzzi, G. Adipose tissue, adipokines, and inflammation. J. Allergy Clin. Immunol. 2005, 115, 911–919. [Google Scholar] [CrossRef]
- Nair, S.; Lee, Y.H.; Rousseau, E.; Cam, M.; Tataranni, P.A.; Baier, L.J.; Bogardus, C.; Permana, P.A. Increased expression of inflammation-related genes in cultured preadipocytes/stromal vascular cells from obese compared with non-obese Pima Indians. Diabetologia 2005, 48, 1784–1788. [Google Scholar] [CrossRef]
- Gutierrez, D.A.; Puglisi, M.J.; Hasty, A.H. Impact of increased adipose tissue mass on inflammation, insulin resistance, and dyslipidemia. Curr. Diabetes Rep. 2009, 9, 26–32. [Google Scholar] [CrossRef]
- Weisberg, S.P.; McCann, D.; Desai, M.; Rosenbaum, M.; Leibel, R.L.; Ferrante, A.W., Jr. Obesity is associated with macrophage accumulation in adipose tissue. J. Clin. Invest. 2003, 112, 1796–1808. [Google Scholar]
- Galic, S.; Oakhill, J.S.; Steinberg, G.R. Adipose tissue as an endocrine organ. Mol. Cell. Endocrinol. 2010, 316, 129–139. [Google Scholar] [CrossRef]
- Xu, H.; Barnes, G.T.; Yang, Q.; Tan, G.; Yang, D.; Chou, C.J.; Sole, J.; Nichols, A.; Ross, J.S.; Tartaglia, L.A.; et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J. Clin. Invest. 2003, 112, 1821–1830. [Google Scholar]
- Libby, P.; Ridker, P.M.; Maseri, A. Inflammation and atherosclerosis. Circulation 2002, 105, 1135–1143. [Google Scholar] [CrossRef]
- Berliner, J.; Leitinger, N.; Watson, A.; Huber, J.; Fogelman, A.; Navab, M. Oxidized lipids in atherogenesis: Formation, destruction and action. Thromb. Haemost. 1997, 78, 195–199. [Google Scholar]
- Sugiyama, S.; Okada, Y.; Sukhova, G.K.; Virmani, R.; Heinecke, J.W.; Libby, P. Macrophage myeloperoxidase regulation by granulocyte macrophage colony-stimulating factor in human atherosclerosis and implications in acute coronary syndromes. Am. J. Pathol. 2001, 158, 879–891. [Google Scholar] [CrossRef]
- Dichtl, W.; Nilsson, L.; Goncalves, I.; Ares, M.P.; Banfi, C.; Calara, F.; Hamsten, A.; Eriksson, P.; Nilsson, J. Very low-density lipoprotein activates nuclear factor-κB in endothelial cells. Circ. Res. 1999, 84, 1085–1094. [Google Scholar] [CrossRef]
- Burzotta, F.; Iacoviello, L.; Di Castelnuovo, A.; Glieca, F.; Luciani, N.; Zamparelli, R.; Schiavello, R.; Donati, M.B.; Maseri, A.; Possati, G.; et al. Relation of the –174 G/C polymorphism of interleukin-6 to interleukin-6 plasma levels and to length of hospitalization after surgical coronary revascularization. Am. J. Cardiol. 2001, 88, 1125–1128. [Google Scholar] [CrossRef]
- Santtila, S.; Savinainen, K.; Hurme, M. Presence of the IL-1RA allele 2 (IL1RN*2) is associated with enhanced IL-1β production in vitro. Scand. J. Immunol. 1998, 47, 195–198. [Google Scholar]
- Terry, C.F.; Loukaci, V.; Green, F.R. Cooperative influence of genetic polymorphisms on interleukin 6 transcriptional regulation. J. Biol. Chem. 2000, 275, 18138–18144. [Google Scholar] [CrossRef]
- Wilson, A.G.; Symons, J.A.; McDowell, T.L.; McDevitt, H.O.; Duff, G.W. Effects of a polymorphism in the human tumor necrosis factor α promoter on transcriptional activation. Proc. Natl. Acad. Sci. USA 1997, 94, 3195–3199. [Google Scholar]
- Yu, Z.; Han, S.; Cao, X.; Zhu, C.; Wang, X.; Guo, X. Genetic polymorphisms in adipokine genes and the risk of obesity: A systematic review and meta-analysis. Obesity 2011, 20, 396–406. [Google Scholar]
- Phillips, C.M. Nutrigenetics and metabolic disease: Current status and implications for personalised nutrition. Nutrients 2013, 5, 32–57. [Google Scholar] [CrossRef]
- Phillips, C.M.; Goumidi, L.; Bertrais, S.; Ferguson, J.F.; Field, M.R.; Kelly, E.D.; Mehegan, J.; Peloso, G.M.; Cupples, L.A.; Shen, J.; et al. Additive effect of polymorphisms in the IL-6, LTA, and TNF-αgenes and plasma fatty acid level modulate risk for the metabolic syndrome and its components. J. Clin. Endocrinol. Metab. 2010, 95, 1386–1394. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 polyunsaturated fatty acids and inflammation: From molecular biology to the clinic. Lipids 2003, 38, 343–352. [Google Scholar] [CrossRef]
- Calder, P.C. n-3 polyunsaturated fatty acids, inflammation, and inflammatory diseases. Am. J. Clin. Nutr. 2006, 83, 1505S–1519S. [Google Scholar]
- Van den Berghe, W.; Vermeulen, L.; Delerive, P.; De Bosscher, K.; Staels, B.; Haegeman, G. A paradigm for gene regulation: Inflammation, NF-κB and PPAR. Adv. Exp. Med. Biol. 2003, 544, 181–196. [Google Scholar]
- Calder, P.C.; Ahluwalia, N.; Brouns, F.; Buetler, T.; Clement, K.; Cunningham, K.; Esposito, K.; Jonsson, L.S.; Kolb, H.; Lansink, M.; et al. Dietary factors and low-grade inflammation in relation to overweight and obesity. Br. J. Nutr. 2011, 106 (Suppl. 3), S5–S78. [Google Scholar] [CrossRef]
- Suganami, T.; Tanimoto-Koyama, K.; Nishida, J.; Itoh, M.; Yuan, X.; Mizuarai, S.; Kotani, H.; Yamaoka, S.; Miyake, K.; Aoe, S.; et al. Role of the Toll-like receptor 4/NF-κB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler. Thromb. Vasc. Biol. 2007, 27, 84–91. [Google Scholar] [CrossRef]
- Fessler, M.B.; Rudel, L.L.; Brown, J.M. Toll-like receptor signaling links dietary fatty acids to the metabolic syndrome. Curr. Opin. Lipidol. 2009, 20, 379–385. [Google Scholar] [CrossRef]
- Weatherill, A.R.; Lee, J.Y.; Zhao, L.; Lemay, D.G.; Youn, H.S.; Hwang, D.H. Saturated and polyunsaturated fatty acids reciprocally modulate dendritic cell functions mediated through TLR4. J. Immunol. 2005, 174, 5390–5397. [Google Scholar]
- Ajuwon, K.M.; Spurlock, M.E. Palmitate activates the NF-κB transcription factor and induces IL-6 and TNFα expression in 3T3-L1 adipocytes. J. Nutr. 2005, 135, 1841–1846. [Google Scholar]
- Weigert, C.; Brodbeck, K.; Staiger, H.; Kausch, C.; Machicao, F.; Haring, H.U.; Schleicher, E.D. Palmitate, but not unsaturated fatty acids, induces the expression of interleukin-6 in human myotubes through proteasome-dependent activation of nuclear factor-κB. J. Biol. Chem. 2004, 279, 23942–23952. [Google Scholar]
- Lee, J.Y.; Plakidas, A.; Lee, W.H.; Heikkinen, A.; Chanmugam, P.; Bray, G.; Hwang, D.H. Differential modulation of Toll-like receptors by fatty acids: Preferential inhibition by n-3 polyunsaturated fatty acids. J. Lipid Res. 2003, 44, 479–486. [Google Scholar] [CrossRef]
- Baum, S.J.; Kris-Etherton, P.M.; Willett, W.C.; Lichtenstein, A.H.; Rudel, L.L.; Maki, K.C.; Whelan, J.; Ramsden, C.E.; Block, R.C. Fatty acids in cardiovascular health and disease: A comprehensive update. J. Clin. Lipidol. 2012, 6, 216–234. [Google Scholar] [CrossRef]
- Pischon, T.; Hankinson, S.E.; Hotamisligil, G.S.; Rifai, N.; Willett, W.C.; Rimm, E.B. Habitual dietary intake of n-3 and n-6 fatty acids in relation to inflammatory markers among US men and women. Circulation 2003, 108, 155–160. [Google Scholar] [CrossRef]
- Bjermo, H.; Iggman, D.; Kullberg, J.; Dahlman, I.; Johansson, L.; Persson, L.; Berglund, J.; Pulkki, K.; Basu, S.; Uusitupa, M.; et al. Effects of n-6 PUFAs compared with SFAs on liver fat, lipoproteins, and inflammation in abdominal obesity: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 95, 1003–1012. [Google Scholar] [CrossRef]
- Kelley, D.S.; Taylor, P.C.; Nelson, G.J.; Schmidt, P.C.; Mackey, B.E.; Kyle, D. Effects of dietary arachidonic acid on human immune response. Lipids 1997, 32, 449–456. [Google Scholar] [CrossRef]
- Thies, F.; Miles, E.A.; Nebe-von-Caron, G.; Powell, J.R.; Hurst, T.L.; Newsholme, E.A.; Calder, P.C. Influence of dietary supplementation with long-chain n-3 or n-6 polyunsaturated fatty acids on blood inflammatory cell populations and functions and on plasma soluble adhesion molecules in healthy adults. Lipids 2001, 36, 1183–1193. [Google Scholar] [CrossRef]
- Thies, F.; Nebe-von-Caron, G.; Powell, J.R.; Yaqoob, P.; Newsholme, E.A.; Calder, P.C. Dietary supplementation with eicosapentaenoic acid, but not with other long-chain n-3 or n-6 polyunsaturated fatty acids, decreases natural killer cell activity in healthy subjects aged >55 y. Am. J. Clin. Nutr. 2001, 73, 539–548. [Google Scholar]
- Erkkila, A.; de Mello, V.D.; Riserus, U.; Laaksonen, D.E. Dietary fatty acids and cardiovascular disease: An epidemiological approach. Prog. Lipid Res. 2008, 47, 172–187. [Google Scholar] [CrossRef]
- Williams, C.M.; Burdge, G. Long-chain n-3 PUFA: Plant v. marine sources. Proc. Nutr. Soc. 2006, 65, 42–50. [Google Scholar] [CrossRef]
- Stulnig, T.M. Immunomodulation by polyunsaturated fatty acids: Mechanisms and effects. Int. Arch. Allergy Immunol. 2003, 132, 310–321. [Google Scholar] [CrossRef]
- Zhao, G.; Etherton, T.D.; Martin, K.R.; Gillies, P.J.; West, S.G.; Kris-Etherton, P.M. Dietary α-linolenic acid inhibits proinflammatory cytokine production by peripheral blood mononuclear cells in hypercholesterolemic subjects. Am. J. Clin. Nutr. 2007, 85, 385–391. [Google Scholar]
- Zhao, G.; Etherton, T.D.; Martin, K.R.; West, S.G.; Gillies, P.J.; Kris-Etherton, P.M. Dietary alpha-linolenic acid reduces inflammatory and lipid cardiovascular risk factors in hypercholesterolemic men and women. J. Nutr. 2004, 134, 2991–2997. [Google Scholar]
- Calder, P.C. Polyunsaturated fatty acids and inflammatory processes: New twists in an old tale. Biochimie 2009, 91, 791–795. [Google Scholar] [CrossRef]
- Bray, G.A.; Lovejoy, J.C.; Smith, S.R.; DeLany, J.P.; Lefevre, M.; Hwang, D.; Ryan, D.H.; York, D.A. The influence of different fats and fatty acids on obesity, insulin resistance and inflammation. J. Nutr. 2002, 132, 2488–2491. [Google Scholar]
- Itariu, B.K.; Zeyda, M.; Hochbrugger, E.E.; Neuhofer, A.; Prager, G.; Schindler, K.; Bohdjalian, A.; Mascher, D.; Vangala, S.; Schranz, M.; et al. Long-chain n-3 PUFAs reduce adipose tissue and systemic inflammation in severely obese nondiabetic patients: A randomized controlled trial. Am. J. Clin. Nutr. 2012, 96, 1137–1149. [Google Scholar] [CrossRef]
- Simopoulos, A.P. Omega-3 fatty acids in inflammation and autoimmune diseases. J. Am. Coll. Nutr. 2002, 21, 495–505. [Google Scholar]
- Maury, E.; Noel, L.; Detry, R.; Brichard, S.M. In vitro hyperresponsiveness to tumor necrosis factor-α contributes to adipokine dysregulation in omental adipocytes of obese subjects. J. Clin. Endocrinol. Metab. 2009, 94, 1393–1400. [Google Scholar] [CrossRef]
- Suganami, T.; Nishida, J.; Ogawa, Y. A paracrine loop between adipocytes and macrophages aggravates inflammatory changes: Role of free fatty acids and tumor necrosis factor α. Arterioscler. Thromb. Vasc. Biol. 2005, 25, 2062–2068. [Google Scholar] [CrossRef]
- Dandona, P.; Weinstock, R.; Thusu, K.; Abdel-Rahman, E.; Aljada, A.; Wadden, T. Tumor necrosis factor-α in sera of obese patients: Fall with weight loss. J. Clin. Endocrinol. Metab. 1998, 83, 2907–2910. [Google Scholar] [CrossRef]
- Wang, B.; Trayhurn, P. Acute and prolonged effects of TNF-α on the expression and secretion of inflammation-related adipokines by human adipocytes differentiated in culture. Pflug. Archiv. Eur. J. Physiol. 2006, 452, 418–427. [Google Scholar]
- Bruun, J.M.; Lihn, A.S.; Verdich, C.; Pedersen, S.B.; Toubro, S.; Astrup, A.; Richelsen, B. Regulation of adiponectin by adipose tissue-derived cytokines: In vivo and in vitro investigations in humans. Am. J. Physiol. Endocrinol. Metab. 2003, 285, E527–E533. [Google Scholar]
- Ruan, H.; Hacohen, N.; Golub, T.R.; Van Parijs, L.; Lodish, H.F. Tumor necrosis factor-α suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: Nuclear factor-κB activation by TNF-α is obligatory. Diabetes 2002, 51, 1319–1336. [Google Scholar] [CrossRef]
- Chen, X.; Xun, K.; Chen, L.; Wang, Y. TNF-α, a potent lipid metabolism regulator. Cell Biochem. Funct. 2009, 27, 407–416. [Google Scholar] [CrossRef]
- Madej, A.; Okopien, B.; Kowalski, J.; Zielinski, M.; Wysocki, J.; Szygula, B.; Kalina, Z.; Herman, Z. Levels of tumor necrosis factor alpha in serum of patients with hyperlipoproteinemia IIB before and after micronized fenofibrate therapy. Pol. Arch. Med. Wewn. 1998, 99, 308–313. [Google Scholar]
- Ascer, E.; Bertolami, M.C.; Venturinelli, M.L.; Buccheri, V.; Souza, J.; Nicolau, J.C.; Ramires, J.A.; Serrano, C.V., Jr. Atorvastatin reduces proinflammatory markers in hypercholesterolemic patients. Atherosclerosis 2004, 177, 161–166. [Google Scholar] [CrossRef]
- Marketou, M.E.; Zacharis, E.A.; Nikitovic, D.; Ganotakis, E.S.; Parthenakis, F.I.; Maliaraki, N.; Vardas, P.E. Early effects of simvastatinversus atorvastatin on oxidative stress and proinflammatory cytokines in hyperlipidemic subjects. Angiology 2006, 57, 211–218. [Google Scholar] [CrossRef]
- Popa, C.; Netea, M.G.; Radstake, T.; van der Meer, J.W.; Stalenhoef, A.F.; van Riel, P.L.; Barerra, P. Influence of anti-tumour necrosis factor therapy on cardiovascular risk factors in patients with active rheumatoid arthritis. Ann. Rheum Dis. 2005, 64, 303–305. [Google Scholar] [CrossRef]
- Feingold, K.R.; Grunfeld, C. Role of cytokines in inducing hyperlipidemia. Diabetes 1992, 41 (Suppl. 2), 97–101. [Google Scholar]
- Feingold, K.R.; Soued, M.; Staprans, I.; Gavin, L.A.; Donahue, M.E.; Huang, B.J.; Moser, A.H.; Gulli, R.; Grunfeld, C. Effect of tumor necrosis factor (TNF) on lipid metabolism in the diabetic rat. Evidence that inhibition of adipose tissue lipoprotein lipase activity is not required for TNF-induced hyperlipidemia. J. Clin. Invest. 1989, 83, 1116–1121. [Google Scholar] [CrossRef]
- Bradley, R.L.; Fisher, F.F.; Maratos-Flier, E. Dietary fatty acids differentially regulate production of TNF-α and IL-10 by murine 3T3-L1 adipocytes. Obesity 2008, 16, 938–944. [Google Scholar] [CrossRef]
- Chandrasekar, B.; Fernandes, G. Decreased pro-inflammatory cytokines and increased antioxidant enzyme gene expression by omega-3 lipids in murine lupus nephritis. Biochem. Biophys. Res. Commun. 1994, 200, 893–898. [Google Scholar] [CrossRef]
- Perez-Matute, P.; Perez-Echarri, N.; Martinez, J.A.; Marti, A.; Moreno-Aliaga, M.J. Eicosapentaenoic acid actions on adiposity and insulin resistance in control and high-fat-fed rats: Role of apoptosis, adiponectin and tumour necrosis factor-α. Br. J. Nutr. 2007, 97, 389–398. [Google Scholar] [CrossRef]
- Caughey, G.E.; Mantzioris, E.; Gibson, R.A.; Cleland, L.G.; James, M.J. The effect on human tumor necrosis factor alpha and interleukin 1 β production of diets enriched in n-3 fatty acids from vegetable oil or fish oil. Am. J. Clin. Nutr. 1996, 63, 116–122. [Google Scholar]
- Endres, S.; Ghorbani, R.; Kelley, V.E.; Georgilis, K.; Lonnemann, G.; van der Meer, J.W.; Cannon, J.G.; Rogers, T.S.; Klempner, M.S.; Weber, P.C.; et al. The effect of dietary supplementation with n-3 polyunsaturated fatty acids on the synthesis of interleukin-1 and tumor necrosis factor by mononuclear cells. N. Engl. J. Med. 1989, 320, 265–271. [Google Scholar] [CrossRef]
- Grimble, R.F.; Howell, W.M.; O’Reilly, G.; Turner, S.J.; Markovic, O.; Hirrell, S.; East, J.M.; Calder, P.C. The ability of fish oil to suppress tumor necrosis factor αa production by peripheral blood mononuclear cells in healthy men is associated with polymorphisms in genes that influence tumor necrosis factor α production. Am. J. Clin. Nutr. 2002, 76, 454–459. [Google Scholar]
- Brand, E.; Schorr, U.; Kunz, I.; Kertmen, E.; Ringel, J.; Distler, A.; Sharma, A.M. Tumor necrosis factor-α: 308 G/A polymorphism in obese Caucasians. Int. J. Obes. Relat. Metab. Disord. 2001, 25, 581–585. [Google Scholar]
- Herrmann, S.M.; Ricard, S.; Nicaud, V.; Mallet, C.; Arveiler, D.; Evans, A.; Ruidavets, J.B.; Luc, G.; Bara, L.; Parra, H.J.; et al. Polymorphisms of the tumour necrosis factor-α gene, coronary heart disease and obesity. Eur. J. Clin. Invest. 1998, 28, 59–66. [Google Scholar] [CrossRef]
- Hoffstedt, J.; Eriksson, P.; Hellstrom, L.; Rossner, S.; Ryden, M.; Arner, P. Excessive fat accumulation is associated with the TNF α –308 G/A promoter polymorphism in women but not in men. Diabetologia 2000, 43, 117–120. [Google Scholar] [CrossRef]
- Nieters, A.; Becker, N.; Linseisen, J. Polymorphisms in candidate obesity genes and their interaction with dietary intake of n-6 polyunsaturated fatty acids affect obesity risk in a sub-sample of the EPIC-Heidelberg cohort. Eur. J. Nutr. 2002, 41, 210–221. [Google Scholar] [CrossRef]
- Pihlajamaki, J.; Ylinen, M.; Karhapaa, P.; Vauhkonen, I.; Laakso, M. The effect of the –308A allele of the TNF-α gene on insulin action is dependent on obesity. Obes. Res. 2003, 11, 912–917. [Google Scholar] [CrossRef]
- Rosmond, R.; Chagnon, M.; Bouchard, C.; Bjorntorp, P. G-308A polymorphism of the tumor necrosis factor α gene promoter and salivary cortisol secretion. J. Clin. Endocrinol. Metab. 2001, 86, 2178–2180. [Google Scholar] [CrossRef]
- Sookoian, S.; Garcia, S.I.; Gianotti, T.F.; Dieuzeide, G.; Gonzalez, C.D.; Pirola, C.J. The G-308A promoter variant of the tumor necrosis factor-α gene is associated with hypertension in adolescents harboring the metabolic syndrome. Am. J. Hypertens. 2005, 18, 1271–1275. [Google Scholar] [CrossRef]
- Dalziel, B.; Gosby, A.K.; Richman, R.M.; Bryson, J.M.; Caterson, I.D. Association of the TNF-α –308 G/A promoter polymorphism with insulin resistance in obesity. Obes. Res. 2002, 10, 401–407. [Google Scholar] [CrossRef]
- Wybranska, I.; Malczewska-Malec, M.; Niedbal, S.; Naskalski, J.W.; Dembinska-Kiec, A. The TNF-α gene NcoI polymorphism at position –308 of the promoter influences insulin resistance, and increases serum triglycerides after postprandial lipaemia in familiar obesity. Clin. Chem. Lab. Med. 2003, 41, 501–510. [Google Scholar]
- Weldon, S.M.; Mullen, A.C.; Loscher, C.E.; Hurley, L.A.; Roche, H.M. Docosahexaenoic acid induces an anti-inflammatory profile in lipopolysaccharide-stimulated human THP-1 macrophages more effectively than eicosapentaenoic acid. J. Nutr. Biochem. 2007, 18, 250–258. [Google Scholar] [CrossRef]
- Davis, J.E.; Gabler, N.K.; Walker-Daniels, J.; Spurlock, M.E. Tlr-4 deficiency selectively protects against obesity induced by diets high in saturated fat. Obesity 2008, 16, 1248–1255. [Google Scholar] [CrossRef]
- Garcia-Escobar, E.; Rodriguez-Pacheco, F.; Garcia-Serrano, S.; Gomez-Zumaquero, J.M.; Haro-Mora, J.J.; Soriguer, F.; Rojo-Martinez, G. Nutritional regulation of interleukin-6 release from adipocytes. Int. J. Obes. (Lond.) 2010, 34, 1328–1332. [Google Scholar] [CrossRef]
- Van Dijk, S.J.; Feskens, E.J.; Bos, M.B.; Hoelen, D.W.; Heijligenberg, R.; Bromhaar, M.G.; de Groot, L.C.; de Vries, J.H.; Muller, M.; Afman, L.A. A saturated fatty acid-rich diet induces an obesity-linked proinflammatory gene expression profile in adipose tissue of subjects at risk of metabolic syndrome. Am. J. Clin. Nutr. 2009, 90, 1656–1664. [Google Scholar] [CrossRef]
- He, K.; Liu, K.; Daviglus, M.L.; Jenny, N.S.; Mayer-Davis, E.; Jiang, R.; Steffen, L.; Siscovick, D.; Tsai, M.; Herrington, D. Associations of dietary long-chain n-3 polyunsaturated fatty acids and fish with biomarkers of inflammation and endothelial activation (from the Multi-Ethnic Study of Atherosclerosis [MESA]). Am. J. Cardiol. 2009, 103, 1238–1243. [Google Scholar] [CrossRef]
- Ferrucci, L.; Cherubini, A.; Bandinelli, S.; Bartali, B.; Corsi, A.; Lauretani, F.; Martin, A.; Andres-Lacueva, C.; Senin, U.; Guralnik, J.M. Relationship of plasma polyunsaturated fatty acids to circulating inflammatory markers. J. Clin. Endocrinol. Metab. 2006, 91, 439–446. [Google Scholar]
- Romeo, S.; Sentinelli, F.; Capici, F.; Arca, M.; Berni, A.; Vecci, E.; Di Mario, U.; Baroni, M.G. The G-308A variant of the Tumor Necrosis Factor-alpha (TNF-α) gene is not associated with obesity, insulin resistance and body fat distribution. BMC Med. Genet. 2001, 2, 10. [Google Scholar]
- Um, J.Y.; Park, J.H.; Kim, H.M. Gene polymorphisms in tumor necrosis factor locus and waist-hip ratio in obese Koreans. Clin. Chim. Acta 2003, 338, 117–122. [Google Scholar] [CrossRef]
- Hedayati, M.; Sharifi, K.; Rostami, F.; Daneshpour, M.S.; Zarif Yeganeh, M.; Azizi, F. Association between TNF-α promoter G-308A and G-238A polymorphisms and obesity. Mol. Biol. Rep. 2011, 39, 825–829. [Google Scholar]
- Walston, J.; Seibert, M.; Yen, C.J.; Cheskin, L.J.; Andersen, R.E. Tumor necrosis factor-α-238 and –308 polymorphisms do not associated with traits related to obesity and insulin resistance. Diabetes 1999, 48, 2096–2098. [Google Scholar] [CrossRef]
- Riikola, A.; Sipila, K.; Kahonen, M.; Jula, A.; Nieminen, M.S.; Moilanen, L.; Kesaniemi, Y.A.; Lehtimaki, T.; Hulkkonen, J. Interleukin-6 promoter polymorphism and cardiovascular risk factors: The Health 2000 Survey. Atherosclerosis 2009, 207, 466–470. [Google Scholar] [CrossRef]
- Huth, C.; Illig, T.; Herder, C.; Gieger, C.; Grallert, H.; Vollmert, C.; Rathmann, W.; Hamid, Y.H.; Pedersen, O.H.; Hansen, T.; et al. Joint analysis of individual participants’ data from 17 studies on the association of the IL6 variant –174G > C with circulating glucose levels, interleukin-6 levels, and body mass index. Ann. Med. 2009, 41, 128–138. [Google Scholar] [CrossRef]
- Qi, L.; Zhang, C.; van Dam, R.M.; Hu, F.B. Interleukin-6 genetic variability and adiposity: Associations in two prospective cohorts and systematic review in 26,944 individuals. J. Clin. Endocrinol. Metab. 2007, 92, 3618–3625. [Google Scholar] [CrossRef]
- Joffe, Y.T.; van der Merwe, L.; Evans, J.; Collins, M.; Lambert, E.V.; September, A.; Goedecke, J.H. Genetic polymorphisms in the Interleukin-6 gene and the risk of obesity and dyslipidaemia in black and white South African women. Genes Nutr. 2013. submitted for publication. [Google Scholar]
- Henningsson, S.; Hakansson, A.; Westberg, L.; Baghaei, F.; Rosmond, R.; Holm, G.; Ekman, A.; Nissbrandt, H.; Eriksson, E. Interleukin-6 gene polymorphism –174G/C influences plasma lipid levels in women. Obesity 2006, 14, 1868–1873. [Google Scholar] [CrossRef]
- Fernandez-Real, J.M.; Broch, M.; Vendrell, J.; Richart, C.; Ricart, W. Interleukin-6 gene polymorphism and lipid abnormalities in healthy subjects. J. Clin. Endocrinol. Metab. 2000, 85, 1334–1339. [Google Scholar] [CrossRef]
- Hulkkonen, J.; Lehtimaki, T.; Mononen, N.; Juonala, M.; Hutri-Kahonen, N.; Taittonen, L.; Marniemi, J.; Nieminen, T.; Viikari, J.; Raitakari, O.; et al. Polymorphism in the IL6 promoter region is associated with the risk factors and markers of subclinical atherosclerosis in men: The Cardiovascular Risk in Young Finns Study. Atherosclerosis 2009, 203, 454–458. [Google Scholar] [CrossRef]
- Fontaine-Bisson, B.; Wolever, T.M.; Chiasson, J.L.; Rabasa-Lhoret, R.; Maheux, P.; Josse, R.G.; Leiter, L.A.; Rodger, N.W.; Ryan, E.A.; Connelly, P.W.; et al. Genetic polymorphisms of tumor necrosis factor-αmodify the association between dietary polyunsaturated fatty acids and fasting HDL-cholesterol and apo A–I concentrations. Am. J. Clin. Nutr. 2007, 86, 768–774. [Google Scholar]
- Fontaine-Bisson, B.; El-Sohemy, A. Genetic polymorphisms of tumor necrosis factor-α modify the association between dietary polyunsaturated fatty acids and plasma high-density lipoprotein-cholesterol concentration in a population of young adults. J. Nutrigenet Nutrigenomics 2008, 1, 215–223. [Google Scholar] [CrossRef]
- Corpeleijn, E.; Petersen, L.; Holst, C.; Saris, W.H.; Astrup, A.; Langin, D.; MacDonald, I.; Martinez, J.A.; Oppert, J.M.; Polak, J.; et al. Obesity-related polymorphisms and their associations with the ability to regulate fat oxidation in obese Europeans: The NUGENOB study. Obesity 2010, 18, 1369–1377. [Google Scholar]
- Razquin, C.; Martinez, J.A.; Martinez-Gonzalez, M.A.; Fernandez-Crehuet, J.; Santos, J.M.; Marti, A. A Mediterranean diet rich in virgin olive oil may reverse the effects of the –174G/C IL6 gene variant on 3-year body weight change. Mol. Nutr. Food Res. 2010, 54 (Suppl. 1), 75–82. [Google Scholar] [CrossRef]
- Joffe, Y.T.; van der Merwe, L.; Evans, J.; Collins, M.; Lambert, E.V.; September, A.; Goedecke, J.H.; UCT/MRC Research Unit for Exercise Science and Sports Medicine, University of Cape Town, Cape Town, South Africa. Unpublished work. 2013.
- Curti, M.L.; Jacob, P.; Borges, M.C.; Rogero, M.M.; Ferreira, S.R. Studies of gene variants related to inflammation, oxidative stress, dyslipidemia, and obesity: Implications for a nutrigenetic approach. J. Obes. 2011, 2011, 497401. [Google Scholar] [CrossRef]
- Harkins, J.M.; Moustaid-Moussa, N.; Chung, Y.J.; Penner, K.M.; Pestka, J.J.; North, C.M.; Claycombe, K.J. Expression of interleukin-6 is greater in preadipocytes than in adipocytes of 3T3-L1 cells and C57BL/6J and ob/ob mice. J. Nutr. 2004, 134, 2673–2677. [Google Scholar]
- Eder, K.; Baffy, N.; Falus, A.; Fulop, A.K. The major inflammatory mediator interleukin-6 and obesity. Inflamm. Res. 2009, 58, 727–736. [Google Scholar] [CrossRef]
- Vozarova, B.; Weyer, C.; Hanson, K.; Tataranni, P.A.; Bogardus, C.; Pratley, R.E. Circulating interleukin-6 in relation to adiposity, insulin action, and insulin secretion. Obes. Res. 2001, 9, 414–417. [Google Scholar] [CrossRef]
- Shoelson, S.E.; Herrero, L.; Naaz, A. Obesity, inflammation, and insulin resistance. Gastroenterology 2007, 132, 2169–2180. [Google Scholar] [CrossRef]
- Rexrode, K.M.; Pradhan, A.; Manson, J.E.; Buring, J.E.; Ridker, P.M. Relationship of total and abdominal adiposity with CRP and IL-6 in women. Ann. Epidemiol. 2003, 13, 674–682. [Google Scholar] [CrossRef]
- Bougoulia, M.; Triantos, A.; Koliakos, G. Plasma interleukin-6 levels, glutathione peroxidase and isoprostane in obese women before and after weight loss. Association with cardiovascular risk factors. Hormones 2006, 5, 192–199. [Google Scholar]
- Unger, R.H. Lipotoxicity in the pathogenesis of obesity-dependent NIDDM. Genetic and clinical implications. Diabetes 1995, 44, 863–870. [Google Scholar]
- Inoguchi, T.; Li, P.; Umeda, F.; Yu, H.Y.; Kakimoto, M.; Imamura, M.; Aoki, T.; Etoh, T.; Hashimoto, T.; Naruse, M.; et al. High glucose level and free fatty acid stimulate reactive oxygen species production through protein kinase C—Dependent activation of NAD(P)H oxidase in cultured vascular cells. Diabetes 2000, 49, 1939–1945. [Google Scholar] [CrossRef]
- Lau, D.C.; Dhillon, B.; Yan, H.; Szmitko, P.E.; Verma, S. Adipokines: Molecular links between obesity and atheroslcerosis. Am. J. Physiol. Heart Circ. Physiol. 2005, 288, H2031–H2041. [Google Scholar] [CrossRef]
- Nonogaki, K.; Fuller, G.M.; Fuentes, N.L.; Moser, A.H.; Staprans, I.; Grunfeld, C.; Feingold, K.R. Interleukin-6 stimulates hepatic triglyceride secretion in rats. Endocrinology 1995, 136, 2143–2149. [Google Scholar] [CrossRef]
- Bastard, J.P.; Maachi, M.; Lagathu, C.; Kim, M.J.; Caron, M.; Vidal, H.; Capeau, J.; Feve, B. Recent advances in the relationship between obesity, inflammation, and insulin resistance. Eur. Cytokine Netw. 2006, 17, 4–12. [Google Scholar]
- Stouthard, J.M.; Romijn, J.A.; van der Poll, T.; Endert, E.; Klein, S.; Bakker, P.J.; Veenhof, C.H.; Sauerwein, H.P. Endocrinologic and metabolic effects of interleukin-6 in humans. Am. J. Physiol. 1995, 268, E813–E819. [Google Scholar]
- Humphries, S.E.; Luong, L.A.; Ogg, M.S.; Hawe, E.; Miller, G.J. The interleukin-6 –174 G/C promoter polymorphism is associated with risk of coronary heart disease and systolic blood pressure in healthy men. Eur. Heart J. 2001, 22, 2243–2252. [Google Scholar] [CrossRef]
- Jenny, N.S.; Tracy, R.P.; Ogg, M.S.; Luong, L.A.; Kuller, L.H.; Arnold, A.M.; Sharrett, A.R.; Humphries, S.E. In the elderly, interleukin-6 plasma levels and the –174G > C polymorphism are associated with the development of cardiovascular disease. Arterioscler. Thromb. Vasc. Biol. 2002, 22, 2066–2071. [Google Scholar] [CrossRef]
- Sie, M.P.; Sayed-Tabatabaei, F.A.; Oei, H.H.; Uitterlinden, A.G.; Pols, H.A.; Hofman, A.; van Duijn, C.M.; Witteman, J.C. Interleukin 6 –174 G/C promoter polymorphism and risk of coronary heart disease: Results from the rotterdam study and a meta-analysis. Arterioscler. Thromb. Vasc. Biol. 2006, 26, 212–217. [Google Scholar] [CrossRef]
- Walston, J.D.; Fallin, M.D.; Cushman, M.; Lange, L.; Psaty, B.; Jenny, N.; Browner, W.; Tracy, R.; Durda, P.; Reiner, A. IL-6 gene variation is associated with IL-6 and C-reactive protein levels but not cardiovascular outcomes in the Cardiovascular Health Study. Hum. Genet. 2007, 122, 485–494. [Google Scholar] [CrossRef]
- Joubert, J.; Norman, R.; Bradshaw, D.; Goedecke, J.H.; Steyn, N.P.; Puoane, T. Estimating the burden of disease attributable to excess body weight in South Africa in 2000. S. Afr. Med. J. 2007, 97, 683–690. [Google Scholar]
- Lovejoy, J.C.; de la Bretonne, J.A.; Klemperer, M.; Tulley, R. Abdominal fat distribution and metabolic risk factors: Effects of race. Metab. Clin. Exp. 1996, 45, 1119–1124. [Google Scholar] [CrossRef]
- Goedecke, J.H.; Levitt, N.S.; Evans, J.; Ellman, N.; Hume, D.; Kotze, L.; Tootla, M.; Victor, H.; Keswell, D. The role of adipose tissue in insulin resistance in women of African Ancestry. J. Obes. 2013, 2013, 952916. [Google Scholar] [CrossRef]
- Punyadeera, C.; Crowther, N.J.; van der Merwe, M.T.; Toman, M.; Immelman, A.R.; Schlaphoff, G.P.; Gray, I.P. Metabolic response to a mixed meal in obese and lean women from two South african populations. Obes. Res. 2002, 10, 1207–1216. [Google Scholar] [CrossRef]
- Wells, J.C. Ethnic variability in adiposity, thrifty phenotypes and cardiometabolic risk: Addressing the full range of ethnicity, including those of mixed ethnicity. Obes. Rev. 2012, 13 (Suppl. 2), 14–29. [Google Scholar] [CrossRef]
- Lovejoy, J.C.; Champagne, C.M.; Smith, S.R.; de Jonge, L.; Xie, H. Ethnic differences in dietary intakes, physical activity, and energy expenditure in middle-aged, premenopausal women: The Healthy Transitions Study. Am. J. Clin. Nutr. 2001, 74, 90–95. [Google Scholar]
- Goedecke, J.H.; Jennings, C.; Lambert, E.V. Obesity in South Africa; Medical Research Council: Cape Town, South Africa, 2006; pp. 65–79. [Google Scholar]
- Adeboye, B.; Bermano, G.; Rolland, C. Obesity and its health impact in Africa: A systematic review. Cardiovasc. J. Afr. 2012, 23, 512–521. [Google Scholar] [CrossRef]
- Despres, J.P.; Couillard, C.; Gagnon, J.; Bergeron, J.; Leon, A.S.; Rao, D.C.; Skinner, J.S.; Wilmore, J.H.; Bouchard, C. Race, visceral adipose tissue, plasma lipids, and lipoprotein lipase activity in men and women: The health, risk factors, exercise training, and genetics (HERITAGE) family study. Arterioscler. Thromb. Vasc. Biol. 2000, 20, 1932–1938. [Google Scholar] [CrossRef]
- Punyadeera, C.; van der Merwe, M.T.; Crowther, N.J.; Toman, M.; Schlaphoff, G.P.; Gray, I.P. Ethnic differences in lipid metabolism in two groups of obese South African women. J. Lipid Res. 2001, 42, 760–767. [Google Scholar]
- Sumner, A.E.; Cowie, C.C. Ethnic differences in the ability of triglyceride levels to identify insulin resistance. Atherosclerosis 2008, 196, 696–703. [Google Scholar] [CrossRef]
- Goedecke, J.H.; Utzschneider, K.; Faulenbach, M.V.; Rizzo, M.; Berneis, K.; Spinas, G.A.; Dave, J.A.; Levitt, N.S.; Lambert, E.V.; Olsson, T.; et al. Ethnic differences in serum lipoproteins and their determinants in South African women. Metabolism 2010, 59, 1341–1350. [Google Scholar] [CrossRef]
- Nazare, J.A.; Smith, J.D.; Borel, A.L.; Haffner, S.M.; Balkau, B.; Ross, R.; Massien, C.; Almeras, N.; Despres, J.P. Ethnic influences on the relations between abdominal subcutaneous and visceral adiposity, liver fat, and cardiometabolic risk profile: The international study of prediction of intra-abdominal adiposity and its relationship with cardiometabolic risk/intra-abdominal adiposity. Am. J. Clin. Nutr. 2012, 96, 714–726. [Google Scholar] [CrossRef]
- Corella, D.; Ordovas, J.M. Single nucleotide polymorphisms that influence lipid metabolism: Interaction with dietary factors. Annu. Rev. Nutr. 2005, 25, 341–390. [Google Scholar] [CrossRef]
- Ness, R.B.; Haggerty, C.L.; Harger, G.; Ferrell, R. Differential distribution of allelic variants in cytokine genes among African Americans and White Americans. Am. J. Epidemiol. 2004, 160, 1033–1038. [Google Scholar] [CrossRef]
- Evans, J.; Goedecke, J.H.; Soderstrom, I.; Buren, J.; Alvehus, M.; Blomquist, C.; Jonsson, F.; Hayes, P.M.; Adams, K.; Dave, J.A.; et al. Depot- and ethnic-specific differences in the relationship between adipose tissue inflammation and insulin sensitivity. Clin. Endocrinol. 2011, 74, 51–59. [Google Scholar] [CrossRef]
- Martin, A.M.; Athanasiadis, G.; Greshock, J.D.; Fisher, J.; Lux, M.P.; Calzone, K.; Rebbeck, T.R.; Weber, B.L. Population frequencies of single nucleotide polymorphisms (SNPs) in immuno-modulatory genes. Hum. Hered. 2003, 55, 171–178. [Google Scholar] [CrossRef]
- Patel, J.V.; Tracey, I.; Hughes, E.A.; Lip, G.Y. Omega-3 polyunsaturated acids and cardiovascular disease: Notable ethnic differences or unfulfilled promise? J. Thromb. Haemost. 2010, 8, 2095–2104. [Google Scholar] [CrossRef]
- Lu, Y.; Feskens, E.J.; Dolle, M.E.; Imholz, S.; Verschuren, W.M.; Muller, M.; Boer, J.M. Dietary n-3 and n-6 polyunsaturated fatty acid intake interacts with FADS1 genetic variation to affect total and HDL-cholesterol concentrations in the Doetinchem Cohort Study. Am. J. Clin. Nutr. 2010, 92, 258–265. [Google Scholar]
- Mathias, R.A.; Fu, W.; Akey, J.M.; Ainsworth, H.C.; Torgerson, D.G.; Ruczinski, I.; Sergeant, S.; Barnes, K.C.; Chilton, F.H. Adaptive evolution of the FADS gene cluster within Africa. PLoS One 2012, 7, e44926. [Google Scholar]
- Sergeant, S.; Hugenschmidt, C.E.; Rudock, M.E.; Ziegler, J.T.; Ivester, P.; Ainsworth, H.C.; Vaidya, D.; Case, L.D.; Langefeld, C.D.; Freedman, B.I.; et al. Differences in arachidonic acid levels and fatty acid desaturase (FADS) gene variants in African Americans and European Americans with diabetes or the metabolic syndrome. Br. J. Nutr. 2012, 107, 547–555. [Google Scholar] [CrossRef]
- 1000 Genomes. Available online: http://browser.1000genomes.org (accessed on 1 February 2013).
- Flicek, P.; Amode, M.R.; Barrell, D.; Beal, K.; Brent, S.; Carvalho-Silva, D.; Clapham, P.; Coates, G.; Fairley, S.; Fitzgerald, S.; et al. Ensembl 2012. Nucl. Acids Res. 2012, 40, D84–D90. [Google Scholar] [CrossRef]
- Caslake, M.J.; Miles, E.A.; Kofler, B.M.; Lietz, G.; Curtis, P.; Armah, C.K.; Kimber, A.C.; Grew, J.P.; Farrell, L.; Stannard, J.; et al. Effect of sex and genotype on cardiovascular biomarker response to fish oils: The FINGEN Study. Am. J. Clin. Nutr. 2008, 88, 618–629. [Google Scholar]
- Ordovas, J.M.; Corella, D.; Cupples, L.A.; Demissie, S.; Kelleher, A.; Coltell, O.; Wilson, P.W.; Schaefer, E.J.; Tucker, K. Polyunsaturated fatty acids modulate the effects of the APOA1 G-A polymorphism on HDL-cholesterol concentrations in a sex-specific manner: The Framingham Study. Am. J. Clin. Nutr. 2002, 75, 38–46. [Google Scholar]
- Phillips, C.M.; Goumidi, L.; Bertrais, S.; Field, M.R.; McManus, R.; Hercberg, S.; Lairon, D.; Planells, R.; Roche, H.M. Dietary saturated fat, gender and genetic variation at the TCF7L2 locus predict the development of metabolic syndrome. J. Nutr. Biochem. 2012, 23, 239–244. [Google Scholar] [CrossRef]
- Phillips, C.M.; Goumidi, L.; Bertrais, S.; Field, M.R.; McManus, R.; Hercberg, S.; Lairon, D.; Planells, R.; Roche, H.M. Gene-nutrient interactions and gender may modulate the association between ApoA1 and ApoB gene polymorphisms and metabolic syndrome risk. Atherosclerosis 2011, 214, 408–414. [Google Scholar] [CrossRef]
© 2013 by the authors; licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution license (http://creativecommons.org/licenses/by/3.0/).
Share and Cite
Joffe, Y.T.; Collins, M.; Goedecke, J.H. The Relationship between Dietary Fatty Acids and Inflammatory Genes on the Obese Phenotype and Serum Lipids. Nutrients 2013, 5, 1672-1705. https://doi.org/10.3390/nu5051672
Joffe YT, Collins M, Goedecke JH. The Relationship between Dietary Fatty Acids and Inflammatory Genes on the Obese Phenotype and Serum Lipids. Nutrients. 2013; 5(5):1672-1705. https://doi.org/10.3390/nu5051672
Chicago/Turabian StyleJoffe, Yael T., Malcolm Collins, and Julia H. Goedecke. 2013. "The Relationship between Dietary Fatty Acids and Inflammatory Genes on the Obese Phenotype and Serum Lipids" Nutrients 5, no. 5: 1672-1705. https://doi.org/10.3390/nu5051672